

Abstract
To compare entire genomes from different species, biologists increasingly need alignment methods that are efficient enough to handle long sequences, and accurate enough to correctly align the conserved biological features between distant species. We present LAGAN, a system for rapid global alignment of two homologous genomic sequences, and Multi-LAGAN, a system for multiple global alignment of genomic sequences. We tested our systems on a data set consisting of greater than 12 Mb of high-quality sequence from 12 vertebrate species. All the sequence was derived from the genomic region orthologous to an ∼1.5-Mb region on human chromosome 7q31.3. We found that both LAGAN and Multi-LAGAN compare favorably with other leading alignment methods in correctly aligning protein-coding exons, especially between distant homologs such as human and chicken, or human and fugu. Multi-LAGAN produced the most accurate alignments, while requiring just 75 minutes on a personal computer to obtain the multiple alignment of all 12 sequences. Multi-LAGAN is a practical method for generating multiple alignments of long genomic sequences at any evolutionary distance. Our systems are publicly available at http://lagan.stanford.edu.
Comparing genomic sequences across related species is a fruitful source of biological insight, because functional elements such as exons tend to exhibit significant sequence similarity, whereas regions that are not functional tend to be less conserved. The first step in comparing genomic sequences is to align them—that is, to map the letters of one sequence to those of the others. There are several categories of alignments: local alignments that identify local similarities between regions of each sequence, and global alignments that find a monotonically increasing map between the letters of each sequence; pairwise alignments that compare two sequences, and multiple alignments that compare several sequences.
Local pairwise alignment methods such as Smith-Waterman (1981), BLAST (Altschul et al. 1990, 1997), BLASTZ (Schwartz et al. 2000), SSAHA (Ning et al. 2001), and BLAT (Kent 2002) are able to pinpoint locations of rearrangements between two sequences, and are suitable for aligning draft sequences or individual reads. Global alignments are important because they reveal the shared order of biological features in the compared species, and produce a more accurate alignment at the base-pair level when the features are in the same order. The best-known global alignment algorithm is Needleman-Wunsch (1970), which requires time proportional to the product of the lengths of the aligned sequences. Unfortunately this algorithm is too inefficient for comparing long genomic sequences. Faster methods have been developed recently: DIALIGN (Morgenstern et al. 1998, Brudno and Morgenstern 2002), MUMmer (Delcher et al. 1999, 2002), GLASS (Batzoglou et al. 2000), WABA (Kent and Zahler 2000), and AVID (Bray et al. 2003). Most of these methods have proven effective in aligning genomic sequences from two closely related organisms, such as human and mouse or Caenorhabditis elegans and C. briggsae, but have not been tested in alignments between distant relatives such as human and fugu.
Multiple alignments, a natural extension of two-sequence comparisons, are a powerful way to study biological sequences. Even weak similarity across several sequences usually reveals an important conserved biological feature (Dubchak et al. 2000; Göttgens et al. 2002). Moreover, multiple alignments enable the computation of local rates of evolution, giving a quantitative measure of the strength of evolutionary constraints and the functional importance of local regions (Simon et al. 2002). Multiple alignments are considerably more difficult to compute than are pairwise alignments: the running time scales as the product of the lengths of all the sequences. Formally, the problem is NP-complete (Wang and Jiang 1994; Bonizzoni and Vedova 2001). For this reason heuristic approaches are usually applied, of which the most widely used is progressive alignment, which constructs a multiple alignment by successive applications of a pairwise alignment algorithm. The best-known system based on progressive alignment is perhaps CLUSTALW (Thompson et al. 1994). Some other systems include MULTALIGN (Barton and Sternberg 1987), MULTAL (Taylor 1988), YAMA (Hardison et al. 1993, 1994), and PRRP (Gotoh 1996). DIALIGN (Morgenstern 1999) does not use progressive alignment; instead it uses another heuristic approach to chain local conserved blocks between several sequences into a multiple alignment. These systems can effectively align proteins and relatively short genomic regions, but are not efficient enough to align entire genomes. MGA (Höhl et al. 2002) is a rapid multiple aligner suitable for comparing very close homologs, such as different strains of a bacterium, but is not designed to align distant homologs.
Here we describe novel systems for pairwise and multiple alignment of genomic sequences: LAGAN (Limited Area Global Alignment of Nucleotides), an efficient and reliable pairwise aligner that is suitable for genomic comparison of distantly related organisms, and MLAGAN (Multi-LAGAN), a multiple aligner based on progressive alignment with LAGAN. We tested our systems on sequence from 12 species generated for the genomic segment harboring the cystic fibrosis transmembrane conductance regulator (CFTR) gene (J.W. Thomas, J.W. Touchman, R.W. Blakesley, G.G. Bouffard, S.M. Beckstrom-Sternberg, E.H. Margulies, M. Blanchette, A.C. Siepel, P.J. Thomas, J.C. McDowell, B. Maskeri, N.F. Hansen, M.S. Schwartz, R.J. Weber, W.J. Kent, D. Karolchik, T.C. Bruen, R. Bevan, D.J. Cutler, S. Schwartz, L. Elnitski, J.R. Idol, A.B. Prasad, S.-Q. Lee-Lin, V.V.B. Maduro, M.E. Portnoy, N.L. Dietrich, N. Akhter, K. Ayele, B. Benjamin, K. Cariaga, C.P. Brinkley, S.Y. Brooks, S. Granite, X. Guan, J. Gupta, P. Haghighi, S-L. Ho, M.C. Huang, E. Karlins, P.L. Laric, R. Legaspi, M.J. Lim, Q.L. Maduro, C.A. Masiello, S.D. Mastrian, J.C. McCloskey, R. Pearson, S. Stantripop, E.E. Tiongson, J.T. Tran, C. Tsurgeon, J.L. Vogt, M.A. Walker, K.D. Wetherby, L.S. Wiggins, A.C. Young, L-H. Zhang, K. Osoegawa, B. Zhu, B. Zhao, C.L. Shu, P.J. De Jong, C.E. Lawrence, A.F. Smit, A. Chakravarti, D. Haussler, P. Green, W. Miller, and E.D. Green, in prep.). Based on comparisons with other available alignment programs and benchmarking on standard desktop computer systems, we conclude that LAGAN and MLAGAN are practical and reliable methods for large-scale pairwise and multiple genomic alignment that should prove useful for obtaining alignments of the entire human, mouse, fugu, rat, and other genomes in the context of a whole-genome alignment pipeline.
RESULTS
Outline of Algorithms
LAGAN is a global alignment system that aligns two genomic sequences in three main steps: (1) generation of local alignments between the two sequences, (2) construction of a rough global map, by chaining an ordered subset of the local alignments, and (3) computation of the final global alignment, by finding the best alignment that stays within a limited area around the rough global map. Based on LAGAN, we developed MLAGAN, a new multiple alignment system that aligns genomic sequences in two main phases: (1) a progressive alignment phase that constructs a multiple alignment by successively aligning two sequences, or intermediate multiple alignments, with the LAGAN algorithm, and (2) an optional iterative improvement phase that successively removes each sequence from the multiple alignment, and realigns it to the rest of the alignment, until no significant improvements are observed.
Global Sequence Alignment, LAGAN
In aligning two long genomic sequences, the Needleman-Wunsch algorithm is too inefficient because it requires time proportional to the product of the sequence lengths. Efficient global aligners such as MUMmer (Delcher et al. 1999, 2002), GLASS (Batzoglou et al. 2000), and AVID (Bray et al. 2003) rely on anchoring. First, they detect local similarities between the two sequences. Second, they select and fix an ordered set of local similarities, the anchors. Finally, they align the interleaving regions.
Anchoring reduces computation time because the initial large alignment problem is subdivided into many smaller, manageable ones. To perform anchoring reliably, the aligner needs a sensitive local-alignment detection method, an accurate procedure for selecting the anchors, and an efficient method for computing the global alignment based on the anchors. Anchoring is easier when the two sequences are highly similar and contain frequent matching words. Existing global aligners based on anchoring are primarily designed for comparing highly similar sequences, such as human and mouse. MUMmer, GLASS, and AVID, for instance, use exact matching words for detecting local alignments.
LAGAN performs anchoring with techniques designed to work well on distant, as well as close organisms. Specifically, LAGAN uses the CHAOS algorithm (Brudno and Morgenstern 2002), a highly sensitive method that detects local alignments using multiple short inexact words instead of longer exact words. LAGAN then constructs the global alignment by first applying CHAOS recursively in areas with sparse anchors, so that each consecutive pair of anchors is separated by a distance smaller than a given maximum, and then performing a limited-area dynamic programming algorithm on the area around the anchors.
LAGAN aligns two sequences in three steps outlined next, and described in Methods.
Algorithm LAGAN
Given a pair of sequences X, Y: A global alignment between the two sequences is a path from the top-left to the bottom-right corner of the alignment matrix X × Y (Fig. 1A)
Step 1.
Generation of local alignments. Compute the local alignments between the two sequences (Fig. 1B). Assign a weight to each local alignment.
Step 2.
Construction of rough global map. Two local alignments can be chained if the end of one precedes the start of the other in both X and Y. Compute the highest-weight chain of local alignments, using the Longest Increasing Subsequence algorithm (Fig. 1C). Call each local alignment in the chain an anchor. Apply Steps 1, 2 recursively between every pair of anchors that are more than a threshold distance apart.
Step 3.
Computation of global alignment. Define a subset of the cells in the alignment matrix:
1. Include all cells at distance at most r from cells of the anchor.
2. Between each consecutive pair of anchors, where the first ends at positions (i, j) and the second begins at positions (k, l) in X, Y, respectively, include the full alignment box (Xi…Xk) × (Yj…Yl) between the two endpoints (Fig. 1D).
Compute the global alignment using Needleman-Wunsch-like dynamic programming within the subset of the cells defined above.
Figure 1.

The LAGAN algorithm. (A) A global alignment between two sequences is a path between the top-left and the bottom-right corner of their alignment matrix. (B) LAGAN first finds all local alignments between the two sequences. (C) LAGAN computes a maximal-scoring ordered subset of the alignments, the anchors, and puts together a rough global map. (D) LAGAN limits the search for an optimal alignment to the area included in the boxes and around the anchors, and computes the optimal Needleman-Wunsch alignment limited to that area. LAGAN uses memory proportional to the area of the largest box plus the memory to hold the optimal alignment.
Multiple Alignment, MLAGAN
MLAGAN is based on progressive alignment: A multiple alignment of K sequences is constructed in K–1 pairwise alignment steps, where in each step two sequences, or intermediate multiple alignments, are aligned. MLAGAN uses LAGAN as the pairwise-alignment subroutine, and introduces new methods for scoring a multiple alignment with affine gaps, and for refining a multiple alignment.
The multiple aligner CLUSTALW infers a phylogenetic tree of the sequences, using hierarchical clustering. Inferring such a tree is useful when aligning protein families with several members within one genome. On the other hand, when aligning orthologous genomic regions from different species (e.g., human, baboon, mouse, rat, and chicken)—which is the problem that we are targeting—the phylogenetic tree is usually known. Accordingly, MLAGAN assumes that the phylogenetic tree is given.
MLAGAN aligns K sequences in three main steps that are outlined next. Step 1, the generation of rough global maps, uses Steps 1 and 2 of LAGAN. Steps 2 and 3 are described in Methods.
Algorithm MLAGAN
Given K sequences X1,…, XK, and a phylogenetic binary tree between them.
Step 1.
Generation of rough global maps. Find the rough global map between each pair of sequences.
Step 2.
Progressive multiple alignment with anchors.
2.1
Perform a global alignment between the two closest sequences according to the phylogenetic tree, using LAGAN.
2.2
Each alignment between two or more sequences produced in step 2 is a new multi-sequence. Find the rough global maps of this multi-sequence to all other multi-sequences. The details of this procedure and of the scoring metric are described in Methods.
2.3
Iterate Steps 2.1 and 2.2, performing at every step a global alignment between the two closest (multi)- sequences, according to the phylogenetic tree. Repeat until left with a multiple alignment of all sequences.
Step 3
(Optional). Iterative refinement with anchors. For each sequence Xi in the multiple alignment:
3.1
Find segments of Xi that align better than a given cutoff, in the existing multiple alignment. These segments are the anchors between Xi and the other sequences.
3.2
Align Xi to the multiple alignment of the other sequences with LAGAN.
Evaluation of Performance
We tested the performance of LAGAN and MLAGAN against that of leading global and local alignment programs, by measuring the ability of each aligner to correctly align protein-coding exons in orthologous sequences. We used the following two datasets: (1) the ROSETTA set (Batzoglou et al. 2000), which contains 129 orthologous annotated genes with complete intron sequences between human and mouse of average length 10 Kbp, and (2) the CFTR region (J.W. Thomas, J.W. Touchman, R.W. Blakesley, G.G. Bouffard, S.M. Beckstrom-Sternberg, E.H. Margulies, M. Blanchette, A.C. Siepel, P.J. Thomas, J.C. McDowell, B. Maskeri, N.F. Hansen, M.S. Schwartz, R.J. Weber, W.J. Kent, D. Karolchik, T.C. Bruen, R. Bevan, D.J. Cutler, S. Schwartz, L. Elnitski, J.R. Idol, A.B. Prasad, S.-Q. Lee-Lin, V.V.B. Maduro, M.E. Portnoy, N.L. Dietrich, N. Akhter, K. Ayele, B. Benjamin, K. Cariaga, C.P. Brinkley, S.Y. Brooks, S. Granite, X. Guan, J. Gupta, P. Haghighi, S-L. Ho, M.C. Huang, E. Karlins, P.L. Laric, R. Legaspi, M.J. Lim, Q.L. Maduro, C.A. Masiello, S.D. Mastrian, J.C. McCloskey, R. Pearson, S. Stantripop, E.E. Tiongson, J.T. Tran, C. Tsurgeon, J.L. Vogt, M.A. Walker, K.D. Wetherby, L.S. Wiggins, A.C. Young, L-H. Zhang, K. Osoegawa, B. Zhu, B. Zhao, C.L. Shu, P.J. De Jong, C.E. Lawrence, A.F. Smit, A. Chakravarti, D. Haussler, P. Green, W. Miller, and E.D. Green, in prep.), which for the studies described here consisted of 12 orthologous sequences from human, chimpanzee, baboon, cat, dog, cow, pig, mouse, rat, chicken, fugu, and zebrafish; these ranged in length from 160 Kbp to 1.8 Mbp, with an average of 1 Mbp. For the CFTR region sequences, we used the human gene annotations to identify the orthologous exons in the other 11 species' sequences. This was done on the unaligned sequences before any of the tests were conducted. We first aligned each known human exon to the other sequences using TBLASTN and filtered out all hits that were <50% identical at the amino-acid level or covered <50% of the human exon. Then we designated the best match for each species as the orthologous exon. Using this method, we annotated 75 or more exons in all species, except chicken (24) and zebrafish (34), whose sequences were grossly incomplete. We recorded the positions of the exons for use in the tests. After producing the alignments of the entire region, we tested whether the exons were correctly aligned: For each exon, we calculated the fraction of its length aligned to the corresponding exon in the orthologous sequences.
Tables 1 and 2 summarize the results for the ROSETTA and CFTR datasets, respectively. In the ROSETTA set, we tested the performance of MUMmer, GLASS, DIALIGN, AVID, BLASTZ, and LAGAN. In the CFTR set, we did not test GLASS or DIALIGN because these methods require too much computation time to align longer sequences. We tested the performance of MLAGAN by projecting the multiple alignment to the 11 pairwise alignments between the human and the other species' sequences. Of the pairwise aligners tested, LAGAN, MLAGAN, and BLASTZ were the most accurate at aligning the close (mammalian) sequences. LAGAN and MLAGAN were the best in aligning distant homologs. MLAGAN was slower than the other aligners, but was also the most accurate.
Table 1.
Performance of Aligners on the ROSETTA Dataset of 1160 Total Exons in Human and Mouse
| Aligner | 100% exons | 90% exons | 70% exons | Time (sec) |
| DIALIGN | 89 | 96 | 98 | 388 |
| MUMmer | 0 | 1 | 3 | 17 |
| GLASS | 91 | 97 | 98 | 154 |
| AVID | 90 | 95 | 97 | 19 |
| BlastZ | 94 | 97 | 98 | 17 |
| LAGAN | 94 | 97 | 98 | 48 |
Columns show the percentage of exons annotated in human that are aligned to the orthologous mouse exon over at least 70%, 90%, and 100% of their length, and the time required to align the 129 sequences.
Table 2.
Performance of Aligners on the CFTR Region
| Baboon | Chimpanzee | Mouse | Rat | Cow | Pig | Cat | Dog | Chicken | Zebrafish | Fugu | Overall | |
| Number of exons | 232 | 176 | 230 | 230 | 224 | 174 | 176 | 182 | 68 | 48 | 150 | 1914 |
| MUMmer | ||||||||||||
| 100% | 100 | 99 | 6 | 7 | 28 | 32 | 38 | 28 | 0 | 0 | 0 | 36 |
| 90%–100% | 100 | 100 | 8 | 9 | 40 | 44 | 47 | 37 | 0 | 0 | 0 | 41 |
| 70%–100% | 100 | 100 | 14 | 16 | 52 | 55 | 56 | 45 | 0 | 0 | 0 | 47 |
| AVID | ||||||||||||
| 100% | 100 | 100 | 94 | 95 | 98 | 97 | 99 | 93 | 66 | 33 | 19 | 88 |
| 90%–100% | 100 | 100 | 98 | 100 | 99 | 100 | 100 | 98 | 79 | 42 | 21 | 91 |
| 70%–100% | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 99 | 85 | 44 | 29 | 92 |
| BlastZ | ||||||||||||
| 100% | 100 | 100 | 97 | 97 | 96 | 97 | 100 | 94 | 96 | 73 | 66 | 94 |
| 90%–100% | 100 | 100 | 100 | 100 | 98 | 100 | 100 | 99 | 97 | 79 | 72 | 97 |
| 70%–100% | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 99 | 97 | 79 | 80 | 98 |
| LAGAN | ||||||||||||
| 100% | 100 | 100 | 97 | 97 | 98 | 97 | 100 | 94 | 96 | 83 | 72 | 95 |
| 90%–100% | 100 | 100 | 100 | 100 | 99 | 100 | 100 | 99 | 99 | 88 | 77 | 98 |
| 70%–100% | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 99 | 100 | 92 | 81 | 98 |
| MLAGAN | ||||||||||||
| 100% | 100 | 100 | 97 | 97 | 98 | 97 | 100 | 94 | 99 | 88 | 73 | 96 |
| 90%–100% | 100 | 100 | 100 | 100 | 99 | 100 | 100 | 99 | 100 | 98 | 84 | 98 |
| 70%–100% | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 99 | 100 | 100 | 90 | 99 |
| Time (sec) | ||||||||||||
| MUMmer | 8 | 6 | 7 | 7 | 8 | 6 | 6 | 6 | 3 | 2 | 2 | 61 |
| AVID | 82 | 57 | 215 | 221 | 165 | 111 | 139 | 131 | 83 | 53 | 76 | 1775 |
| BlastZ | 31 | 24 | 46 | 43 | 40 | 36 | 34 | 33 | 7 | 5 | 6 | 305 |
| LAGAN | 56 | 50 | 78 | 82 | 60 | 68 | 62 | 78 | 338 | 158 | 133 | 1135 |
| MLAGAN | — | — | — | — | — | — | — | — | — | — | — | 4547 |
| Max memory (MB) | ||||||||||||
| MUMmer | 40 | 39 | 40 | 40 | 40 | 39 | 39 | 39 | 39 | 38 | 38 | 40 |
| AVID | 598 | 551 | 581 | 584 | 578 | 498 | 522 | 502 | 387 | 340 | 360 | 598 |
| BlastZ | 239 | 276 | 202 | 212 | 204 | 200 | 208 | 206 | 188 | 185 | 185 | 276 |
| LAGAN | 90 | 90 | 90 | 89 | 90 | 87 | 88 | 87 | 88 | 87 | 89 | 90 |
| MLAGAN | — | — | — | — | — | — | — | — | — | — | — | 670 |
The annotated human exons were aligned with TBLASTN to create pseudo-annotations of exons in the other organisms. The table reports the percentage of exons covered by alignments over at least 70%, 90%, and 100% of their length, and the time and memory required to obtain the alignments. The last column reports the percentages of exons aligned out of total number of exons, the total time required for the 11 alignments (for the single 12-sequence multiple alignment in the case of MLAGAN), and the maximum memory required.
LAGAN and MLAGAN can be applied with a translated anchoring option, according to which one or more of the local-alignment-finding steps (the calls to CHAOS during LAGAN Step 1) are performed on the translated amino-acid level, rather than on the nucleotide level (as explained in Methods). We tested the performance of translated anchoring on the CFTR dataset. Table 3 summarizes the performance of LAGAN and MLAGAN, when applied under our default translated anchoring parameters (see Methods for details on the parameters). LAGAN's performance changes in the more distant alignments: it worsens in human/chicken, and improves significantly in human/fugu, and human/zebrafish. MLAGAN's performance is only affected in the human/fugu alignment, where it improves. Overall the performance improves when translated anchoring is used, and this option may be significant when aligning very distant species such as human/fugu. However, translated anchoring in principle biases the aligners to perform best in detecting homologies in protein-coding regions, and consequently could perform worse in non-coding regions. For that reason, we disabled translated anchoring in the default parameters of the programs.
Table 3.
Changes in Performance of LAGAN and MLAGAN on the CFTRDataset When Translated Anchoring Is Enabled
| Alignment | Aligner | 100% | 90%+ | 70%+ | Time diff. |
| Human/chicken | LAGAN | −2% | −3% | −3% | +25% |
| MLAGAN | 0% | 0% | 0% | — | |
| Human/fugu | LAGAN | +6% | +8% | +8% | +93% |
| MLAGAN | +4% | +3% | +1% | — | |
| Human/zebrafish | LAGAN | +7% | +6% | +4% | +29% |
| MLAGAN | 0% | 0% | 0% | — | |
| Total for all 11 | LAGAN | +1% | 0% | +1% | +60% |
| alignments | MLAGAN | 0% | +1% | 0% | +66% |
We report the differences in the percentage of exons correctly aligned with translated anchoring vs. with the default options where translated anchoring is disabled. No changes were observed in the alignments between mammalian sequences.
In anchoring-based global alignment, accuracy of the final alignment depends on the accuracy of the anchors. We found that the majority of human exons are covered correctly during the anchoring phase (calls to CHAOS, Steps 1 and 2 of LAGAN; Table 4).
Table 4.
Percentage of Exons Correctly Aligned During the CHAOS-Based Anchoring Phase of LAGAN (Steps 1,2)
| Alignment human vs. | 90%–100% | 50%–89% | 10%–49% | 0%–9% | ||||
| default | transl. | default | transl. | default | transl. | default | transl. | |
| Chimpanzee | 97 | 97 | 3 | 3 | 0 | 0 | 0 | 0 |
| Baboon | 77 | 77 | 21 | 21 | 3 | 3 | 0 | 0 |
| Cow | 21 | 21 | 43 | 43 | 36 | 36 | 0 | 0 |
| Pig | 19 | 19 | 33 | 30 | 47 | 50 | 1 | 1 |
| Dog | 20 | 20 | 37 | 37 | 41 | 42 | 2 | 1 |
| Cat | 20 | 20 | 41 | 41 | 39 | 39 | 0 | 0 |
| Mouse | 10 | 10 | 35 | 37 | 52 | 51 | 2 | 2 |
| Rat | 13 | 10 | 47 | 46 | 40 | 42 | 1 | 2 |
| Chicken | 9 | 9 | 26 | 41 | 62 | 47 | 3 | 3 |
| Fugu | 12 | 29 | 19 | 29 | 57 | 35 | 12 | 7 |
| Zebrafish | 27 | 33 | 29 | 42 | 38 | 25 | 4 | 0 |
Percentage of human exons that have a given proportion of their lengths covered (90%–100%, 50%–89%, 10%–49%, and 0%–9%). Translated anchoring performs better at covering a greater proportion of exons. However, we do not have measurements on whether translated anchoring performs worse in covering biologically significant noncoding features.
Pairwise alignments produced by LAGAN can be visualized using the VISTA (Mayor et al. 2000) program. Multiple alignments produced by MLAGAN can also be visualized with VISTA, by projecting them into the pairwise alignments with respect to one reference sequence. For example, Figure 2A shows the VISTA plot of a multiple alignment between human, chimpanzee, cow, mouse, chicken, and fugu, projected into the human/chimpanzee, human/cow, human/mouse, human/chicken, and human/fugu alignments.
Figure 2.

Visualization of a multiple alignment using VISTA. (A) MLAGAN alignments can be visualized using VISTA, if they are projected to pairwise alignments with respect to one reference sequence. This plot shows the conservation between human and chimpanzee, cow, mouse, and fugu around the first intron of the cMet gene. The human/chimpanzee conservation is uniformly very high; human/cow and human/mouse show varying levels of conservation. The human/chicken alignment also shows some conservation in the non-coding areas. The human/fugu alignment shows conservation only within the first coding exon, and to a lesser degree within the regions upstream and downstream of that exon. (B) First introns of cMet, comparison of CLUSTALW and MLAGAN alignments. We compared the alignment generated by LAGAN and CLUSTALW for the first intron of the cMet gene in eight mammalian sequences (human, baboon, cat, dog, cow, pig, mouse, and rat). The alignments between all of the species except rodents were similar. VISTA plots of the projections to human and mouse are shown. CLUSTALW (top) misaligned the mouse sequence around 4 Kb and 10 Kb, whereas MLAGAN (bottom) found significant conservation in these regions.
We compared the alignment generated by LAGAN and CLUSTALW for the first intron of the cMet gene in eight mammalian sequences (human, baboon, cat, dog, cow, pig, mouse, and rat). Although the alignments between all of the species besides rodents were similar, CLUSTALW misaligned the mouse sequence around 4 kb and 10 kb, as is shown in Figure 2B. CLUSTALW required ∼40 minutes to compute the eight species alignment after it generated the guide tree, whereas MLAGAN required ∼2 minutes. We did not compare the performance of MLAGAN and CLUSTALW in longer sequences, because the running time of CLUSTALW scales quadratically with sequence length.
MLAGAN's accuracy in aligning known exons permits identification of potential misannotations, and can provide evidence for an alternative annotation. For example, consider exon 10 of the cMet gene, as annotated by RefSeq as well as Ensembl and Twinscan gene predictions. According to our alignment, the RefSeq-annotated start position is not well conserved among the mammals analyzed (Fig. 3). There is also a large gap (∼250 positions) in the alignment after the first 20 nucleotides of this exon, generated by sequence in cat that has no clear homolog in any of the other species aligned. However, a consensus splice acceptor site and the start position of an Acembly gene prediction exon (exon 9 in Met.f and Met.e, exon 10 in Met.a and Met.h), beginning 54 nucleotides downstream of the RefSeq exon 10, is neatly conserved across all the mammals studied. Additionally, no human ESTs correspond to the RefSeq annotation, whereas all ESTs (as annotated in the UCSC genome browser) align precisely with the Acembly gene-prediction exon. The comparative information provided by MLAGAN thus complements the experimental evidence from the human ESTs in identifying this as a potential misannotation.
Figure 3.

Multiple alignment of a misannotated exon of the cMet gene. According to the MLAGAN alignment, the RefSeq-annotated start position of the exon is not well conserved among the mammals. There is also a large gap (∼250 positions) in the alignment after the first 20 nucleotides of this exon, generated by sequence in cat that has no clear homolog in any of the other species aligned. However, a consensus splice acceptor site and the start position of an Acembly gene prediction exon (exon 9 in Met.f and Met.e, exon 10 in Met.a and Met.h), beginning 54 nucleotides downstream of the RefSeq exon 10, is neatly conserved across all the mammals studied. Additionally, no human ESTs correspond to the RefSeq annotation, whereas all ESTs (as annotated in the UCSC genome browser) align precisely with the Acembly gene prediction exon.
Finally, we investigated whether the availability of genomic sequence of intermediate distances can help align distant sequences correctly. We applied MLAGAN to the human, fugu, and mouse sequences of the CFTR region, and compared the projected human/fugu alignment with the corresponding alignment produced by LAGAN. The MLAGAN alignment resulted in a 3% increase in the number of exons perfectly aligned (from 72%–75%), and a 6% increase in the number of exons aligned over at least 70% of their length (from 81%–87%), compared to the LAGAN human/fugu alignment. The well-aligning regions of human and mouse effectively attract the conserved regions in fugu, making it more likely that those will be correctly aligned to human.
In summary, our results demonstrate that LAGAN and MLAGAN are capable of efficiently solving difficult multiple sequence alignment problems.
DISCUSSION
Sequence alignment is one of the oldest and most successful applications of Computer Science to Biology. Despite the considerable advances achieved after several decades of research in this area, many important challenges remain (Miller 2001). One of these challenges is the development of systems for aligning multiple long genomic sequences efficiently and reliably. This problem is becoming increasingly important as several entire genomes from related species become available.
MLAGAN is suitable for high-throughput reliable multiple alignment of genomic sequences. MLAGAN is based on LAGAN, which is designed for rapid and reliable alignment of a pair of genomic sequences that may be very similar (e.g., human and chimpanzee), or very distant (e.g., human and fugu). Both programs are available for use on a public web server, http://lagan.stanford.edu, and are compatible with the VISTA visualization tool. The source code for LAGAN and MLAGAN is available from the authors.
LAGAN and MLAGAN assume that one has already identified apparent orthologous regions between two species, and that there are no genomic rearrangements. Identifying orthologous regions is challenging in the context of an automatic pipeline for whole-genome alignment. While genome rearrangements have been researched at the level of genes (Sankoff 1999), aligning sequences in the presence of rearrangements is a direction for future research.
An interesting question in research on sequence comparisons has been whether local or global alignment algorithms are more appropriate for a given application. When genomic sequences come in several unordered fragments, or when genomic rearrangements are frequent, local alignments are more appropriate, and less likely to miss true conservation. At the same time, genomes such as human, mouse, rat, and even fugu, share large blocks of essentially uninterrupted synteny. As the number of sequences increases, pairwise local alignments may be increasingly difficult to reconcile into an overall picture of conservation. We believe that multiple global alignments of orthologous blocks provide a much cleaner picture of the genome evolution, and will become increasingly important as the number of available genomes increases.
Our results suggest that multiple alignments are better than pairwise alignments at aligning conserved exons between distant species. For that reason, a three-way global alignment of the human, mouse, and fugu genomes may be a good way to obtain accurate alignments of the human and fugu genes.
Some of the most promising methods for annotating biological features such as genes and regulatory elements are based on two-species sequence comparisons. Such annotation methods use sequence conservation, plus the coincidence of signature of the biological feature in the two species, as signals to detect the feature. Characteristic signatures such as open reading frames and splice-site consensus sequences in genes, nested nucleotide complementarities in non-coding RNA genes, and motifs in regulatory elements, should be much easier to recognize whenever they are conserved across multiple species. Multiple-comparison-based annotation methods may prove powerful in annotating biological features within a multiple alignment.
Multiple sequence alignment will become an increasingly important tool for biological discovery as several genomes suitable for cross-species comparison become available. Our systems should enable researchers to align long orthologous regions from several species, and design multiple alignment pipelines on a whole-genome scale.
METHODS
Global Pairwise Alignment, LAGAN
LAGAN aligns a pair of genomic sequences in three main steps: (1) generation of local alignments, (2) construction of a rough global map, and (3) computation of the final global alignment. These three steps are described in detail next.
Generation of Local Alignments
To generate local alignments between the two sequences, LAGAN uses CHAOS, a method that finds local homologies between two sequences, and chains them into a rough global map (Brudno and Morgenstern 2002). Instead of CHAOS, any efficient local alignment method can be used (details on how to incorporate a different local aligner into LAGAN can be found in our website, http://lagan.stanford.edu). Here, we summarize CHAOS and describe rapid rescoring, an improvement we have made to this algorithm.
The CHAOS algorithm works by chaining short words, the seeds, which match between the two sequences. Given a word length k, and a degeneracy c, a (k, c)-seed is a pair of k-long words (k-mers) that match with at most c differences between the two sequences. Given a maximum distance d, and maximum shift s, two seeds that are x- and y-letters apart in the first and second sequences, respectively, can be chained together if x ≤ d, y ≤ d, and |x − y| ≤ s. A seed is chained to the single previous seed that creates the highest scoring chain among all chains that end with this seed. CHAOS also supports a translation option, in which both nucleic sequences are translated in all six coding frames (three forward and three reverse), and all combinations of frames are compared in turn. Amino acids are grouped (Stanfel 1996), and all amino acids in the same group are considered equal.
Scoring of Chains
After computing the maximal chains, CHAOS scores each chain by using match and mismatch penalties for the letters of each seed, and gap penalties proportional to |x − y| for each pair of chained seeds. CHAOS throws away chains that score below some threshold t. We augment this scoring method, by adding a rapid rescoring step: Chains that score below t are immediately thrown away. Chains that score above t are rescored by performing ungapped extensions in both directions from each seed, and finding the optimal location to insert exactly one gap of size |x − y|. If the alignment is done on amino acid sequences, the rescoring is done using a BLOSUM (Henikoff and Henikoff 1992) matrix.
Construction of a Rough Global Map
LAGAN orders the local alignments produced by CHAOS into a rough global map (Fig. 1B,C). A local alignment is a vector (b, e, b‘, e‘, s), representing begin and end positions of the alignment in each sequence, and the score of the alignment. Given two local alignments A1 = (b1, e1, b1‘, e1‘, s1), A2 = (b2, e2, b2‘, e2‘, s2), we say A1 < A2 if and only if e1 < b2 and e1′ < b2′. A chain of local alignments A1 < A2 < … < Ak, has score s1 + s2 + … + sk. The optimal rough global map is the highest-scoring chain, which can be computed using Sparse Dynamic Programming in time O(n log n) where n is the total number of local alignments (Eppstein et al. 1992).
Recursive Anchoring
When computing the local alignments with CHAOS, the choice of parameters k (length of seeds), c (maximum degeneracy of seeds), and t (score threshold) presents a tradeoff between speed (more restrictive parameters: higher k, lower c), and sensitivity to detecting all significant alignments (more permissive parameters: lower k, higher c, lower t). LAGAN uses a recursive method similar to the one used in GLASS (Batzoglou et al. 2000), to achieve a combination of speed and sensitivity. During a recursive anchoring step, LAGAN calls CHAOS with a restrictive set of parameters, computes a rough global map based on the resulting local alignments after the latter are rescored, and then recursively calls CHAOS with a more permissive set of parameters in the regions between each anchor of the global map.
Translated Anchoring
As an option, some of the recursive anchoring steps can be translated. This option biases the aligner to perform best in protein-coding regions and potentially worse in other conserved regions, and therefore as a default translated anchoring is disabled.
Computation of Global Alignment
To compute the final global alignment, LAGAN uses the rough global map to limit the search area of dynamic programming. In standard Needleman-Wunsch, the search area consists of a (M+1) × (N+1) matrix, where M and N are the lengths of sequences X and Y, respectively. The algorithm produces the optimal global alignment, which is the highest-scoring path from cell (0, 0) to cell (M, N); each cell in the path is greater than the previous cell, by 1 in the x coordinate, y coordinate, or both.
The Needleman-Wunsch algorithm evaluates the optimal intermediate alignment score for all (M+1) × (N+1) cells in the alignment matrix, and therefore requires time O(MN). If, however, it is known that the alignment passes through a point (i, j), then there is no need to evaluate cells in the rectangles from (i+1, 0) to (M, j−1), and from (0, j+1) to (i−1, N). LAGAN uses this idea: for every anchor in the rough global map, starting at (i, j) and ending at (i‘, j‘), LAGAN limits the computation of Needleman-Wunsch within the following three areas (where r is a parameter, typically 15): (1) the rectangle (0, 0) to (i + r, j + r), (2) the rectangle (i‘ − r, j‘ − r) to (M, N), (3) the band enclosed by the two diagonals from (i − r, j + r) to (i‘ − r, j‘ +r), and from (i + r, j − r) to (i‘ + r, j‘ − r; Fig. 4). In this sense the anchors in LAGAN are more flexible than the anchors in MUMmer, AVID, and GLASS; in these programs, the anchors are fixed, whereas in LAGAN they provide only approximate locations by which the alignment should pass.
Figure 4.

Limited area of dynamic programming around an anchor. An anchor between (i, j) and (i‘, j‘) in the rough global alignment limits the search area of Needleman-Wunsch. The alignment is required to pass through the diagonal from (i − r, j + r) to (i + r, j − r), stay within the diagonals from (i − r, j + r) to (i‘ − r, j‘ + r) and from (i + r, j − r) to (i‘ + r, j‘ − r), and exit through the diagonal from (i‘ − r, j‘ + r) to (i‘ + r, j‘ − r).
Memory-efficient Implementation
The search area of the limited-area dynamic programming step consists of necks that enclose the anchors, and rectangles that connect two consecutive anchors (Fig. 1C). In practice, LAGAN performs the entire computation with memory proportional to the size of the largest rectangle. LAGAN achieves this memory efficiency as follows: First, it allocates working memory for one rectangle and the neck that follows it, and computes the Needleman-Wunsch matrix within this rectangle and neck. Second, LAGAN traces back all optimal alignments ending in the cells at the rightmost column of the neck; in practice, these alignments quickly converge upon a single optimal alignment. Finally LAGAN deallocates all working memory, except the memory necessary to keep the traced-back alignments. These three steps are then repeated for the next rectangle and neck.
Running Time Analysis
The running time of LAGAN is dominated by the total number of cells in the rectangles between consecutive anchors. The number of cells in the necks of constant width r is O[r · (M + N)] and is linear in the sequence lengths.
Suppose the anchoring step yielded n anchors, and let (x0, y0),…,(xn, yn) be the dimensions of each of the n + 1 rectangles. Let
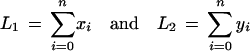 |
denote the total length of the inter-anchor segments in each sequence. At this stage we are effectively working with sequences of length L1 and L2, as we can assume the anchors will be aligned in linear time and therefore ignore their length.
The total number of cells in these rectangles is
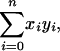 |
which can be rewritten as
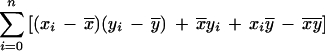 |
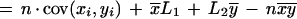 |
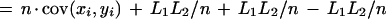 |
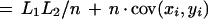 |
where x̄ and ȳ are the means of each distribution, and cov(xi,yi) is the covariance.
The first term of this expression depends only on the effective lengths of the sequences and the total number of anchors. If we assume a lower bound on acceptable anchor density, then L1L2/n behaves linearly in sequence length because L1/n and L2/n are O(1). The second term is at most n ςxςy, where ς denotes the standard deviation of the corresponding distribution. The upper bound is achieved in case of perfect correlation in the spacing of anchors in the two sequences. Assuming constant anchor density—a reasonable assumption for a fixed pair of organisms—this upper bound is linear in sequence length provided the standard deviation ς of each distribution is constant. Thus, if the anchors are spaced approximately evenly, and with a constant density, the running time will be linear in sequence length.
Global Multiple Alignment, MLAGAN
MLAGAN aligns multiple genomic sequences in two main phases, namely (1) a progressive alignment phase based on LAGAN pairwise alignments, and (2) an optional iterative improvement phase. Next we describe in detail the progressive alignment phase, the scoring method used in MLAGAN, and the iterative improvement phase.
Progressive Multiple Alignment with Anchors
During progressive alignment, MLAGAN aligns the sequences in the order of the given phylogenetic tree. For example, MLAGAN aligns sequences from human, chimpanzee, mouse, rat, and chicken, in the following order: (1) (human, chimpanzee), (2) (mouse, rat), (3) (human/chimpanzee, mouse/rat), (4) (human/chimpanzee/mouse/rat, chicken). Each alignment step merges two sequences or alignments into a larger alignment, effectively building a profile of all the sequences. The scoring method used is similar to CLUSTALW’s method, with some significant differences in the treatment of gaps. We explain our scoring method in detail in the next subsection.
In Step 2.1, MLAGAN aligns two (multi-)sequences X and Y, to generate multi-sequence X/Y, by performing limited-area dynamic programming (LAGAN Step 3). This step assumes that the rough global map between the two (multi-) sequences is known, as guaranteed by Step 2.2. In Step 2.2, MLAGAN recomputes the rough global map between the newly generated multi-sequence X/Y and each (multi)sequence Z, in two steps:
The anchors between X/Y and Z are computed as follows: First, all anchors in the rough global maps between X and Z, and between Y and Z, become anchors between X/Y and Z, with score equal to their original score. Second, each anchor between X and Z that overlaps an anchor between Y and Z, is reweighed with score equal to (s1 + s2) × I/U, where s1, s2 are the scores of the (X, Z) and (Y, Z) anchors, respectively, I is the length of intersection, and U is the length of union of the anchors (summed in X/Y and Z; Fig. 5).
The rough global map between X/Y and Z is the highest-weight chain of these anchors, computed using the Longest Increasing Subsequence algorithm.
Figure 5.

Generation of anchors during progressive alignment. Multi-sequence X/Y is aligned to sequence Z. Anchors between X and Z (top) and anchors between Y and Z (middle) are remapped to coordinates in the X/Y multi-sequence, and given a new score. Then, the Longest Increasing Subsequence algorithm is applied to select a subset of the remapped anchors, as the anchors between X/Y and Z.
In Step 2.3, MLAGAN selects the next two closest (multi)sequences in the phylogenetic tree, and goes to Step 2.1.
Multiple Alignment Scoring with Affine Gaps
Scoring schemes for multiple alignments is an open research area, with some innovative solutions published recently, such as T-COFFEE (Notredame et al. 2000). A full probabilistic framework for modeling multiple sequence alignments was implemented in HANDEL (Holmes and Bruno 2001). Two classical models are sum-of-pairs, and consensus. The sum-of-pairs model scores the multiple alignment according to the sum of scores of all pairwise alignments. The consensus model creates a consensus string by a (weighted) majority vote at each position, and scores the multiple alignment according to the sum of pairwise scores between the consensus sequence and each individual sequence (Gusfield 1999). Each scoring model has advantages and disadvantages. The sum-of-pairs model, for example, penalizes each additional gapped sequence less than previous ones. Given a gap penalty of g, in a K-sequence alignment the first gap in a given position gets penalized by (K−1) × g, the second gap by (K−3) × g, and the ith gap by (K−2i+1) × g. As a result, once a few gaps are already open it becomes advantageous to open more gaps whenever they result in small improvements in the number of matches. An alternative is to penalize gaps per sequence, but this is also problematic because a deletion in one sequence is significantly favored over an insertion in one sequence, leading to artificial compression of the multiple alignment. The consensus model does not properly model different lineages: A column with seven ‘T’s and five ‘A’s aligned, scores equally to one with seven ‘T’s, two ‘C’s, two ‘G’s, and one ‘A’. We use a combination of the approaches: sum-of-pairs for scoring substitutions, and consensus (scaled appropriately) for scoring gaps. Our approach is most similar to CLUSTALW, which uses a sum-of-pairs approach for scoring substitutions, and heuristically weighted per-sequence penalties to score gaps.
A straightforward implementation of the consensus affine-gap model in multiple alignments leads to the stacking effect: Because gap-open penalties are large compared to match and mismatch scores, often it is favorable to artificially open additional gaps in order to stack the gap openings (Fig. 6). To overcome this problem we use a gap-end penalty equal to the gap-open penalty (effectively, we divide equally the penalty for opening a gap into gap open and gap end). As can be seen from Figure 7, introducing such a gap-end penalty eliminates the stacking effect.
Figure 6.

The stacking effect. (A) The correct multiple alignment of seven sequences. (B) The multiple alignment of the same seven sequences, with the stacking effect. When only gap-open and gap-continue penalties are used, the stacked alignment (B) incurs one gap-open penalty less than the correct alignment (A), and therefore is optimal. When gap-end penalties are used, the correct alignment (A) is optimal because it incurs two gap-end penalties less than the stacked alignment (B).
Figure 7.

Part of a multiple alignment between eight mammalian sequences, before and after iterative refinement. Refinement improves the overall alignment by correctly placing the two T’s in the fifth and sixth sequence, and changing a 1-bp deletion in sequence 5 into an insertion in sequence 6.
We define the multiple alignment score precisely as follows. Given a multiple alignment of K sequences, let (Aij) be the K × L alignment matrix: Ai1 … AiL is the ith aligned sequence, where Aij ∈ {A, C, G, T, –} (‘–’ denotes a gap). Define the K × L matrix (Bij) over the four letters ‘N’ (nucleotide), ‘O’ (gap open), ‘G’ (gap continue), and ‘C’ (gap close): Bij = ‘G’ in all gapped positions (where Aij = ‘–’), except the ones opening a gap, in which case Bij = ‘O’; Bij = ‘N’ in all nucleotide positions (where Aij ≠ ‘–’), except the ones closing a gap, in which case Bij = ‘C’.
Let m, d, g, and c be the match, mismatch, gap-open, and gap-continue penalties, respectively. Define the function S(x, y), where x, y ∈ {A, C, G, T, –}, as follows: S(x, y) = 0, if x = ‘–’ or y = ‘–’; otherwise, S(x, y) = m if x = y, and S(x, y) = d if x ≠ y. The function S(x, y) will be used for the sum-of-pairs match and mismatch component of the score.
Let Nj be the number of ‘N’s in the jth column of (Bij). Similarly, define Oj, Gj, and Cj. Define the function T(i) = min(Oi, K−Oi) × (g + c) + min(Gi, K−Gi) × c + min(Ci, K−Ci) × g. The function T(i) will be used for the consensus-based gap component of the score. The multiple alignment score is then:
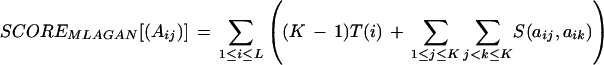 |
Iterative Refinement With Anchors
One shortcoming of progressive alignment is that the initial pairwise alignments are fixed, and early errors cannot be corrected later, when more information is available. A simple example of this situation is shown in Figure 7, where sequences 5 and 6 are initially misaligned. To fix such errors we use a technique known as iterative refinement.
In standard iterative refinement, as used by CLUSTALW, each sequence is removed from the multiple alignment and re-aligned, yielding a better alignment. This process is repeated for a number of iterations, or until the score cannot be further improved (a local maximum is reached). Anson and Myers (1997) introduced a limited-area version of iterative refinement: Each sequence is realigned under the constraint that the realignment stays within radius r from the original alignment. This procedure improves the alignment locally, but does not allow large-scale changes. MLAGAN introduces a limited-area version of iterative refinement (Step 3 of Algorithm MLAGAN) that has the advantages of performing more work in the areas that need it, and of allowing larger-scale adjustments: Each sequence is removed iteratively, and every region of the removed sequence that improved significantly the score of the multiple alignment becomes an anchor. For each position i in the sequence, the cumulative contribution si of positions 1,…,i to the existing multiple alignment score is calculated by summing the score of position i to si–1. When si falls below 0 at position i, si is set to 0. When si reaches a threshold T, si is set to 0, and an anchor is created at position i. The removed sequence is then re-aligned to the remaining ones using the LAGAN algorithm with these anchors. LAGAN allows for small changes around the anchors, while doing a full alignment search in between the anchors. Therefore, the above procedure makes small improvements in well-aligning regions and allows for larger adjustments between them.
Training, Testing, and Default Parameters
We trained the parameters of CHAOS, LAGAN, and MLAGAN on the stem cell leukemia (SCL) region from human, mouse, chicken, fugu, and zebrafish (Göttgens et al. 2002). During training we did not use the ROSETTA dataset, or the CFTR region, where we tested. One exception is a number of software bug fixes that were specific to the size of the dataset, such as memory leaks, which were corrected after applying LAGAN and MLAGAN to the CFTR sequences.
LAGAN and MLAGAN compute the rough global map using recursive calls to CHAOS with parameters (k, c, t), where k is the k-mer size, c is the allowed degeneracy, and t is the threshold (see Methods: Generation of Local Alignments). The default recursive steps are (12, 0, 30), (13, 1, 30), (8, 1, 30), and (7, 1, 30), on sequences masked using RepeatMasker (Smit and Green). These steps are followed by a (7, 1, 30) step on the unmasked sequences in order to anchor the conserved repeats. An optional (5, 0, 50) step on the translated sequences can be inserted between the (13, 1, 30) step and the (8, 1, 30) step. The translated anchoring step is scored using a BLOSUM62 matrix. This step is disabled by default, and was disabled during the testing reported in Tables 1 and 2. The distance d parameter is 20 for genomic sequences and 8 for comparison of peptides (translated). The shift s is 5 for all runs.
During the limited-area computation of global alignment (Step 3 of LAGAN, Fig. 4), the default radius is r = 15.
The limited-area dynamic programming pass (Step 3 of LAGAN) uses match m = +12 (or +18 for MLAGAN), mismatch d = −8, gapopen g = −100, and gapcontinue c = −5. In MLAGAN, the gapopen parameter gets split equally, as explained in Section 3.2.2, into gapopen = −50 and gapend = −50.
All of the tests were run on a 2.3-GHz Pentium IV machine with 500MB of RAM, except for AVID on the CFTR dataset, which due to the memory requirements was run on a 2-GHz Pentium IV machine with 1GB of RAM.
Most of the other programs we tested can take many different parameters. For all programs we used the parameters that were either suggested by the authors, or gave the best results for that particular program. Specifically: BLASTZ was run with a cutoff score of 2200 (K = 2200); DIALIGN used CHAOS anchoring, and was run with the ‘−nt’ (compare at the protein-coding level) option; AVID version 0.91 was used, as it performed better than AVID 1.2 on the CFTR dataset. (AVID 1.2 performed slightly better on the ROSETTA dataset.).
WEB SITE REFERENCES
http://lagan.stanford.edu; LAGAN alignment server.
http://ftp.genome.washington.edu/RM/RepeatMasker.html; RepeatMasker (Smit, A.F.A. and Green, P.).
Acknowledgments
M.B. was supported by an NSF Graduate Fellowship. G.M.C. was supported by a Howard Hughes Medical Institute predoctoral fellowship. E.D.G. and the NISC Comparative Sequencing Program were supported by funds from the National Human Genome Research Institute. The following individuals were key contributors within the NISC Comparative Sequencing Program: Jim Thomas (BAC isolation and mapping); Jeff Touchman and Bob Blakesley (BAC sequencing); Gerry Bouffard, Steve Beckstrom-Sternberg, Pam Thomas, Jenny McDowell, and Baishali Maskeri (computational analyses). We thank Berthold Göttgens and Michael Chapman for providing us with the stem cell leukemia region, Burkhard Morgenstern for help with developing the CHAOS program, and Inna Dubchak and Alex Poliakov for testing LAGAN and providing us with feedback.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
E-MAIL serafim@cs.stanford.edu; FAX (650) 725-1449.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.926603. Article published online before print in March 2003.
REFERENCES
- 1.Altschul S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- 2.Altschul S.F., Madden, T.L., Schoffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anson E.L. and Myers, E.W. 1997. Re-Aligner: A program for refining DNA sequence multialignments. J. Comp. Biol. 4: 369-383. [DOI] [PubMed] [Google Scholar]
- 4.Barton G.J. and Sternberg, M.J.E. 1987. A strategy for the rapid multiple alignment of protein sequences. J. Mol. Biol. 198: 327-337. [DOI] [PubMed] [Google Scholar]
- 5.Batzoglou S., Pachter, L., Mesirov, J., Berger, B., and Lander, E.S. 2000. Human and mouse gene structure: Comparative analysis and application to exon prediction. Genome Res. 10: 950-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonizzoni P. and Vedova, G.D. 2001. The complexity of multiple sequence alignment with SP-score that is a metric. Theoretical Computer Science 259: 63-79. [Google Scholar]
- 7.Bray N., Dubchak, I., and Pachter, L. 2003. AVID: A global alignment program. Genome Res. 13: 97-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brudno M. and Morgenstern, B., 2002. Fast and sensitive alignment of large genomic sequences. In Proceeding of the IEEE Computer Society Bioinformatics Conference (CSB).. [PubMed]
- 9.Delcher A.L., Kasif, S., Fleischman, R., Peterson, J., White, O., and Salzberg, S.L. 1999. Alignment of whole genomes. Nucleic Acids Res. 27: 2369-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delcher A.L., Phillippy, A., Carlton, J., and Salzberg, S.L. 2002. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 30: 2478-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubchak I., Brudno, M., Loots, G.G., Pachter, L., Mayor, C., Rubin, E.M., and Frazer, K.A. 2000. Active conservation of noncoding sequences revealed by three-way species comparisons. Genome Res. 10: 1304-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eppstein D., Galil, Z., Giancarlo, R., and Italiano, G.F. 1992. Sparse dynamic programming I: Linear cost functions. JACM 39: 546-567. [Google Scholar]
- 13.Gotoh O. 1996. Significant improvement in accuracy of multiple protein sequence alignments by iterative refinement as assessed by reference to structural alignments. J. Mol. Biol. 264: 823-838. [DOI] [PubMed] [Google Scholar]
- 14.Göttgens B., Barton, L.M., Chapman, M.A., Sinclair, A.M., Knudsen, B., Grafham, D., Gilbert, J.G.R., Rogers, J., Bentley, D.R., and Green, A.R. 2002. Transcriptional regulation of the stem cell leukemia gene (SCL)—Comparative analysis of five vertebrate SCL loci. Genome Res. 12: 749-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gusfield D., 1999. Algorithms on strings, trees, and sequences: Computer science and computational biology, pp. 351–353. Cambridge University Press, Cambridge, UK.
- 16.Hardison R., Chao, K.-M, Adamkiewicz, M., Price, D., Jackson, J., Zeigler, T., Stojanovic, N., and Miller, W. 1993. Positive and negative regulatory elements of the rabbit ε-globin gene revealed by an improved multiple alignment program and functional analysis. DNA Seq. 4: 163-176. [DOI] [PubMed] [Google Scholar]
- 17.Hardison R., Chao, K.-M., Schwartz, S., Stojanovic, N., Ganetsky, M., and Miller, W. 1994. Globin Gene Server: A prototype E-mail database server featuring extensive multiple alignments and data compilation for electronic genetic analysis. Genomics 21: 344-353. [DOI] [PubMed] [Google Scholar]
- 18.Henikoff S. and Henikoff, J.G. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89: 10,915-10,919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Höhl M., Kurtz, S., and Ohlebusch, E. 2002. Efficient multiple genome alignment. Bioinformatics 18: 312-320. [DOI] [PubMed] [Google Scholar]
- 20.Holmes I. and Bruno, W.J. 2001. Evolutionary HMMs: A Bayesian approach to multiple alignment. Bioinformatics 17: 803-820. [DOI] [PubMed] [Google Scholar]
- 21.Kent W.J. 2002. BLAT—The BLAST-like alignment tool. Genome Res. 12: 656-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kent W.J. and Zahler, A.M. 2000. Conservation, regulation, synteny, and introns in a large-scale C. briggsae–C. elegans genomic alignment. Genome Res. 10: 1115-1125. [DOI] [PubMed] [Google Scholar]
- 23.Mayor C., Brudno, M., Schwartz, J.R., Poliakov, A., Rubin, E.M., Frazer, K.A., Pachter, L.S., and Dubchak, I. 2000. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16: 1046. [DOI] [PubMed] [Google Scholar]
- 24.Miller W. 2001. Comparison of genomic DNA sequences: Solved and unsolved problems. Bioinformatics 17: 391-397. [DOI] [PubMed] [Google Scholar]
- 25.Morgenstern B. 1999. DIALIGN 2: Improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics 15: 211-218. [DOI] [PubMed] [Google Scholar]
- 26.Morgenstern B., French, K., Dress, A., and Werner, T. 1998. DIALIGN: Finding local similarities by multiple sequence alignment. Bioinformatics 14: 290-294. [DOI] [PubMed] [Google Scholar]
- 27.Needleman S.B. and Wunsch, C.D. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48: 443-453. [DOI] [PubMed] [Google Scholar]
- 28.Ning Z., Cox, A.J., and Mullikin, J.C. 2001. SSAHA: A fast search method for large DNA databases. Genome Res. 11: 1725-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Notredame C., Higgins, D.G., and Heringa, J. 2000. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302: 205-217. [DOI] [PubMed] [Google Scholar]
- 30.Sankoff D. 1999. Genome rearrangement with gene families. Bioinformatics 15: 909-917. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz S., Zhang, Z., Frazer, K.A., Smit, A., Riemer, C., Bouck, J., Gibbs, R., Hardison, R., and Miller, W. 2000. PipMaker—A web server for aligning two genomic DNA sequences. Genome Res. 10: 577-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simon A., Stone, E.A., and Sidow, A. 2002. Inference of functional regions in proteins by quantification of evolutionary constraints. Proc. Natl. Acad. Sci. 99: 2912-2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147: 195-197. [DOI] [PubMed] [Google Scholar]
- 34.Stanfel L.E. 1996. A new approach to clustering the amino acids. J. Theor. Biol. 183: 195-205. [DOI] [PubMed] [Google Scholar]
- 35.Taylor W. 1988. A flexible method to align large numbers of biological sequences. J. Mol. Evol. 28: 161-169. [DOI] [PubMed] [Google Scholar]
- 36.Thompson J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTALW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L. and Jiang, T. 1994. On the complexity of multiple sequence alignment. J. Comp. Biol. 1: 337-348. [DOI] [PubMed] [Google Scholar]