

Abstract
Amplification of human papillomaviruses (HPV) is dependent on the ATM DNA damage pathway. In cells with impaired p53 activity, DNA damage repair requires the activation of p38MAPK along with MAPKAP kinase 2 (MK2). In HPV-positive cells, phosphorylation of p38 and MK2 proteins was induced along with relocalization to the cytoplasm. Treatment with MK2 or p38 inhibitors blocked HPV genome amplification, identifying the p38/MK2 pathway as a key regulator of the HPV life cycle.
TEXT
Human papillomaviruses (HPV) infect stratified epithelium and induce a variety of lesions (1). A subset of HPV types, referred to as high risk, are the causative agents of cervical and other anogenital malignancies as well as many oral cancers (2–4). Papillomaviruses infect basal epithelial cells of stratified epithelium that become exposed through microwounds and establish a latent infection, in which viral genomes are maintained in the nucleus as low-copy-number episomes. As infected basal cells divide and one daughter cell leaves the basal layer, the cell differentiation program is initiated, which results in activation of viral gene expression and replication. HPV genomes do not encode DNA polymerases or replication factors except for the DNA helicase E1, and viral replication is dependent largely on host factors (5). It is, therefore, necessary for HPV-positive cells to retain the ability to reenter S/G2 upon differentiation (6). This process is mediated through the action of the E6 and E7 proteins that modulate the function of p53, Rb, and a number of other cell cycle regulators (7–9). In addition to maintaining cells active in the cell cycle, HPV genome amplification requires activation of the ATM (ataxia telengiectasia mutated) DNA damage pathway (10–14).
The ATM pathway is responsible for the DNA damage response (DDR) to double-strand DNA breaks and is mediated through the action of downstream kinases, such as CHK2 (15). The ATR pathway is activated by single-strand breaks as well as replication fork collapse and functions through CHK1 (16). While these two pathways generally act independently of each other, some overlap exists, such as when one pathway is deficient or compromised. A third DDR pathway has recently been described in cells with reduced or impaired p53 activity, and this pathway is mediated by the p38MAPK kinases (17–21). The p38MAPK pathway is activated in response to a variety of stress-induced signals, including DNA damage, osmotic shock, or cytokine signaling. p38MAPK phosphorylates a number of downstream effectors, such as c-Myc, c-Jun, and ATF2, but it specifically induces DDR through the phosphorylation of mitogen-activated protein (MAP) kinase-activated protein kinase 2 (MAPKAPK 2, or MK2) (20, 22–26). Activation of MK2 leads to phosphorylation of a series of downstream targets that results in G2/M arrest and DNA repair (21). While the DNA repair portion of the p38MAPK/MK2 pathway is activated by ATM or ATR kinases, it functions independently and in parallel to the activities of CHK1 and CHK2 (19, 21). Furthermore, p38MAPK/MK2 has been shown to be important for DDR in cells that have impaired p53 function, such as U2OS, HeLa, or immortalized fibroblasts with diminished p53 function (18). MK2 is relocalized to the cytoplasm upon activation, while activated CHK1 and CHK2 are retained in the nucleus (18). Keratinocytes that stably maintain high-risk HPV genomes have reduced levels of p53 through the action of E6/E6AP complexes, as well as altered levels of acetylated p53 through E6 modulation of p300 activity (8). We therefore investigated if p38MAPK/MK2 played any role in the genome amplification of HPV-positive cells.
To determine what effect, if any, the p38/MK2 pathway plays in HPV replication, we first examined the levels and activation status of p38 and MK2 in normal and HPV-positive human keratinocytes in undifferentiated cells grown as monolayer cultures. For this analysis, the levels of p38/MK2 proteins were examined in a series of HPV-positive human keratinocyte cell lines that were generated by transfection with recircularized genomes from HPV-16, -18, and -31, as previously described (27), as well as a biopsy sample-derived cell line that is HPV-31 positive, CIN-612. All these HPV-positive lines stably maintain episomal copies of HPV genomes in undifferentiated monolayer cultures. As shown in Fig. 1A, the levels of total p38 were similar in both HPV-positive and normal keratinocyte cells. In contrast, the phosphorylated form of p38 (p-p38) was only detected in HPV-positive cells. Slightly increased levels of total MK2 were seen in HPV-positive cells, with minimal levels of phosphorylated MK2 (p-MK2) detected in undifferentiated cultures of either normal or HPV-positive cells.
FIG 1.
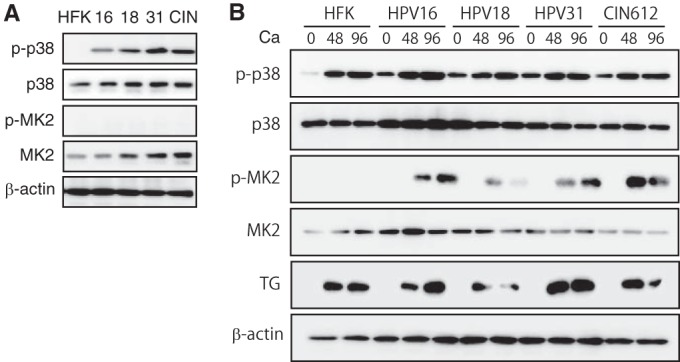
p-38 and p-MK2 levels are increased in HPV-positive cells. (A) Western blot analysis for p-p38, p38, p-MK2, and MK2 in undifferentiated monolayer cultures of normal human foreskin keratinocytes (HFK), HPV-16, HPV-18, HPV-31, and CIN-612 cells. The HPV-16, -18, and -31 cells were generated by transfection of HFKs with recircularized HPV genomes as previously described (11, 27), followed by selection for a cotransfected drug resistance marker. β-Actin was used as a loading control. The following antibodies were used: p-p38(T180/Y182, D3F9; catalog number 4511), p38(D13E1; catalog number 8690), p-MK2(T334, 27B7; catalog number 3007), and MK2 (catalog number 3042) (all from Cell Signaling Technologies, San Diego, CA). Secondary antibodies included horseradish peroxidase-linked anti-rabbit (Santa Cruz Biotechnology, Santa Cruz, CA). Levels of p-p38, p38, p-MK2, and MK2 following differentiation in high-calcium medium for 0, 48, and 96 h were determined by Western blotting. Calcium-induced differentiation was performed as described previously (27). The 0-h data represent results for undifferentiated cells. TG (transglutaminase) was used as a differentiation marker. β-Actin was used as a loading control.
It was next important to investigate if the levels of p38/MK2 proteins were altered upon differentiation. For this analysis, we used calcium-induced differentiation, as this method allows for isolation of uniformly differentiated populations of cells (27). Amplification of viral genomes begins within 48 h and plateaus by 96 h after the calcium switch. In both normal and HPV-positive keratinocytes, the levels of phosphorylated p38 increased upon differentiation, while the total amounts were largely unchanged (Fig. 1B). Slightly higher levels of MK2 total proteins were seen in HPV-positive cells and remained unchanged upon differentiation. In contrast to the minimal levels p-MK2 observed in undifferentiated HPV-positive cells, significant induction of p-MK2 was seen upon differentiation. No activation of p-MK2 was seen in either undifferentiated or differentiated normal keratinocytes (Fig. 1B). This indicated that phosphorylation of MK2 is induced upon differentiation of HPV-positive cells.
We previously demonstrated that the ATM DNA damage pathway is constitutively activated in HPV-positive cells and that it plays a critical role in controlling amplification upon differentiation (11). To investigate if ATM activation is linked to p38/MK2 activation in HPV-positive cells, the levels of ATM and its phosphorylated form (p-ATM) were examined by Western blotting. Consistent with previous studies, total ATM levels were relatively unchanged between normal and HPV-positive cells, while p-ATM was detected only in HPV-positive cells (Fig. 2A). The ATM inhibitor KU-55933 blocks phosphorylation of ATM along with its downstream targets and is specific (28). To determine if ATM was responsible for activating phosphorylation of MK2, HPV-positive cells were induced to differentiate in high-calcium medium and treated with KU-55933, and the levels of phosphorylated MK2 were examined by Western analysis. As seen in Fig. 2B, treatment of cells with KU-55933 blocked phosphorylation of p-p38 without affecting total levels of p38. Similarly, phosphorylation of MK2 was blocked following treatment of HPV-positive cells that had been induced to differentiate (Fig. 2B). Treatment of cells with KU-55933 also inhibited p-ATM (Fig. 2C). This demonstrated that phosphorylation of both p38 and MK2 is dependent on ATM in HPV-positive cells. Cells were also treated with an inhibitor of p38 (Fig. 2D), and MK2 phosphorylation was significantly reduced, demonstrating a dependence on p-p38 activity. Neither the p38 nor MK2 inhibitors had any effect on the expression of transglutaminase, a marker of epithelial differentiation (Fig. 2C) or proliferation (data not shown).
FIG 2.

The ATM pathway is activated in HPV-positive cells, and addition of ATM inhibitors blocks induction of p-p38 and p-MK2. (A) Levels of total and phosphorylated p-ATM upon differentiation of HPV-positive cells as well as HFKs. Cells were seeded as monolayer cultures, followed by the addition of medium containing 1.5 mM Ca2+ for 96 h (27), as previously described (11). Extracts were harvested at the indicated times and examined by Western blotting using the corresponding antibodies. Anti-ATM (S1981, D6H9) was obtained from Cell Signaling Technologies. (B) Levels of p-p38, p38, p-MK2, and MK2 following differentiation in high-calcium medium in the presence or absence of an ATM inhibitor. Undifferentiated cells were grown as monolayer cultures in the presence or absence of 5 μM ATM inhibitor KU-55933, and protein levels were determined by Western blotting. Lanes designated with Ca were induced to differentiate by the addition of high-calcium medium for 96 h with or without the presence of 5 μM KU-55933, and protein levels were examined by Western blotting. (C) Levels of phosphorylated ATM and total ATM following treatment with an ATM inhibitor. (D) Levels of p-MK2 in the absence or presence of a p38 inhibitor. Cells were seeded as monolayer cultures (left) or exposed to medium containing 1.5 mM Ca2+ for 96 h (right) (11). The cells were left untreated or incubated with 10 μM p-p38 inhibitor SB203580 for 96 h. Extracts were harvested and examined by Western blotting using the corresponding antibodies. TG (transglutaminase) was used as a differentiation marker. β-Actin was used as a loading control.
Upon DNA damage, the p38/MK2 complex relocalizes from the nucleus to the cytoplasm to target a series of factors, some of which regulate the stability of a number of mRNAs (18, 29). We therefore investigated the cellular localization of p-MK2 during the differentiation of HPV-positive cells by using immunofluorescence with cells induced to differentiate in high-calcium medium. As shown in Fig. 3, very low levels of p-MK2 were found in undifferentiated cells. Upon differentiation, we observed induction of MK2 phosphorylation and localization of p-MK2 to the cytoplasm (Fig. 3). Similar localization was seen with p-p38, which formed a complex with p-MK2. One target of cytoplasmic p-MK2 kinase activity is the chaperone protein HSP-27 (30, 31), and we observed via Western blotting that it became phosphorylated upon differentiation of HPV-positive cells (Fig. 4). Furthermore, the addition of MK2 inhibitors blocked phosphorylation of HSP-27 (Fig. 4). This confirmed that p-MK2 is active in differentiated HPV-positive cells. Many of the factors described as MK2 targets, including HSP-27, contribute to cell cycle arrest in G2/M (32) and work in parallel with factors targeted by the nuclear DNA damage kinases CHK1 and CHK2 (19).
FIG 3.
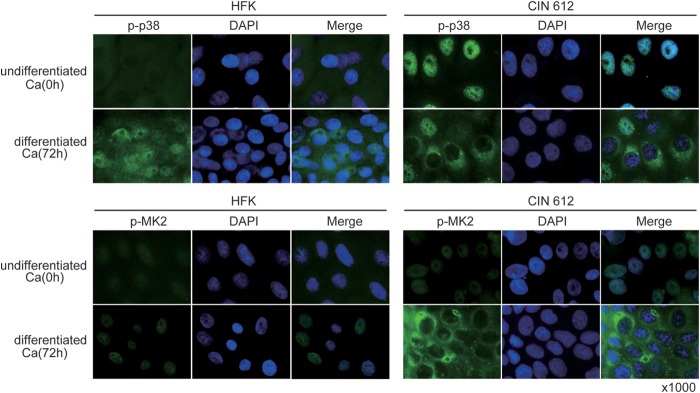
Immunofluoresence of p-p38 and p-MK2 in HPV-positive cells upon differentiation. Undifferentiated cells were stained for p-p38 or p-MK2 using appropriate antibodies. 4′,6-Diamidino-2-phenylindole (DAPI) staining was used to identify nuclei (ProLong Gold antifade reagent with DAPI; catalog number P36931; Life Technologies, Thermo Fisher Scientific, Waltham, MA). Induction and localization of p-MK2 as well as of p-p38 to the cytoplasm following Ca2+-induced differentiation for 72 h is shown. Normal human keratinocytes (HFK) and HPV-31-positive CIN 612 cells were examined.
FIG 4.
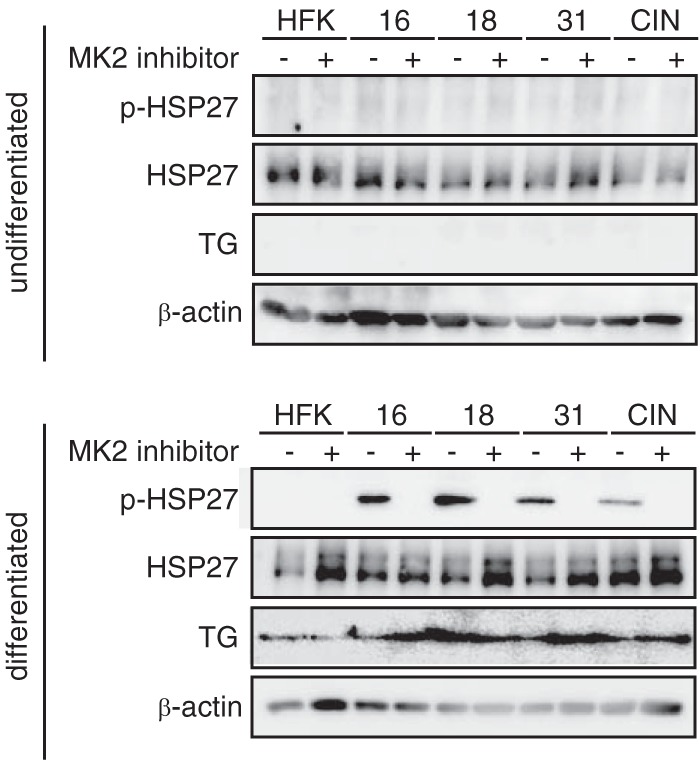
HSP27 phosphorylation is induced by p-MK2 upon differentiation. Cell lysates of HPV-16, HPV-18, HPV-31, and CIN 612 cells were induced to differentiate in high-calcium medium, and levels of p-HSP27 as well as total HSP-27 were examined by Western blotting. Cells were treated with 10 μM MK2 inhibitor (MK2 inhibitor III), and loss of p-HSP27 was observed. Antibodies used were anti-p-HSP-27 and anti-HSP27 (both from Cell Signaling Technologies). TG (transglutaminase) was used as a differentiation marker. β-Actin was used as a loading control.
Our results indicate the p38/MK2 pathway is activated upon differentiation of HPV-positive cells but not normal cells. Next, it was important to investigate if the p38/MK2 pathway had any role in the regulation of HPV replication in undifferentiated and differentiated cells. In undifferentiated HPV-positive cells, treatment with either p38 or MK2 inhibitors showed no effect on genome copy number (Fig. 5A), indicating the p38/MK2 pathway is not involved in the stable maintenance of HPV genomes, even though ATM and p38 are activated. Upon calcium-induced differentiation, only about a quarter of cells amplify viral DNA, and the total amount of HPV DNA is increased (11). When HPV-positive cells were differentiated in the presence of either a p38 or MK2 inhibitor, genome amplification was significantly suppressed (Fig. 5B and C). Similar effects were seen in multiple independent experiments. Treatment of cells with these inhibitors did not affect the phosphorylation of ATM (data not shown). These results indicated that the p38/MK2 pathway is activated in HPV-positive cells and plays an important role in the differentiation-specific genome amplification of HPV.
FIG 5.
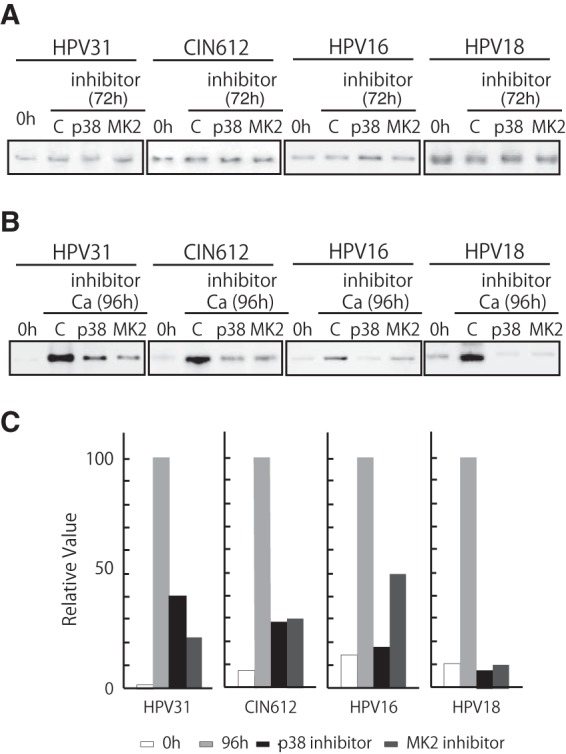
Inhibition of p38 or MK2 blocks HPV genome amplification. (A) Southern blot analysis of undifferentiated HPV-31 cells, CIN 612 cells, HPV-16 cells, and HPV-18 cells treated with 10 μM p38 inhibitor (SB203580) or 10 μM MK2 inhibitor (MK2 inhibitor III). Cells were treated for 72 h prior to analysis. The 0-h data are results without inhibitor; C designates results after 72-h treatment with DMSO vehicle alone. p38 and MK2 indicate the inhibitor used. Viral episomes are shown. (B) Southern analysis was performed as previously described (41), using the digoxigenin (DIG)-High Prime DNA labeling and detection system (Roche Diagnostics, Mannheim, Germany). The chemiluminescent signal was visualized by using a chemiluminescent image analyzer (Fc Imager; Odyssey, Lincoln, NE) to visualize DNA. Southern blot analysis results are shown for HPV-positive cells that stably maintained viral episomes treated and untreated with inhibitors of p38 or MK2 during differentiation induced by high-calcium medium for 96 h. The viral episomes are shown. (C) Quantification of the effect on amplification shown in panel B. The signal strengths were quantified by using ImageJ software (NIH, Bethesda, MD), and the results are shown in the bar chart. Each relative value was determined based on setting the signal strength of 96-h incubation with calcium as 100.
Our studies identified a novel role of the p38/MK2 pathway in the control of HPV amplification. The p38/MK2 pathway was found to be activated in HPV-positive cells upon differentiation, and it was observed to be critical for genome amplification. MK2 is a Ser/Thr kinase that is regulated through phosphorylation by p38 MAPK and ATM. The MK2 kinase has been reported to be involved in many cellular processes, including stress and inflammatory responses (33, 34), nuclear export (29), gene expression regulation (33, 35, 36), and cell proliferation (37). Recently, the p38/MK2 pathway was identified as an alternative pathway in the DNA damage response (17–21). One of the important downstream effectors of the DDR is the p53 tumor suppressor protein, whose activation can mediate cell cycle arrest to repair DNA damage or to induce apoptotic cell death (38). In p53-deficient cells, p38/MK2 functions as a third member of the DDR pathway (21). In our studies, we determined that MK2 phosphorylation is specifically induced upon differentiation of HPV-positive cells. As such, it is one of only three DNA damage factors that have been identified to be induced upon differentiation. In addition to p-MK2, γ-H2AX and p-NBS1 are two members of the ATM pathway (39) whose activation increases upon differentiation; this contrasts with ATM and CHK2, which are activated in both differentiated and undifferentiated cells (11). Since the DDR has no role in stable or transient HPV replication but only affects differentiation-dependent genome amplification, MK2 along with γ-H2Ax and NBS1 are likely to be critical regulators of this process.
The MK2 pathway activates the genome amplification of HPV but not stable maintenance replication, which is consistent with our observation that active MK2 kinases are only detected upon differentiation. MK2 has a broad range of downstream effectors and is involved in various cellular events. Cytoplasmic MK2 activity is critical for checkpoint maintenance, and it acts in part by stabilizing a number of mRNAs (5), so it is possible that MK2 plays a role in regulating the stabilities of a subset of viral transcripts. MK2 also phosphorylates several transcriptional factors, such as SRF and CREB (40), whose function might be critical for regulating the late viral promoter. One known target of p-MK2 is the heat shock protein HSP27 (30, 31), and in our studies we observed phosphorylation of HSP27 upon differentiation of HPV-positive cells that was dependent upon p-MK2 action. What role HSP27 plays in the HPV life cycle will be a focus of future studies. Finally, MK2 appears to act in parallel to CHK1 and CHK2 to activate DNA repair factors that are necessary for HPV genome amplification. Given the different cellular localizations of MK2 and CHK1/CHK2, these kinases seem to act coordinately to induce the full spectrum of DNA damage factors.
ACKNOWLEDGMENTS
This work was supported by a grant to L.L. from the National Cancer Institute (CA142861).
We thank the many colleagues who provided technical assistance and were involved in manuscript preparation.
REFERENCES
- 1.Stubenrauch F, Laimins LA. 1999. Human papillomavirus life cycle: active and latent phases. Semin Cancer Biol 9:379–386. doi: 10.1006/scbi.1999.0141. [DOI] [PubMed] [Google Scholar]
- 2.Durst M, Gissmann L, Ikenberg H, zur Hausen H. 1983. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci U S A 80:3812–3815. doi: 10.1073/pnas.80.12.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.zur Hausen H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 4.Leemans CR, Braakhuis BJ, Brakenhoff RH. 2011. The molecular biology of head and neck cancer. Nat Rev Cancer 11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 5.Kajitani N, Satsuka A, Kawate A, Sakai H. 2012. Productive lifecycle of human papillomaviruses that depends upon squamous epithelial differentiation. Front Microbiol 3:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longworth MS, Laimins LA. 2004. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev 68:362–372. doi: 10.1128/MMBR.68.2.362-372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 8.Moody CA, Laimins LA. 2010. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 9.Helt AM, Funk JO, Galloway DA. 2002. Inactivation of both the retinoblastoma tumor suppressor and p21 by the human papillomavirus type 16 E7 oncoprotein is necessary to inhibit cell cycle arrest in human epithelial cells. J Virol 76:10559–10568. doi: 10.1128/JVI.76.20.10559-10568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reinson T, Toots M, Kadaja M, Pipitch R, Allik M, Ustav E, Ustav M. 2013. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J Virol 87:951–964. doi: 10.1128/JVI.01943-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5:e1000605. doi: 10.1371/journal.ppat.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kadaja M, Isok-Paas H, Laos T, Ustav E, Ustav M. 2009. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog 5:e1000397. doi: 10.1371/journal.ppat.1000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J. 2011. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J Virol 85:8996–9012. doi: 10.1128/JVI.00542-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallace NA, Galloway DA. 2014. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin Cancer Biol 26:30–42. doi: 10.1016/j.semcancer.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavin MF. 2008. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 16.Flynn RL, Zou L. 2011. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci 36:133–140. doi: 10.1016/j.tibs.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morandell S, Reinhardt HC, Cannell IG, Kim JS, Ruf DM, Mitra T, Couvillon AD, Jacks T, Yaffe MB. 2013. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep 5:868–877. doi: 10.1016/j.celrep.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MA, Wang X, Linding R, Ong SE, Weaver D, Carr SA, Yaffe MB. 2010. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol Cell 40:34–49. doi: 10.1016/j.molcel.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reinhardt HC, Yaffe MB. 2009. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 21:245–255. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB. 2005. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell 17:37–48. doi: 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 21.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. 2007. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11:175–189. doi: 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waas WF, Lo HH, Dalby KN. 2001. The kinetic mechanism of the dual phosphorylation of the ATF2 transcription factor by p38 mitogen-activated protein (MAP) kinase alpha. Implications for signal/response profiles of MAP kinase pathways. Int J Biol Sci 276:5676–5684. doi: 10.1074/jbc.M008787200. [DOI] [PubMed] [Google Scholar]
- 23.Marderosian M, Sharma A, Funk AP, Vartanian R, Masri J, Jo OD, Gera JF. 2006. Tristetraprolin regulates cyclin D1 and c-Myc mRNA stability in response to rapamycin in an Akt-dependent manner via p38 MAPK signaling. Oncogene 25:6277–6290. doi: 10.1038/sj.onc.1209645. [DOI] [PubMed] [Google Scholar]
- 24.Thornton TM, Rincon M. 2009. Non-classical p38 MAP kinase functions: cell cycle checkpoints and survival. Int J Biol Sci 5:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cannell IG, Kong YW, Johnston SJ, Chen ML, Collins HM, Dobbyn HC, Elia A, Kress TR, Dickens M, Clemens MJ, Heery DM, Gaestel M, Eilers M, Willis AE, Bushell M. 2010. p38 MAPK/MK2-mediated induction of miR-34c following DNA damage prevents Myc-dependent DNA replication. Proc Natl Acad Sci U S A 107:5375–5380. doi: 10.1073/pnas.0910015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loesch M, Zhi HY, Hou SW, Qi XM, Li RS, Basir Z, Iftner T, Cuenda A, Chen G. 2010. p38γ MAPK cooperates with c-Jun in trans-activating matrix metalloproteinase 9. J Biol Chem 285:15149–15158. doi: 10.1074/jbc.M110.105429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fehrmann F, Laimins LA. 2005. Human papillomavirus type 31 life cycle: methods for study using tissue culture models. Methods Mol Biol 292:317–330. doi: 10.1385/1-59259-848-x:317. [DOI] [PubMed] [Google Scholar]
- 28.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 29.Engel K, Kotlyarov A, Gaestel M. 1998. Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J 17:3363–3371. doi: 10.1093/emboj/17.12.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rane MJ, Coxon PY, Powell DW, Webster R, Klein JB, Pierce W, Ping P, McLeish KR. 2001. p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J Biol Chem 276:3517–3523. doi: 10.1074/jbc.M005953200. [DOI] [PubMed] [Google Scholar]
- 31.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR. 1994. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 32.Gaestel M. 2006. MAPKAP kinases – MKs - two's company, three's a crowd. Nat Rev Mol Cell Biol 7:120–130. doi: 10.1038/nrm1834. [DOI] [PubMed] [Google Scholar]
- 33.Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. 1999. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J 18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. 1999. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol 1:94–97. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- 35.McCormick C, Ganem D. 2005. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 307:739–741. doi: 10.1126/science.1105779. [DOI] [PubMed] [Google Scholar]
- 36.Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW. 2004. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J Biol Chem 279:10176–10184. doi: 10.1074/jbc.M310486200. [DOI] [PubMed] [Google Scholar]
- 37.Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J. 1997. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci 110:357–368. [DOI] [PubMed] [Google Scholar]
- 38.Sengupta S, Harris CC. 2005. p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol 6:44–55. doi: 10.1038/nrm1546. [DOI] [PubMed] [Google Scholar]
- 39.Xu Y, Xu C, Price BD. 2012. Mechanistic links between ATM and histone methylation codes during DNA repair. Prog Mol Biol Transl Sci 110:263–288. doi: 10.1016/B978-0-12-387665-2.00010-9. [DOI] [PubMed] [Google Scholar]
- 40.Heidenreich O, Neininger A, Schratt G, Zinck R, Cahill MA, Engel K, Kotlyarov A, Kraft R, Kostka S, Gaestel M, Nordheim A. 1999. MAPKAP kinase 2 phosphorylates serum response factor in vitro and in vivo. J Biol Chem 274:14434–14443. doi: 10.1074/jbc.274.20.14434. [DOI] [PubMed] [Google Scholar]
- 41.Satsuka A, Yoshida S, Kajitani N, Nakamura H, Sakai H. 2010. Novel human papillomavirus type 18 replicon and its application in screening the antiviral effects of cytokines. Cancer Sci 101:536–542. doi: 10.1111/j.1349-7006.2009.01411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]