

Abstract
Evidence indicates that for most human cancers the problem is not that gene mutations occur but is more dependent upon how the body deals with damaged cells. It has been estimated that only about 1% of human cancers can be accounted for by unmistakable hereditary cancer syndromes, only up to 5% can be accounted for due to high-penetrance, single-gene mutations, and in total only 5%–15% of all cancers may have a major genetic component. The predominant contribution to the causation of most sporadic cancers is considered to be environmental factors contributing between 58% and 82% toward different cancers. A nutritionally poor lifestyle is associated with increased risk of many cancers, including those of the breast. As nutrition, energy balance, macronutrient composition of the diet, and physical activity levels are major determinants of insulin-like growth factor (IGF-I) bioactivity, it has been proposed that, at least in part, these increases in cancer risk and progression may be mediated by alterations in the IGF axis, related to nutritional lifestyle. Localized breast cancer is a manageable disease, and death from breast cancer predominantly occurs due to the development of metastatic disease as treatment becomes more complicated with poorer outcomes. In recent years, epithelial-to-mesenchymal transition has emerged as an important contributor to breast cancer progression and malignant transformation resulting in tumor cells with increased potential for migration and invasion. Furthermore, accumulating evidence suggests a strong link between components of the IGF pathway, epithelial-to-mesenchymal transition, and breast cancer mortality. Here, we highlight some recent studies highlighting the relationship between IGFs, IGF-binding protein 3, and epithelial-to-mesenchymal transition.
Keywords: IGF-I, IGFBP-3, EMT, breast cancer
Insulin-like growth factor axis
The activity of insulin-like growth factors (IGFs) within any tissue is due to a combination of some locally expressed components together with IGFs and IGF-binding proteins (IGFBPs) delivered to the tissue from the circulation where high levels are maintained. This provides a mechanism for integrating systemic and local regulation systems. The circulating IGF system is under the influence of growth hormones, insulin, nutrition and systemic disease status, such as cancer, whereas the locally expressed components are controlled by factors specific to each individual tissue.
The cellular effects of the IGFs are mediated by a number of cell surface receptors including the type I and type II receptors, insulin receptor (IR), and IR–IGF-I receptor hybrids. Both IGF-I and IGF-II can mediate their actions on cell growth and survival via the IGF-IR, which is a transmembrane, tyrosine kinase that is structurally and functionally homologous to the IR. IGF-I can act via the IR but only at supraphysiological doses.1 IGF-II can bind with high affinity to the IGF-II receptor/mannose-6-phosphate receptor (IGF-IIR), a non-tyrosine kinase receptor considered to play an important role in the clearance and degradation of IGF-II. Additionally, the IGF-IIR is critically involved in the cellular trafficking of lysosomal enzymes and also acts as a high-affinity binding site for latent transforming growth factor-beta (TGFβ)-I, II, and III and retinoids.2 It was originally believed that IGF-II did not bind to the IR with any meaningful affinity until mice knockout studies revealed that IGF-II could act via this receptor during development.3 There are two alternatively spliced isoforms of the IR, A and B, and it has further been demonstrated that IGF-II binds with high affinity to the IRA but not the IRB isoform.4 The IR and IGF-IR can dimerize forming hybrid receptors5 and each IR isoform is equally able to form hybrids with the IGF-IR. Hybrid IGF-IR/IRB receptors have a high affinity for IGF-I and a lower affinity for insulin, whereas hybrid IGF-IR/IRA receptors not only have an even higher affinity for IGF-I but also bind IGF-II and insulin6 (Figure 1). Activation of the IGF-IR by IGFs results in oligomerization, autophosphorylation, and activation of the intrinsic tyrosine kinase.1 The IGF-IR tyrosine kinase directly phosphorylates a number of intracellular substrates including IR substrates (IRS)-1, 2, and 4,7–9 src homology domain containing (Shc),10,11 phosphatidylinositol-3 kinase (PI3-K),12 growth factor receptor–bound protein 10,13 focal adhesion kinase,14 and C-Src kinase.15
Figure 1.

Schematic representation of the insulin-like growth factor system.
Notes: The IGF system consists of peptides (IGF-I, IGF-II and insulin), receptors (the IGF-I receptor, the IR, and the IGF-II), a family of six high-affinity IGFBPs, as well as a group of proteases that are responsible for IGFBP degradation. Ligand binding to the receptor results in activation of different intracellular signaling pathways that regulate cell survival, proliferation differentiation, and metabolism.
Abbreviations: IGF, insulin-like growth factor; IGFBP, insulin-like growth factor–binding protein; IR, insulin receptor.
There are six high-affinity IGFBPs 1–6 that all have greater affinity for binding to the IGFs, than the IGF-IR, and can modulate IGF actions in many cell types. The IGFBPs slow the clearance of the IGFs, particularly IGFBP-3 and IGFBP-5, which also bind to a glycoprotein called the acid labile subunit forming ternary complexes, which have very long half-lives in the circulation. It is these complexes that maintain very high concentrations of IGFs in the body. Accumulating evidence indicates that most of the IGFBPs can also act in an intrinsic manner, independent of IGF binding, affecting various aspects of cell function including growth, apoptosis, migration, and attachment.16–18 The nature and complexity of the IGF axis during development and in disease states have been defined through the use of advanced molecular techniques, transgenic and knockout mouse models, which are beyond the scope of this review but have been elegantly summarized elsewhere.19–22
Metabolic disturbance and breast cancer
In all Western societies, women who present with breast cancer are increasingly likely to also suffer from comorbid conditions such as diabetes and obesity due to the very high prevalence of these conditions in the general population. In a study of over 1000 women treated for breast cancer at the MD Anderson Cancer Center in Houston, 30% were found to be obese and a further 32% were overweight.23 In addition, the prevalence of metabolic syndrome in patients with breast cancer has been reported to be up to 50%.24,25
Evidence derived from epidemiology, in vitro and in vivo studies suggest that metabolic disturbances adversely affect breast cancer cell survival and progression. In women with breast cancer, having metabolic syndrome also was associated with more aggressive tumor characteristics24 and being obese was associated with worse overall survival.23 In an orthotopic syngeneic mouse model of triple-negative breast cancer, a high-energy diet promoted tumor growth and increased the number of metastases to the lungs.26 In vitro, it has been shown that breast cancer cells exposed to high levels of glucose, such as those associated with type 2 diabetes causes resistance to chemotherapy.27 As IGF is metabolically regulated and mediates the effects of nutrition on cell growth,28 it is not surprising that evidence indicates that IGF-I plays a critical role in also affecting the impact of altered metabolism on tumor growth and survival. Calorie restriction has long been known to reduce obesity, improve metabolic status, and reduce the propensity for cancer development.29 Calorie restriction studies using the 4T1 mammary tumor model showed a reduction in tumor growth and metastasis that was associated with decreased circulating IGF-I levels.30 Furthermore, using orthotopically transplanted mammary tumors in control and calorie-restricted mice, it was concluded that a reduction in mammary tumor growth could be accounted for in part by reduced levels of IGF-I.31
Most cancer cells have an increased ability to enhance their glucose uptake and rely on aerobic glycolysis, a phenomenon termed “the Warburg effect” in order to generate energy, in contrast to normal differentiated cells, which use mitochondrial oxidative phosphorylation to generate the energy needed for cellular processes. Such strategies employed by cancer cells to ensure progression also have an impact on other aspects of tumorigenesis: artificially stimulating the Warburg effect in animal models and in cell culture has been found to induce epithelial-to-mesenchymal transition (EMT). For example, genetic silencing of the respiratory enzyme citrate synthase, resulted in greatly increased glycolytic metabolism that was reported to induce EMT and increase metastases in a mouse xenograft model.32 In addition, reversal of the Warburg effect by silencing of expression of pyruvate dehydrogenase kinase 1 in breast cancer cells restored their sensitivity to anoikis when detached and reduced lung metastasis when they were inoculated in a mouse model.33 It has been reported that developmental EMT can be induced by exposure to hyperglycemia, and it has recently been demonstrated that exposure to hyperglycemia can similarly induce EMT of human lung adenocarcinoma cells.34
EMT in breast cancer progression and metastasis
Metastasis is the spread of cancer cells to distant sites in the body. It is the ultimate cause of death in more than 90% of breast cancer patients and therefore represents one of the biggest challenges in cancer research. In about 6%–10% of breast cancer diagnoses, cancer has already metastasized to the other parts of the body and approximately 30% of patients with early-stage breast cancer have a recurrent or metastatic disease.35
The seeding and growth of breast cancer cells at sites distinct from the primary tumor is a complex and multistage process that involves the ability to detach and invade through the basement membrane, intravasation into blood vessels or lymphatics, ability to transit through the vasculature, and extravasation from the circulation to establish new secondary tumors typically in organs such as bone, lung, liver, or brain (Figure 2). Emerging evidence suggests that in the initial stage, in the so-called invasion–metastasis cascade, cancer cells undergo a dramatic phenotypic change from an epithelial-to-mesenchymal phenotype via a process referred to as EMT. The plasticity of the epithelial phenotype is illustrated by the occurrence of the reverse process, mesenchymal-to-epithelial transition, during which cancer cells regain the epithelial properties of the primary tumor to promote outgrowth at metastatic sites. The EMT process was originally identified as being essential for embryonic development, mesoderm and neural crest formation, and also during wound healing.36 It is, however, now increasingly clear that inappropriate activation of EMT is a critical component of the progression of many cancers including breast.37 Cellular switching from an epithelial into mesenchymal cell requires a large number of complex modifications that include loss of cell adhesion, phenotypic change from typical cuboidal to an elongated spindle shape, rearrangement in the cytoskeletal architecture, and adoption of migratory and invasive phenotype (Figure 3). Epithelial cells are typically arranged as sheets of cells exhibiting apical–basal polarity. These cells are held together through a variety of structures including adherent junctions, tight junctions, and desmosomes and are separated from the underlying tissues by a thin layer of specialized extracellular matrix called the basement membrane, all of which help the epithelium to fulfill its function as a protective barrier. Mesenchymal cells on the other hand form disorganized cellular layers that are neither polarized nor tightly adherent, which allows for increased migratory capacity. They provide support and structure to the epithelial cells through the production of extracellular matrix components. A defining feature of EMT is a loss of epithelial cell adhesion molecule E-cadherin and a gain of mesenchymal cell markers such as vimentin, N-cadherin, and fibronectin.38
Figure 2.
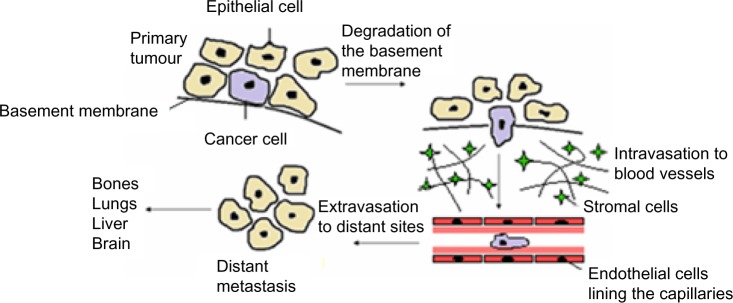
Schematic outline of breast cancer progression.
Notes: To begin the process of metastasis a malignant cell must migrate away from the primary tumor; this requires the degradation of the basement membrane. Following entry to the stroma, a cancer cell must invade the circulatory system via intravasation into existing or newly formed blood or lymph vessels. The cells need to survive the transport through the vasculature and then extravasate from the circulation to establish secondary tumors in distant sites; bone, lungs, liver, and brain are the most common sites for metastasis of breast cancer.
Figure 3.
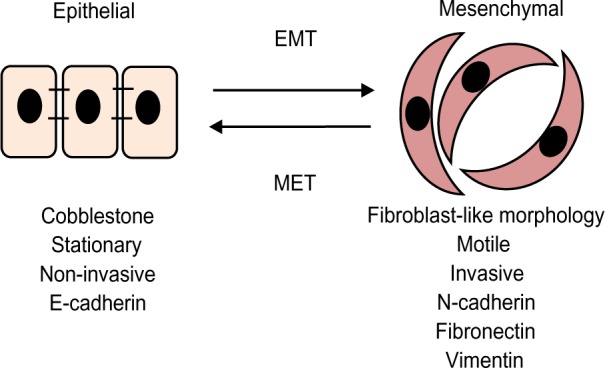
Epithelial-to-mesenchymal transitions.
Notes: The epithelial cells are closely packed, highly polarized cells that are not particularly motile. During EMT, signals provided by the stromal cells cause epithelial cells to lose their cell characteristics and acquire a mesenchymal cell phenotype characterized by enhanced migratory capacity and invasiveness and high expression of mesenchymal markers.
Abbreviations: EMT, epithelial-to-mesenchymal transition; MET, mesenchymal-to-epithelial transition.
A number of studies have shown a strong correlation between the EMT and high invasive and metastatic behavior of breast cancer. Particularly interesting is the recent finding that the EMT process appears to be associated with a basal-like breast cancer phenotype, which tends to be more aggressive and carry a poor prognosis.39 In the clinical setting, studies indicate that circulating tumor cells (CTCs) in patients with metastatic breast cancer frequently express mesenchymal markers, whereas mesenchymal markers are only found in rare cells within the corresponding primary tumours.40 In breast cancer, CD24−CD44+ cells or cells with high aldehyde dehydrogenase activity41 have been shown to be enriched in breast cancer stem cells. Intriguingly, it has also been shown that a major proportion of CTCs found in the blood samples of breast cancer patients also express the stem cell marker aldehyde dehydrogenase 142. A small subset of cells have been prospectively identified and characterized in the primary tumor that exhibited the exclusive capacity to form colonies, using a colony formation assay, and to form tumors in mice; these cells exhibited a stem cell phenotype.43 Abraham identified that breast cancer stem cells only accounted for 10%–20% of the cell population in a primary tumour44 that supports the concept of an exclusive subset of cells in the primary tumor with stem cell properties that have the propensity to metastasize. These findings support the notion that EMT is associated with “stemness” properties and that activation of EMT is critical for cancer cells to disseminate.
EMT is triggered and orchestrated by several growth factors released by the stromal cells including TGFβ, hepatocyte growth factor, platelet-derived growth factor, fibroblast growth factor, and Wnt and Notch ligands. Among these, TGFβ is undoubtedly the most potent driver of EMT that elicits its actions through activation of Smad and non-Smad signaling pathways.45 In fact, simple treatment with exogenous TGFβ can induce EMT in various epithelial cells. TGFβ plays a dual role in cancer: it is tumor suppressive during the early stages of tumor growth, but paradoxically, it becomes pro-oncogenic and promotes EMT and metastasis during late stages of malignancy.46 This switch in TGFβ function during advanced breast cancer is particularly reflected in increased expression of phosphorylated Smad2 in breast tumours.47 TGFβ activates a series of EMT-inducing transcription factors including Snail, Slug, Twist, Zeb1, Zeb2, and FoxC2, which in turn are under the control of multiple micro-RNAs.48–51 Most attention has been directed at the study of Snail. This zinc finger transcription factor was first noted during mesoderm formation in Drosophila melanogaster and was subsequently found to be a potent repressor of E-cadherin and a major EMT inducer.52,53 It is known that Snail binds to E-boxes in the proximal promoter region of E-cadherin and by recruiting histone deacetylases represses its expression.53 The significance of Snail in EMT has been shown in a variety of in vitro and in vivo models. It has been shown that Snail is required for tumor growth and lymph node metastasis of human breast carcinoma MDA–MB-231 cells and that knockdown of Snail increases the chemosensitivity of these cells to gemcitabine and docetaxel.54 Similarly using a mammary-specific, inducible HER2/Neu transgenic mouse model, Moody et al found high expression of Snail in recurrent breast tumours.55 Snail also interacts with other transcription factors as exemplified by a recent study, which shows that Snail cooperates with Twist to induce Zeb1 expression during EMT.56 It has also been reported that Snail co-immunoprecipitates with a Smad3/4 complex to promote TGFβ-mediated repression of E-cadherin, occludin, and coxsackievirus and adenovirus receptor and thereby cause EMT.57
In addition, an increasing number of studies demonstrate that EMT not only facilitates metastasis but also contributes to drug resistance. Using several drug-resistant sublines of MCF-7, Iseri et al reported an increase in the expression of EMT-associated genes (Slug, N-cadherin, and vimentin) together with loss of E-cadherin and the estrogen receptor (ER) α.58
IGF axis, EMT, and breast cancer
The conclusions drawn from a recent case control study nested within The European Prospective Investigation into Cancer and Nutrition cohort confirmed that a wealth of previous epidemiology studies that indicated higher levels of IGF-I within the normal range increase the risk of estrogen receptor–positive (ER+ve) breast cancers in pre- and postmenopausal women but are not related to the risk of developing ER-negative (ER−ve) tumors.59–62 Such a relationship of IGF-I with ER+ve tumors is consistent with the vast amount of literature indicating the synergy that is known to exist between IGF and estrogen-induced signaling pathways in promoting breast cancer progression (see Hawsawi et al63 and Tsonis et al64 for reviews).
For the purposes of this review, we are focusing on the role of IGF-I/II and IGFBP-3 in EMT for which the majority of evidence has been obtained: in terms of circulating IGFBP-3, there has been both positive and negative associations of IGFBP-3 with the risk of breast cancer,65–67 but when Roddam62 pooled 17 prospective trials, he concluded that the risk of breast cancer was not associated with circulating levels of IGFBP-3. These data suggest that for IGFBP-3, either circulating levels do not play a role or they are not reflective of the local situation in the tumor itself.
It is becoming increasingly clear that IGFs are important for inducing EMT by enhancing growth, survival, motility, migration, and metastasis of breast cancer cells. IGF-I and IGF-II are expressed in many tumors and IGF-II in particular has been observed to be overexpressed in a number of different tumors, including the breast,68 and increased expression of either IGF-I69 or IGF-II70–72 has been reported to be associated with more aggressive tumor phenotypes. Similarly increased expression of IGF-II and the IGF-IR has been associated with a more metastatic phenotype.73 Increased expression of the IGF-IR has been reported in many cancer cell lines and in human tumor biopsies.74,75 The IGF-IR appears to play a critical role in malignant transformation and in the maintenance of a transformed cell phenotype76 due to its ability to maintain cell survival and protect cells from apoptosis via multiple signaling pathways.77 In human mammary epithelial cells, overexpression of a constitutively active IGF-IR caused their transformation and growth in immunocompromised mice that was associated with the initiation of EMT, via upregulation of Snail and downregulation of E-cadherin.78 In keeping with this, overexpression of the IGF-IR has been reported to be associated with an aggressive phenotype of a variety of tumours.79,80 An in vivo experimental brain metastasis model demonstrated that IGF-IR silencing in brain-seeking breast cancer cells reduced the propensity of these cells to develop brain metastases.81
As mentioned previously, EMT has been associated with features of “stemness”, and recently, Chang et al82 reported that expression of activated IGF-IR was greater in cancer stem cells than normal stem cells. They also reported that those cells expressing high levels of activated IGF-IR exhibited factors associated with EMT that were lost upon silencing or inhibiting the IGF-IR. Even in normal breast epithelial cells, MCF-10A, IGF-I was able to induce EMT when Akt1 was downregulated but expression of Akt2 was maintained.83 To become motile, cells have to depolarize and work has indicated that IGF-I depolarizes breast cancer cells in a phosphatidylinositol-3 kinase–dependent manner.84 MEMO is a protein recognized as being a critical mediator of motility in response to receptor tyrosine kinase activation and can specifically affect IGF-IR–dependent signaling in breast cancer cell lines, via binding to IR substrate-1 and activating phosphatidylinositol-3 kinase/Akt signaling, which leads to upregulation of the transcription factor, Snail, that is key to effecting EMT.85 IGF-I has also been shown to upregulate Zeb1, another key transcription factor involved in EMT, to induce invasion of MDA–MB-231 cells.86 The ability of IGF-I to drive EMT in another hormone-responsive cancer, the prostate, is also dependent upon its ability to upregulate Zeb-1.87 In order for breast cancer cells to metastasize, they have to travel in the circulation to distant sites, and it has recently been shown that the IGF-IR is present on CTCs of breast cancer patients.88 High circulating IGF-I has been correlated with breast cancer mortality89 and lymph node metastasis in patients with ER+ve disease.90 These data suggest that one way in which serum IGF-I may influence metastasis may be by activating the IGF-IR present on CTCs and maintaining survival of these detached cells in the circulation. Although not in breast cancer cells, a complementary DNA microarray was employed to assess gene expression patterns specifically regulated by IGF-I, differential over those regulated by insulin and found that a key transcription factor upregulated in EMT, Twist, was one of the most upregulated genes and that Twist was key in mediating the antiapoptotic effects of IGF-I in NWTb3 fibroblast cells.91,92 In addition to the IGF-IR, the IR, particularly the IRA isoform, that has a higher affinity for IGF-II similar to that for insulin, together with the hybrid IR/IGF-IR receptors may also be present and play a role in mediating the actions of the IGFs in certain tumors.93,94 IR isoform assessment from formalin-fixed, paraffin-embedded sections, showed that the IRA was more abundant than the IRB isoform in breast tumours.95 In contrast to the IGF-I receptor, the IGF-IIR serves to limit IGF-II actions and hence reduce its growth-promoting and cell survival potential. A number of genetic disruptions resulting in loss of IGF-IIR have been described in various tumor types including missense mutations, loss of heterozygosity, and microsatellite instability.96,97 Loss of the IGF-IIR has been associated with increased tumor growth potential,98 decreasing IGF-IIR expression has a similar effect,99 whereas introduction of the IGF-IIR into cancer cells reduces growth and increases apoptosis.100
The IGFBPs have the potential to either inhibit or enhance IGF actions in many cell types. Most evidence suggests that IGFBPs generally restrict tumor growth and progression by limiting IGF-mitogenic and cell survival actions. The actions of many antiproliferative agents appear to operate, at least in part, via upregulation of endogenous IGFBPs produced by the tumor cells, including TGFβ,101 retinoids,102 vitamin D,103 tamoxifen,104 and butyrate.105
In vitro studies suggest that the more aggressive ER−ve, compared with ER+ve, cell lines secrete higher levels of IGFBP-3.106 These reports are consistent with the observation that higher concentrations of IGFBP-3 in breast tissue are associated with increased risk of mortality and107 are correlated with tumor size and poor prognostic characteristics.108,109 Accumulating evidence indicates that most of the IGFBPs can also act in an intrinsic manner, independent of IGF binding, affecting various aspects of cell function. Growth inhibition and modulation of apoptosis have been described in a variety of cancer cell lines.17,18,110–112
Studies conducted in our laboratory and others have demonstrated that rhIGFBP-3 enhances the cell cycle arrest and apoptotic effects of paclitaxel and radiation in human breast cancer cells in vitro.110,113 In addition, our studies suggest that when normal breast epithelial cells are challenged with exogenous ceramide, rhIGFBP-3 has the opposite effect and acts as a survival factor.114 In preliminary in vivo studies, subcutaneous administration of rhIGFBP-3 to mice bearing MCF-7 breast tumor xenografts enhanced the tumor growth inhibition effect of paclitaxel from 33% to 61%.115 However, in vitro studies clearly indicate that the ability of IGFBP-3 to intrinsically affect breast cancer cell growth, survival, and adhesion is dependent upon the cellular environment. In conditions that reflect a more advanced tumor environment, such as increased fibronectin, IGFBP-3 acts in an opposite manner and promotes tumor progression by acting as a survival factor and growth promoter, whereas in early stages of the disease, IGFBP-3 enhances triggers of apoptosis and inhibits cell growth thus acting as a tumor suppressor.16,116,117 The association of high local expression of IGFBP-3 with poor prognosis could be explained by the above observations that the actions of IGFBP-3 on cancer cells can be switched when the cells are exposed to a more advanced tumor environment such as increased fibronectin.118 There are a number of theories as to the mechanisms underlying the intrinsic actions of IGFBP-3 and these have been extensively reviewed recently (refer Baxter119 and Johnson and Firth120).
TGFβ clearly plays a significant role in breast cancer progression, and it has been shown that IGF-I can activate latent TGFβ in breast cancer cells.121 Like IGFBP-3, TGFβ acts as a tumor suppressor in the early stages, but in later stages of the disease, it switches function and acts to promote tumorigenesis. TGFβ can differentially regulate IGFBP-3 depending on the cell type. With nonmalignant breast epithelial cells, where IGFBP-3 promotes growth and TGFβ is a growth inhibitor, TGFβ was found to downregulate IGFBP-3. Whereas in breast cancer cells, where TGFβ and IGFBP-3 both act as growth inhibitors, TGFβ was found to upregulate IGFBP-3.114 It is well documented that in the later stages of disease, TGFβ enhances the production of extracellular matrix production, such as fibronectin122 as part of its role in inducing EMT. Having shown that IGFBP-3 can act as a tumor promoter in the presence of fibronectin, it would be interesting to see whether TGFβ-induced IGFBP-3 in this context could mediate the pro-tumorigenic actions of TGFβ to induce EMT.
Breast cancer treatment: is the IGF-IR a viable target?
Breast cancer can be treated with different combinations of therapies: surgery, radiation therapy, chemotherapy, and hormone therapy. This depends upon the classification of the tumor and whether it remains localized in the breast or has spread to other sites in the body. Tumors can be classified histologically, of which 21 distinct histological types have been identified.123 Through the use of molecular profiling, breast cancers are now also categorized into four main subtypes: basal-like, HER2+, Luminal A, or Luminal B. Other less common molecular subtypes have also been described including normal breast-like, apocrine molecular type, and claudin-low type. Breast cancers that do not fall into any of these subtypes are often listed as unclassified.124
Rigorous laboratory and preclinical research identified that the IGF-IR played a key role in the progression of a number of different cancers, including the breast and as a result, a number of agents targeting the IGF-IR were developed and taken forward into clinical trials: these included antibodies against the receptor itself, against the receptor ligands, IGF-I and II, in addition to IGF-IR tyrosine kinase inhibitors (see Refs 125–127 for reviews). Despite encouraging preliminary data, the advanced clinical trials failed to show clinical benefit, which culminated in most drug companies terminating current programs targeting the IGF-IR. These trials were undertaken on unselected patients and no other targeted therapy has yet been found to work generally in cohorts of unselected patients. The results of the trials so far suggest that perhaps this receptor will only be an effective target in a subset of patients and to identify them will require a greater understanding of the context in terms of the complement of receptors present: levels of IRA and hybrid receptors and also the cooperative signaling events initiated by receptor signaling.
There have also been suggestions that IGFBP-3 could be developed as a cancer therapeutic due to its ability to inhibit IGF-actions and its IGF-independent pro-apoptotic actions. Although preclinical studies have provided some promising evidence,128 these have not progressed to clinical trials. This is partly due to not only pharmaco-formulation issues but also the increasingly conflicting clinical observations of both positive and negative effects of IGFBP-3, which have added caution to its development as a clinical intervention.
Summary
EMT is clearly important in effecting cancer progression; a better characterization of the role of the IGF axis in this process may lead to a greater understanding of its activity and co-operativity with other signaling pathways that are involved. This may lead to more effective interventions for metastatic disease and help to determine what causes the emergence of resistance to current therapeutic agents and provide potential critical biomarkers of response.
Acknowledgments
We thank the “Aiming to Beat Cancer Fund”, Bristol Oncology Centre, for supporting our work.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.LeRoith D, Werner H, Beitner-Johnson D, Roberts CT., Jr Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- 2.Braulke T. Type-2 IGF receptor: a multi-ligand binding protein. Horm Metab Res. 1999;31:242–246. doi: 10.1055/s-2007-978725. [DOI] [PubMed] [Google Scholar]
- 3.Rother KI, Accili D. Role of insulin receptors and IGF receptors in growth and development. Pediatr Nephrol. 2000;14(7):558–561. doi: 10.1007/s004670000351. [DOI] [PubMed] [Google Scholar]
- 4.Frasca F, Pandini G, Scalia P, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999;19:3278–3288. doi: 10.1128/mcb.19.5.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seely BL, Reichart DR, Takata Y, Yip C, Olefsky JM. A functional assessment of insulin/insulin-like growth factor-I hybrid receptors. Endocrinology. 1995;136:1635–1641. doi: 10.1210/endo.136.4.7895674. [DOI] [PubMed] [Google Scholar]
- 6.Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem. 2002;277:39684–39695. doi: 10.1074/jbc.M202766200. [DOI] [PubMed] [Google Scholar]
- 7.Myers MG, Jr, Grammer TC, Wang LM, et al. Insulin receptor substrate-1 mediates phosphatidylinositol 3′-kinase and p70S6k signaling during insulin, insulin-like growth factor-1, and interleukin-4 stimulation. J Biol Chem. 1994;269:28783–28789. [PubMed] [Google Scholar]
- 8.He W, Craparo A, Zhu Y, et al. Interaction of insulin receptor substrate-2 (IRS-2) with the insulin and insulin-like growth factor I receptors. Evidence for two distinct phosphotyrosine-dependent interaction domains within IRS-2. J Biol Chem. 1996;271:11641–11645. doi: 10.1074/jbc.271.20.11641. [DOI] [PubMed] [Google Scholar]
- 9.Fantin VR, Sparling JD, Slot JW, Keller SR, Lienhard GE, Lavan BE. Characterization of insulin receptor substrate 4 in human embryonic kidney 293 cells. J Biol Chem. 1998;273:10726–10732. doi: 10.1074/jbc.273.17.10726. [DOI] [PubMed] [Google Scholar]
- 10.Pelicci G, Lanfrancone L, Grignani F, et al. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-l. [DOI] [PubMed] [Google Scholar]
- 11.Giorgetti S, Pelicci PG, Pelicci G, Van Obberghen E. Involvement of Src-homology/collagen (SHC) proteins in signaling through the insulin receptor and the insulin-like-growth-factor-I-receptor. Eur J Biochem. 1994;223:195–202. doi: 10.1111/j.1432-1033.1994.tb18983.x. [DOI] [PubMed] [Google Scholar]
- 12.Lamothe B, Bucchini D, Jami J, Joshi RL. Interaction of p85 subunit of PI 3-kinase with insulin and IGF-1 receptors analysed by using the two-hybrid system. FEBS Lett. 1995;373:51–55. doi: 10.1016/0014-5793(95)01011-3. [DOI] [PubMed] [Google Scholar]
- 13.Morrione A, Valentinis B, Li S, Ooi JY, Margolis B, Baserga R. Grb10: A new substrate of the insulin-like growth factor I receptor. Cancer Res. 1996;56:3165–3167. [PubMed] [Google Scholar]
- 14.Baron V, Calleja V, Ferrari P, Alengrin F, Van Obberghen E. p125Fak focal adhesion kinase is a substrate for the insulin and insulin-like growth factor-I tyrosine kinase receptors. J Biol Chem. 1998;273:7162–7168. doi: 10.1074/jbc.273.12.7162. [DOI] [PubMed] [Google Scholar]
- 15.Arbet-Engels C, Tartare-Deckert S, Eckhart W. C-terminal Src kinase associates with ligand-stimulated insulin-like growth factor-I receptor. J Biol Chem. 1999;274:5422–5428. doi: 10.1074/jbc.274.9.5422. [DOI] [PubMed] [Google Scholar]
- 16.McCaig C, Perks CM, Holly JM. Intrinsic actions of IGFBP-3 and IGFBP-5 on Hs578T breast cancer epithelial cells: inhibition or accentuation of attachment and survival is dependent upon the presence of fibronectin. J Cell Sci. 2002;115:4293–4303. doi: 10.1242/jcs.00097. [DOI] [PubMed] [Google Scholar]
- 17.Perks CM, Bowen S, Gill ZP, Newcomb PV, Holly JM. Differential IGF-independent effects of insulin-like growth factor binding proteins (1–6) on apoptosis of breast epithelial cells. J Cell Biochem. 1999;75:652–664. doi: 10.1002/(sici)1097-4644(19991215)75:4<652::aid-jcb11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 18.Oh Y, Müller HL, Lamson G, Rosenfeld RG. Insulin-like growth factor (IGF)-independent action of IGF-binding protein-3 in Hs578T human breast cancer cells. Cell surface binding and growth inhibition. J Biol Chem. 1993;268:14964–14971. [PubMed] [Google Scholar]
- 19.Dupont J, Holzenberger M. Biology of insulin-like growth factors in development. Birth Defects Res C Embryo Today. 2003;69:257–271. doi: 10.1002/bdrc.10022. [DOI] [PubMed] [Google Scholar]
- 20.Yakar S, Sun H, Zhao H, et al. Metabolic effects of IGF-I deficiency: lessons from mouse models. Pediatr Endocrinol Rev. 2005;3:11–19. [PubMed] [Google Scholar]
- 21.Silha JV, Murphy LJ. Insights from insulin-like growth factor binding protein transgenic mice. Endocrinology. 2002;143:3711–3714. doi: 10.1210/en.2002-220116. [DOI] [PubMed] [Google Scholar]
- 22.Holly JM, Perks CM. Insulin-like growth factor physiology: what we have learned from human studies. Endocrinol Metab Clin North Am. 2012;41:249–263. doi: 10.1016/j.ecl.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Litton JK, Gonzalez-Angulo AM, Warneke CL, et al. Relationship between obesity and pathologic response to neoadjuvant chemotherapy among women with operable breast cancer. J Clin Oncol. 2008;26:4072–4077. doi: 10.1200/JCO.2007.14.4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Healy LA, Ryan AM, Carroll P, et al. Metabolic syndrome, central obesity and insulin resistance are associated with adverse pathological features in postmenopausal breast cancer. Clin Oncol (R Coll Radiol) 2010;22:281–288. doi: 10.1016/j.clon.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Stebbing J, Sharma A, North B, et al. A metabolic phenotyping approach to understanding relationships between metabolic syndrome and breast tumour responses to chemotherapy. Ann Oncol. 2012;23:860–866. doi: 10.1093/annonc/mdr347. [DOI] [PubMed] [Google Scholar]
- 26.Phoenix KN, Vumbaca F, Fox MM, Evans R, Claffey KP. Dietary energy availability affects primary and metastatic breast cancer and metformin efficacy. Breast Cancer Res Treat. 2010;123:333–344. doi: 10.1007/s10549-009-0647-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng L, Biernacka KM, Holly JM, et al. Hyperglycaemia confers resistance to chemotherapy on breast cancer cells: the role of fatty acid synthase. Endocr Relat Cancer. 2010;17:539–551. doi: 10.1677/ERC-09-0221. [DOI] [PubMed] [Google Scholar]
- 28.Narasimhan SD, Yen K, Tissenbaum HA. Converging pathways in lifespan regulation. Curr Biol. 2009;19:R657–R666. doi: 10.1016/j.cub.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN. Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research. Carcinogenesis. 2010;31:83–89. doi: 10.1093/carcin/bgp280. [DOI] [PubMed] [Google Scholar]
- 30.De Lorenzo MS, Baljinnyam E, Vatner DE, Abarzua P, Vatner SF, Rabson AB. Caloric restriction reduces growth of mammary tumors and metastases. Carcinogenesis. 2011;32:1381–1387. doi: 10.1093/carcin/bgr107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nogueira LM, Lavigne JA, Chandramouli GV, Lui H, Barrett JC, Hursting SD. Dose-dependent effects of calorie restriction on gene expression, metabolism, and tumor progression are partially mediated by insulin-like growth factor-1. Cancer Med. 2012;1:275–288. doi: 10.1002/cam4.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin CC, Cheng TL, Tsai WH, et al. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep. 2012;2:785. doi: 10.1038/srep00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamarajugadda S, Stemboroski L, Cai Q, et al. Glucose oxidation modulates anoikis and tumor metastasis. Mol Cell Biol. 2012;32:1893–1907. doi: 10.1128/MCB.06248-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alisson-Silva F, Freire-de-Lima L, Donadio JL, et al. Increase of O-glycosylated oncofetal fibronectin in high glucose-induced epithelial-mesenchymal transition of cultured human epithelial cells. PLoS One. 2013;8:e60471. doi: 10.1371/journal.pone.0060471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Shaughnessy J. Extending survival with chemotherapy in metastatic breast cancer. Oncologist. 2005;10(Suppl 3):20–29. doi: 10.1634/theoncologist.10-90003-20. [DOI] [PubMed] [Google Scholar]
- 36.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 38.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68:989–997. doi: 10.1158/0008-5472.CAN-07-2017. [DOI] [PubMed] [Google Scholar]
- 40.Yu M, Bardia A, Wittner BS, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009;11:R46. doi: 10.1186/bcr2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abraham BK, Fritz P, McClellan M, Hauptvogel P, Athelogou M, Brauch H. Prevalence of CD44+/CD24−/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clin Cancer Res. 2005;11:1154–1159. [PubMed] [Google Scholar]
- 45.Miyazono K. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:314–323. doi: 10.2183/pjab.85.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bachman KE, Park BH. Duel nature of TGF-beta signaling: tumor suppressor vs tumor promoter. Curr Opin Oncol. 2005;17:49–54. doi: 10.1097/01.cco.0000143682.45316.ae. [DOI] [PubMed] [Google Scholar]
- 47.Xie W, Mertens JC, Reiss DJ, et al. Alterations of Smad signaling in human breast carcinoma are associated with poor outcome: a tissue microarray study. Cancer Res. 2002;62:497–505. [PubMed] [Google Scholar]
- 48.Fendrich V, Waldmann J, Feldmann G, et al. Unique expression pattern of the EMT markers Snail, Twist and E-cadherin in benign and malignant parathyroid neoplasia. Eur J Endocrinol. 2009;160:695–703. doi: 10.1530/EJE-08-0662. [DOI] [PubMed] [Google Scholar]
- 49.Fang X, Cai Y, Liu J, et al. Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and cancer stem-like cell self-renewal. Oncogene. 2011;30:4707–4720. doi: 10.1038/onc.2011.181. [DOI] [PubMed] [Google Scholar]
- 50.Mani SA, Yang J, Brooks M, et al. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A. 2007;104:10069–10074. doi: 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo F, Parker Kerrigan BC, Yang D, et al. Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J Hematol Oncol. 2014;7:19. doi: 10.1186/1756-8722-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alberga A, Boulay JL, Kempe E, Dennefeld C, Haenlin M. The snail gene required for mesoderm formation in Drosophila is expressed dynamically in derivatives of all three germ layers. Development. 1991;111:983–992. doi: 10.1242/dev.111.4.983. [DOI] [PubMed] [Google Scholar]
- 53.Peinado H, Ballestar E, Esteller M, Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol. 2004;24:306–319. doi: 10.1128/MCB.24.1.306-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olmeda D, Moreno-Bueno G, Flores JM, Fabra A, Portillo F, Cano A. SNAI1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer Res. 2007;67:11721–11731. doi: 10.1158/0008-5472.CAN-07-2318. [DOI] [PubMed] [Google Scholar]
- 55.Moody SE, Perez D, Pan TC, et al. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005;8:197–209. doi: 10.1016/j.ccr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 56.Dave N, Guaita-Esteruelas S, Gutarra S, et al. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. 2011;286:12024–12032. doi: 10.1074/jbc.M110.168625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vincent T, Neve EP, Johnson JR, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11:943–950. doi: 10.1038/ncb1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iseri OD, Kars MD, Arpaci F, Atalay C, Pak I, Gunduz U. Drug resistant MCF-7 cells exhibit epithelial-mesenchymal transition gene expression pattern. Biomed Pharmacother. 2011;65:40–45. doi: 10.1016/j.biopha.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 59.Kaaks R, Johnson T, Tikk K, et al. Insulin-like growth factor I and risk of breast cancer by age and hormone receptor status-A prospective study within the EPIC cohort. Int J Cancer. 2014;134:2683–2690. doi: 10.1002/ijc.28589. [DOI] [PubMed] [Google Scholar]
- 60.Schernhammer ES, Holly JM, Hunter DJ, Pollak MN, Hankinson SE. Insulin-like growth factor-I, its binding proteins (IGFBP-1 and IGFBP-3), and growth hormone and breast cancer risk in The Nurses Health Study II. Endocr Relat Cancer. 2006;13:583–592. doi: 10.1677/erc.1.01149. [DOI] [PubMed] [Google Scholar]
- 61.Baglietto L, English DR, Hopper JL, Morris HA, Tilley WD, Giles GG. Circulating insulin-like growth factor-I and binding protein-3 and the risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:763–768. doi: 10.1158/1055-9965.EPI-06-0960. [DOI] [PubMed] [Google Scholar]
- 62.Endogenous Hormones and Breast Cancer Collaborative Group. Key TJ, Appleby PN, Reeves GK, Roddam AW. Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: pooled individual data analysis of 17 prospective studies. Lancet Oncol. 2010;11:530–542. doi: 10.1016/S1470-2045(10)70095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hawsawi Y, El-Gendy R, Twelves C, Speirs V, Beattie J. Insulin-like growth factor – oestradiol crosstalk and mammary gland tumourigenesis. Biochim Biophys Acta. 2013;1836:345–353. doi: 10.1016/j.bbcan.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 64.Tsonis AI, Afratis N, Gialeli C, et al. Evaluation of the coordinated actions of estrogen receptors with epidermal growth factor receptor and insulin-like growth factor receptor in the expression of cell surface heparan sulfate proteoglycans and cell motility in breast cancer cells. FEBS J. 2013;280:2248–2259. doi: 10.1111/febs.12162. [DOI] [PubMed] [Google Scholar]
- 65.Keinan-Boker L, Bueno De Mesquita HB, Kaaks R, et al. Circulating levels of insulin-like growth factor I, its binding proteins-1, -2, -3, C-peptide and risk of postmenopausal breast cancer. Int J Cancer. 2003;106:90–95. doi: 10.1002/ijc.11193. [DOI] [PubMed] [Google Scholar]
- 66.Gronbaek H, Flyvbjerg A, Mellemkjaer L, et al. Serum insulin-like growth factors, insulin-like growth factor binding proteins, and breast cancer risk in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2004;13:1759–1764. [PubMed] [Google Scholar]
- 67.Renehan AG, Zwahlen M, Minder C, O’Dwyer ST, Shalet SM, Egger M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet. 2004;363:1346–1353. doi: 10.1016/S0140-6736(04)16044-3. [DOI] [PubMed] [Google Scholar]
- 68.Livingstone C. IGF2 and cancer. Endocr Relat Cancer. 2013;20:R321–R339. doi: 10.1530/ERC-13-0231. [DOI] [PubMed] [Google Scholar]
- 69.Hirano H, Lopes MB, Laws ER, Jr, et al. Insulin-like growth factor-1 content and pattern of expression correlates with histopathologic grade in diffusely infiltrating astrocytomas. Neuro-Oncology. 1999;1:109–119. doi: 10.1093/neuonc/1.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sohda T, Oka Y, Iwata K, et al. Co-localisation of insulin-like growth factor II and the proliferation marker MIB1 in hepatocellular carcinoma cells. J Clin Pathol. 1997;50:135–137. doi: 10.1136/jcp.50.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kawamoto K, Onodera H, Kondo S, et al. Expression of insulin-like growth factor-2 can predict the prognosis of human colorectal cancer patients: correlation with tumor progression, proliferative activity and survival. Oncology. 1998;55:242–248. doi: 10.1159/000011858. [DOI] [PubMed] [Google Scholar]
- 72.Rogler CE, Yang D, Rossetti L, et al. Altered body composition and increased frequency of diverse malignancies in insulin-like growth factor-II transgenic mice. J Biol Chem. 1994;269:13779–13784. [PubMed] [Google Scholar]
- 73.Guerra FK, Eijan AM, Puricelli L, et al. Varying patterns of expression of insulin-like growth factors I and II and their receptors in murine mammary adenocarcinomas of different metastasizing ability. Int J Cancer. 1996;65:812–820. doi: 10.1002/(SICI)1097-0215(19960315)65:6<812::AID-IJC18>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 74.Papa V, Gliozzo B, Clark GM, et al. Insulin-like growth factor-I receptors are overexpressed and predict a low risk in human breast cancer. Cancer Res. 1993;53:3736–3740. [PubMed] [Google Scholar]
- 75.Jammes H, Peyrat JP, Ban E, et al. Insulin-like growth factor 1 receptors in human breast tumour: localisation and quantification by histo-autoradiographic analysis. Br J Cancer. 1992;66:248–253. doi: 10.1038/bjc.1992.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Valentinis B, Baserga R. IGF-I receptor signalling in transformation and differentiation. Mol Pathol. 2001;54:133–137. doi: 10.1136/mp.54.3.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peruzzi F, Prisco M, Dews M, et al. Multiple signaling pathways of the insulin-like growth factor 1 receptor in protection from apoptosis. Mol Cell Biol. 1999;19:7203–7215. doi: 10.1128/mcb.19.10.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim HJ, Litzenburger BC, Cui X, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol. 2007;27:3165–3175. doi: 10.1128/MCB.01315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xie Y, Skytting B, Nilsson G, Brodin B, Larsson O. Expression of insulin-like growth factor-1 receptor in synovial sarcoma: association with an aggressive phenotype. Cancer Res. 1999;59:3588–3591. [PubMed] [Google Scholar]
- 80.Long L, Rubin R, Brodt P. Enhanced invasion and liver colonization by lung carcinoma cells overexpressing the type 1 insulin-like growth factor receptor. Exp Cell Res. 1998;238:116–121. doi: 10.1006/excr.1997.3814. [DOI] [PubMed] [Google Scholar]
- 81.Saldana SM, Lee HH, Lowery FJ, et al. Inhibition of type I insulin-like growth factor receptor signaling attenuates the development of breast cancer brain metastasis. PLoS One. 2013;8:e73406. doi: 10.1371/journal.pone.0073406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chang L, Graham PH, Hao J, et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875. doi: 10.1038/cddis.2013.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Irie HY, Pearline RV, Grueneberg D, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guvakova MA, Surmacz E. The activated insulin-like growth factor I receptor induces depolarization in breast epithelial cells characterized by actin filament disassembly and tyrosine dephosphorylation of FAK, Cas, and paxillin. Exp Cell Res. 1999;251:244–255. doi: 10.1006/excr.1999.4566. [DOI] [PubMed] [Google Scholar]
- 85.Sorokin AV, Chen J. MEMO1, a new IRS1-interacting protein, induces epithelial-mesenchymal transition in mammary epithelial cells. Oncogene. 2013;32:3130–3138. doi: 10.1038/onc.2012.327. [DOI] [PubMed] [Google Scholar]
- 86.Lorenzatti G, Huang W, Pal A, Cabanillas AM, Kleer CG. CCN6 (WISP3) decreases ZEB1-mediated EMT and invasion by attenuation of IGF-1 receptor signaling in breast cancer. J Cell Sci. 2011;124:1752–1758. doi: 10.1242/jcs.084194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Graham TR, Zhau HE, Odero-Marah VA, et al. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008;68:2479–2488. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- 88.Pizon M, Zimon D, Carl S, Pachmann U, Pachmann K, Camara O. Heterogeneity of circulating epithelial tumour cells from individual patients with respect to expression profiles and clonal growth (sphere formation) in breast cancer. Ecancermedicalscience. 2013;7:343. doi: 10.3332/ecancer.2013.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Duggan C, Wang CY, Neuhouser ML, et al. Associations of insulin-like growth factor and insulin-like growth factor binding protein-3 with mortality in women with breast cancer. Int J Cancer. 2013;132:1191–1200. doi: 10.1002/ijc.27753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morgillo F, De Vita F, Antoniol G, et al. Serum insulin-like growth factor 1 correlates with the risk of nodal metastasis in endocrine-positive breast cancer. Curr Oncol. 2013;20:e283–e288. doi: 10.3747/co.20.1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dupont J, Khan J, Qu BH, Metzler P, Helman L, LeRoith D. Insulin and IGF-1 induce different patterns of gene expression in mouse fibroblast NIH-3T3 cells: identification by cDNA microarray analysis. Endocrinology. 2001;142:4969–4975. doi: 10.1210/endo.142.11.8476. [DOI] [PubMed] [Google Scholar]
- 92.Dupont J, Fernandez AM, Glackin CA, Helman L, LeRoith D. Insulin-like growth factor 1 (IGF-1)-induced twist expression is involved in the anti-apoptotic effects of the IGF-1 receptor. J Biol Chem. 2001;276:26699–26707. doi: 10.1074/jbc.M102664200. [DOI] [PubMed] [Google Scholar]
- 93.Sciacca L, Costantino A, Pandini G, et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999;18:2471–2479. doi: 10.1038/sj.onc.1202600. [DOI] [PubMed] [Google Scholar]
- 94.Pandini G, Vigneri R, Costantino A, et al. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res. 1999;5:1935–1944. [PubMed] [Google Scholar]
- 95.Harrington SC, Weroha SJ, Reynolds C, Suman VJ, Lingle WL, Haluska P. Quantifying insulin receptor isoform expression in FFPE breast tumors. Growth Horm IGF Res. 2012;22:108–115. doi: 10.1016/j.ghir.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oates AJ, Schumaker LM, Jenkins SB, et al. The mannose 6-phosphate/insulin-like growth factor 2 receptor (M6P/IGF2R), a putative breast tumor suppressor gene. Breast Cancer Res Treat. 1998;47:269–281. doi: 10.1023/a:1005959218524. [DOI] [PubMed] [Google Scholar]
- 97.De Souza AT, Hankins GR, Washington MK, Fine RL, Orton TC, Jirtle RL. Frequent loss of heterozygosity on 6q at the mannose 6-phosphate/insulin-like growth factor II receptor locus in human hepatocellular tumors. Oncogene. 1995;10:1725–1729. [PubMed] [Google Scholar]
- 98.Byrd JC, Devi GR, de Souza AT, Jirtle RL, MacDonald RG. Disruption of ligand binding to the insulin-like growth factor II/mannose 6-phosphate receptor by cancer-associated missense mutations. J Biol Chem. 1999;274:24408–24416. doi: 10.1074/jbc.274.34.24408. [DOI] [PubMed] [Google Scholar]
- 99.O’Gorman DB, Costello M, Weiss J, Firth SM, Scott CD. Decreased insulin-like growth factor-II/mannose 6-phosphate receptor expression enhances tumorigenicity in JEG-3 cells. Cancer Res. 1999;59:5692–5694. [PubMed] [Google Scholar]
- 100.Souza RF, Wang S, Thakar M, et al. Expression of the wild-type insulin-like growth factor II receptor gene suppresses growth and causes death in colorectal carcinoma cells. Oncogene. 1999;18:4063–4068. doi: 10.1038/sj.onc.1202768. [DOI] [PubMed] [Google Scholar]
- 101.Oh Y, Muller HL, Ng L, Rosenfeld RG. Transforming growth factor-beta-induced cell growth inhibition in human breast cancer cells is mediated through insulin-like growth factor-binding protein-3 action. J Biol Chem. 1995;270:13589–13592. doi: 10.1074/jbc.270.23.13589. [DOI] [PubMed] [Google Scholar]
- 102.Shemer J, Yaron A, Werner H, et al. Regulation of insulin-like growth factor (IGF) binding protein-5 in the T47D human breast carcinoma cell line by IGF-I and retinoic acid. J Clin Endocrinol Metab. 1993;77:1246–1250. doi: 10.1210/jcem.77.5.7521344. [DOI] [PubMed] [Google Scholar]
- 103.Colston KW, Perks CM, Xie SP, Holly JM. Growth inhibition of both MCF-7 and Hs578T human breast cancer cell lines by vitamin D analogues is associated with increased expression of insulin-like growth factor binding protein-3. J Mol Endocrinol. 1998;20:157–162. doi: 10.1677/jme.0.0200157. [DOI] [PubMed] [Google Scholar]
- 104.Karas M, Kleinman D, Danilenko M, et al. Components of the IGF system mediate the opposing effects of tamoxifen on endometrial and breast cancer cell growth. Prog Growth Factor Res. 1995;6:513–520. doi: 10.1016/0955-2235(95)00033-x. [DOI] [PubMed] [Google Scholar]
- 105.Nishimura A, Fujimoto M, Oguchi S, Fusunyan RD, MacDermott RP, Sanderson IR. Short-chain fatty acids regulate IGF-binding protein secretion by intestinal epithelial cells. Am J Physiol. 1998;275:E55–E63. doi: 10.1152/ajpendo.1998.275.1.E55. [DOI] [PubMed] [Google Scholar]
- 106.Clemmons DR, Camacho-Hubner C, Coronado E, Osborne CK. Insulin-like growth factor binding protein secretion by breast carcinoma cell lines: correlation with estrogen receptor status. Endocrinology. 1990;127:2679–2686. doi: 10.1210/endo-127-6-2679. [DOI] [PubMed] [Google Scholar]
- 107.Yu H, Levesque MA, Khosravi MJ, Papanastasiou-Diamandi A, Clark GM, Diamandis EP. Insulin-like growth factor-binding protein-3 and breast cancer survival. Int J Cancer. 1998;79:624–628. doi: 10.1002/(sici)1097-0215(19981218)79:6<624::aid-ijc12>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 108.Rocha RL, Hilsenbeck SG, Jackson JG, et al. Insulin-like growth factor binding protein-3 and insulin receptor substrate-1 in breast cancer: correlation with clinical parameters and disease-free survival. Clin Cancer Res. 1997;3:103–109. [PubMed] [Google Scholar]
- 109.Vestey S, Perks C, Sen C, Calder C, Holly J, Winters Z. Immunohistochemical expression of insulin-like growth factor binding protein-3 in invasive breast cancers and ductal carcinoma in situ: implications for clinicopathology and patient outcome. Breast Cancer Res. 2005;7:R119–R129. doi: 10.1186/bcr963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hollowood AD, Lai T, Perks CM, Newcomb PV, Alderson D, Holly JM. IGFBP-3 prolongs the p53 response and enhances apoptosis following UV irradiation. Int J Cancer. 2000;88:336–341. [PubMed] [Google Scholar]
- 111.Gill ZP, Perks CM, Newcomb PV, Holly JM. Insulin-like growth factor-binding protein (IGFBP-3) predisposes breast cancer cells to programmed cell death in a non-IGF-dependent manner. J Biol Chem. 1997;272:25602–25607. doi: 10.1074/jbc.272.41.25602. [DOI] [PubMed] [Google Scholar]
- 112.Williams AC, Collard TJ, Perks CM, et al. Increased p53-dependent apoptosis by the insulin-like growth factor binding protein IGFBP-3 in human colonic adenoma-derived cells. Cancer Res. 2000;60:22–27. [PubMed] [Google Scholar]
- 113.Fowler CA, Perks CM, Newcomb PV, Savage PB, Farndon JR, Holly JM. Insulin-like growth factor binding protein-3 (IGFBP-3) potentiates paclitaxel-induced apoptosis in human breast cancer cells. Int J Cancer. 2000;88:448–453. [PubMed] [Google Scholar]
- 114.McCaig C, Fowler CA, Laurence NJ, et al. Differential interactions between IGFBP-3 and transforming growth factor-beta (TGF-beta) in normal vs cancerous breast epithelial cells. Br J Cancer. 2002;86:1963–1969. doi: 10.1038/sj.bjc.6600355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yu Q, Banerjee K, Paterson J, Alami N, Shiry L, Leyland-Jones B. IGFBP-3: Single Agent and Synergistic Effects with Chemotherapeutic Drugs on Solid Tumor Models, AACR; Proceedings of the AACR, 172: Abstract 755; Washington DC: 2003. [Google Scholar]
- 116.McIntosh J, Dennison G, Holly JM, et al. IGFBP-3 can either inhibit or enhance EGF-mediated growth of breast epithelial cells dependent upon the presence of fibronectin. J Biol Chem. 2010;285:38788–38800. doi: 10.1074/jbc.M110.177311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Burrows C, Holly JM, Laurence NJ, et al. Insulin-like growth factor binding protein 3 has opposing actions on malignant and nonmalignant breast epithelial cells that are each reversible and dependent upon cholesterol-stabilized integrin receptor complexes. Endocrinology. 2006;147:3484–3500. doi: 10.1210/en.2006-0005. [DOI] [PubMed] [Google Scholar]
- 118.Ioachim E, Charchanti A, Briasoulis E, et al. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression. Eur J Cancer. 2002;38:2362–2370. doi: 10.1016/s0959-8049(02)00210-1. [DOI] [PubMed] [Google Scholar]
- 119.Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer. 2014;14:329–341. doi: 10.1038/nrc3720. [DOI] [PubMed] [Google Scholar]
- 120.Johnson MA, Firth SM. IGFBP-3: a cell fate pivot in cancer and disease. Growth Horm IGF Res. 2014;24(5):164–173. doi: 10.1016/j.ghir.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 121.Walsh LA, Damjanovski S. IGF-1 increases invasive potential of MCF 7 breast cancer cells and induces activation of latent TGF-β1 resulting in epithelial to mesenchymal transition. Cell Commun Signal. 2011;9:10. doi: 10.1186/1478-811X-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reiss M, Barcellos-Hoff MH. Transforming growth factor-beta in breast cancer: a working hypothesis. Breast Cancer Res Treat. 1997;45:81–95. doi: 10.1023/a:1005865812918. [DOI] [PubMed] [Google Scholar]
- 123.Dieci MV, Orvieto E, Dominici M, Conte P, Guarneri V. Rare breast cancer subtypes: histological, molecular, and clinical peculiarities. Oncologist. 2014;19:805–813. doi: 10.1634/theoncologist.2014-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kittaneh M, Montero AJ, Gluck S. Molecular profiling for breast cancer: a comprehensive review. Biomarkers Cancer. 2013;5:61–70. doi: 10.4137/BIC.S9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Boone DN, Lee AV. Targeting the insulin-like growth factor receptor: developing biomarkers from gene expression profiling. Crit Rev Oncog. 2012;17:161–173. doi: 10.1615/critrevoncog.v17.i2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee AV, Yee D. Targeting IGF-1R: at a crossroad. Oncology (Williston Park) 2011;25:535–536. discussion 551. [PMC free article] [PubMed] [Google Scholar]
- 127.Chen HX, Sharon E. IGF-1R as an anti-cancer target – trials and tribulations. Chin J Cancer. 2013;32:242–252. doi: 10.5732/cjc.012.10263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Alami N, Page V, Yu Q, et al. Recombinant human insulin-like growth factor-binding protein 3 inhibits tumor growth and targets the Akt pathway in lung and colon cancer models. Growth Horm IGF Res. 2008;18:487–496. doi: 10.1016/j.ghir.2008.04.002. [DOI] [PubMed] [Google Scholar]