

Abstract
Since loss of VHL is frequently detected early phase genetic event in human renal cell carcinoma, pVHL is assumed to be indispensable for suppression of tumor initiation step. However, induction of HIF-1α, target of pVHL E3 ligase, is more adequate to angiogenesis step after tumor mass formation. Concerning this, it has been reported that pVHL is involved in centrosome location during metaphase and regulates ER-α signaling. Here, we provide the evidences that pVHL-mediated ER-α suppression is critical for microtubule organizing center (MTOC) maintaining and elevated ER-α promotes MTOC amplification through disruption of BRCA1-Rad51 interaction. In fact, numerous MTOC in VHL- or BRCA1-deficient cells are reduced by Fulvestrant, inhibitor of ER-α expression as well as antagonist. In addition, we reveal that activation of ER signaling can increase γ-tubulin, core factor of TuRC and render the resistance to Taxol. Thus, Fulvestrant but not Tamoxifen, antagonist against ER-α, can restore the Taxol sensitivity in VHL- or BRCA1-deficient cells. Our results suggest that pVHL-mediated ER-α suppression is important for regulation of MTOC as well as drug resistance.
Introduction
The number of centrosome should be tightly regulated through entire cell cycle for proper chromosome segregation and prevention of aneuploidy. Thus, centrosomal amplification or mis-location have been reported in several kinds of cancer with chromosomal instability (CIN), which is one of strong driving force for mutation accumulation during cancer initiation, with microsatellite instability (MIN). For example, APC (adenomatous polypus coli) [1], frequently mutated tumor suppressor in colon cancer, is involved in chromosomal stability as well as β-catenin signaling [2]. BRCA1 is also known to be involved in chromosomal stability, coupled with DNA repair system [3], [4]. However, despite importance of CIN, regulation mechanisms in other cancers including renal cell carcinoma (RCC) is not clearly demonstrated until now.
VHL (von Hippel-Lindau syndrome gene) is mutated over 70% of clear cell type renal cell carcinoma from early stage [5], [6], implying that pVHL is critical for cancer initiation processes such as cell cycle regulation and maintaining of genetic materials against mutation. However, pVHL, as the E3 ligase, promotes HIF-1α degradation, sensing the oxygen level [7], [8], [9], suggesting that there would be additional pVHL role in tumor initiation, because HIF-1α-mediated angiogenesis is required at late stage. Concerning this, it has been reported that pVHL regulates centrosome localization [10], senescence [11], and ER-α signaling [12]. In addition, pVHL involves in primary cilium maintaining, providing new insight that pVHL would be related with microtubule regulation [13]. However, until now, cancer-initiation-related function of pVHL in RCC has not been clearly demonstrated.
One of notorious feature of RCC is drug resistance, which reduces survival of metastatic RCC below to 10% [14], [15]. However, detail molecular mechanism about the drug resistance has not been suggested. In recent, we revealed that loss of pVHL can suppress p14/ARF activity through increase of progerin [16] that would be one of reason for drug resistance, because senescent cells are not responding to extracellular stresses as well as apoptosis [17]. Despite this, the resistance to Taxol, widely used microtubule de-polymerization inhibitor for anti-cancer drug, is not explained by p14/ARF-progerin network.
Therefore, in this study, we tried to reveal the tumor suppressive role of pVHL in tumor initiation. In addition to cell proliferation, elevated ER-α in VHL-deficient cells induced MTOC amplification via disruption of BRCA1-Rad51 binding. These results indicate that proper regulation of ER-α expression by pVHL or BRCA1 is important for MTOC regulation and prevention of chromosomal instability.
Materials and Methods
Cell Cultures and Reagents
HEK293, MCF-7, and MDA-MB-468 (DMEM) were purchased from ATCC (Manassas, VA). HCT116 p53 −/− cell line was provided by Dr. Vogelstein B (Johns Hopkins University). ACHN, A498, A704 (DMEM) and HCC1937 (RPMI) cells were obtained from Korea cell line bank. Other Cell lines (UMRC2; C2, UMRC2/VHL; C2V), provided by Dr. Jung, YJ (Pusan National University). Cells were maintained in DMEM. All kinds of cell lines were maintained in liquid medium containing 10% FBS and 1% antibiotics at 37°C growth chamber. General chemical inhibitors including Adriamycin (324380) and Colcemid (234109) were purchased from Calbiochem. B02 (SML0364), Estrogen (250155), Fulvestrant (I4409), Taxol (T7402), Tamoxifen (T5648) and 4-OHT (H7904) were purchased from Sigma. Antibodies against GST (sc-138), Actin (sc-1616), ER-α (sc-8002), β-tubulin (sc-9104) and HA (sc-7392) were purchased from Santa Cruz. Anti-γ-tubulin (T6557) and Myc (M5546) were provided by Sigma, anti-pVHL Ab (2738) was obtained from Cell signaling. Rad51 (05–530), BRCA1 (07–434) were purchased from Milliopore.
Vectors and Transfection
The Myc-fused BRCA1-wild type, F6 and F6 M1775R vector were presented from Dr. Livingston, DM (Harvard Medical School). pVHL mammalian expression vectors were obtained from Dr. Jung, YJ (Pusan National University). The HA-tagged HIF-1α expression vector was generously provided by Dr. Kim, YJ (Pusan National University). The HA-fused VHL-L158S, C162F, R167W [18] and TALEN (TAL2302, 2303, 2384, 2385) [19] vector were purchased from Addgene. For the in vitro gene knock out, Si-RNAs against target proteins were generated. The Si-RNA target sequences are as follows: VHL: 5′-ACA CAG GAG CGC ATT GCA CAT-3′; Rad51: 5′-GAG CTT GAC AAA CTA CTT-3′; HIF-1α: 5′-TAC GTT GTG AGT GGT ATT ATT-3′; HIF-2α: 5′-CAG CAT CTT TGA TAG CAG T-3′. For mammalian expression of these vectors, transfection was performed using Jetpei transfection agent (Polyplus). In brief, the vector (1.5 μg) was mixed with 1.5 μl of Jetpei reagent in 150 nM NaCl solution. The mixture is incubated for 15 min at RT. After incubation, the mixture was added to the cell. After 3 hours, the serum-free medium was replaced to 10% FBS contained medium.
Western Blot Analysis and Protein Interaction Studies
Proteins were extracted from cells with RIPA buffer. Samples were applied to SDS-PAGE, and western blot analysis was performed by means of a general protocol. Blotted membranes were incubated with primary antibodies for 1 hour to overnight at 4°C and HRP-conjugate species-matched secondary antibodies for 1 hour at RT. For immunoprecipitation (IP) analysis, whole-cell lysates were incubated first with the proper antibodies for 4 hours at 4°C and then with protein A/G agarose beads (Invitrogen) for 2 hours at 4°C. After centrifugation and washing with RIPA, the precipitated immunocomplexes were subjected to SDS-PAGE and western blot analysis. To determine the direct interaction between proteins, agarose bead-conjugated GST-γ-tubulin (Novus) was incubated with cell lysates for 4 hours at 4°C. Using the same procedure with IP, precipitated proteins were analyzed by western blot analysis.
Immunofluorescence Staining
Cells were seeded on a cover glass and transfected with the indicated vectors or treated with the indicated chemicals. After fixing with Me-OH for 30 min, the cells were incubated with blocking buffer [PBS + anti-human-Ab (1:500)] for 1 hours. After washing with PBS, the cells were incubated with anti-γ-tubulin or β-tubulin antibodies in blocking buffer (1: 100 to 200) for 4 hours and subsequently with FITC-conjugated or Rhodamine-conjugated secondary antibodies in blocking buffer (1: 500) for 2 hours. The nucleus was stained by DAPI. After washing with PBS, cover glasses were mounted with mounting solution (Vector Laboratories). The immunofluorescence signal was detected through fluorescence microscopy (Zeiss).
MTT Assay
To measure the cell viability, cells were treated with the indicated chemicals for 4 days. For the MTT assay, cells were incubated with 0.5 mg/ml of MTT solution (Calbiochem) for 4 hours at 37°C. After removing the excess solution, the precipitated materials were dissolved in 200 μl DMSO and quantified by measuring the absorbance at 540 nm.
Statistical Analysis
To obtain the statistical significance, we performed the Student's t test.
Results
pVHL Determines Taxol Resistance
Since RCC is resistant to chemo-therapy [20], we checked the sensitivity to Taxol in several RCC cell lines and found that VHL-positive cell lines were sensitive to Taxol-induced cytotoxicity (Figure 1A). Cleavage of PARP and Cytochrome C release assay [21], [22] indicated that Taxol-induced cytotoxicity in VHL-positive cells was mediated by apoptosis (Figure 1, B and C). To clarify the involvement of pVHL on Taxol sensitivity, we monitored the cytotoxicity in UMRC2 (C2) and its isogenic VHL expressed UMRC2V (C2V), after transfection with VHL or si-VHL, respectively, (Supplementary Figure S1A and B). Re-expression of VHL in C2 could make be sensitized to taxol (Figure 1D), whereas VHL knockdown provided resistance to Taxol in C2V, indicating that pVHL was critical for determining the Taxol-sensitivity (Figure 1D). However, loss of VHL provided only the tolerance but not complete resistance against Taxol (Supplementary Figure S1C), so that high concentration of Taxol could reduce cell viability even in VHL-deficient cell. Since Taxol blocked the depolymerization of tubulin fiber during M-phase [23], we speculated that increase of microtubule might provide resistance to Taxol and checked the expression of γ-tubulin in RCC cell lines. Interestingly, expression of γ-tubulin showed the reverse-relationship with VHL except HCC1937, human breast cancer cell line (Figure 1E). Moreover, γ-tubulin positive MTOCs were increased in VHL-deficient RCC cell lines such as C2 and A498 (Figure 1F and Supplementary Figure S1D). To know how γ-tubulin increase provide Taxol resistance, we monitored β-tubulin in VHL-deficient A498, comparing with VHL-intact ACHN. Taxol blocked β-tubulin fiber formation in ACHN (Supplementary Figure S1E). In contrast, A498 still showed the β-tubulin fiber formation, despite Taxol-treatment (Supplementary Figure S1E). These results suggest that increase of γ-tubulin may produce additional MTOC, which serve for microtubule formation.
Figure 1.
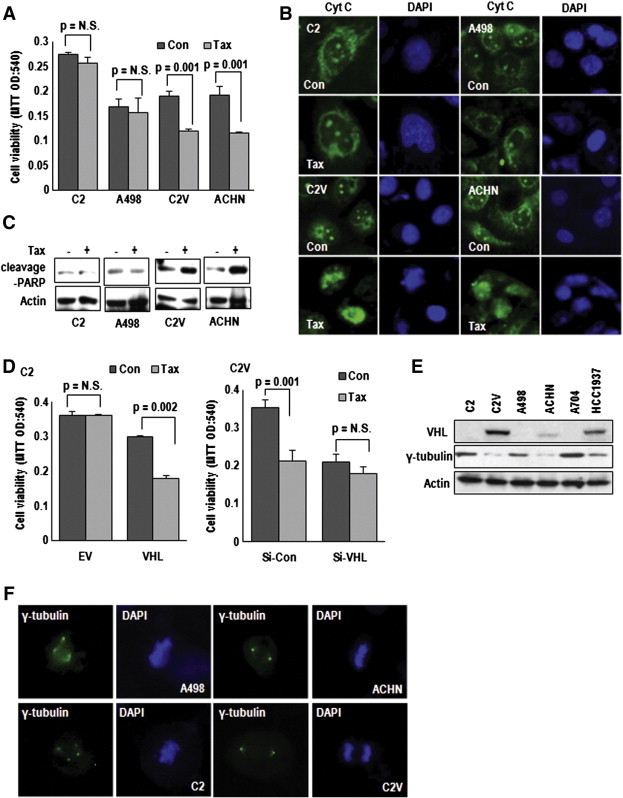
VHL determines Taxol resistance. (A) VHL-positive cell lines are sensitive to Taxol (Tax) treatment. VHL-positive (ACHN and C2V) and VHL-negative (C2 and A498) cells were treated with Tax (3 μM) for 72 hours. The cell viability was monitored by MTT assay. (B and C) Tax-induced cytotoxicity is achieved by apoptosis. Cells were treated with Tax for 72 hours. After fixation, cells were stained with cytochrome C (Cyt C; green) and DAPI (blue) for cyt C release from mitochondria to cytoplasm and nucleus (B). Under same conditions, cleavage-PARP was monitored. Increase of cleavage PARP in VHL-positive cells (C2V and ACHN) was detected (C). (D) pVHL reduces Tax resistance. C2 (left panel) and C2V cells (right panel) were transfected with VHL expression vector or Si-VHL, respectively. After transfection, Tax was treated for 72 hours. The cell viability was monitored by MTT assay. EV, Empty vector; Si-Con, Si-control. (E) Differential expression of γ-tubulin by VHL-status. γ-tubulin was highly expressed in VHL-negative RCC cell lines (C2, A498 and A704), comparing to VHL-positive RCC (C2V and ACHN). HCC1937 is human breast cancer cell line. (F) γ-tubulin positive centrosome is increased in VHL-negative cell lines. Cells were immunofluorescence (IF) stained with γ-tubulin (green) and DAPI (blue).
pVHL Regulates MTOC Number
On the basis of MTOC amplification in VHL-deficient cells, we measured MTOC number and observed the increase of it by si-VHL in C2V (Figure 2A and Supplementary Figure S2A) and restoration by VHL transfection into C2 (Figure 2B, Supplementary Figure S2B, and Figure 2C). Consistently, γ-tubulin expression was reduced by pVHL and increased by si-VHL (Figure 2D). Since VHL is also mutated in RCC, we checked the effect of mutant VHL (L158S, C162F, and R167W) on MTOC amplification and Taxol-resistance. Only WT-VHL, but not mutated-VHL, did suppress γ-tubulin expression (Figure 2E). Moreover, VHL mutants did not show favorable effects on Taxol-induced cell death and MTOC amplification in VHL-null cell lines (Figure 2, F and G and Supplementary Figure S2C). Considering that these mutants are E3 ligase defective form [18], E3 ligase activity is required for maintaining of MTOC.
Figure 2.
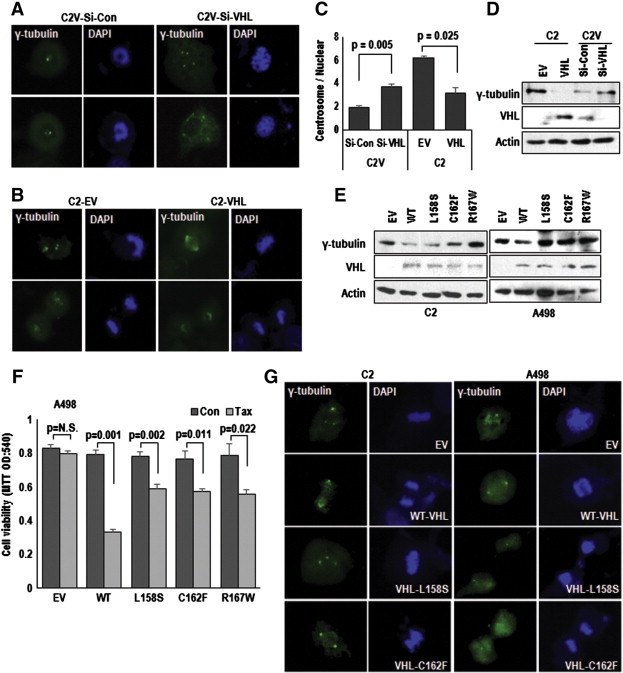
VHL regulates centrosome number. (A−C) VHL down-regulates centrosome number. Elimination of VHL in C2V increased centrosome number (A), whereas VHL-transfection into C2 could reduce centrosome number (B). Cells were transfected with indicated vectors or Si-RNA for 24 hours and incubated for 72 hours. After transfection, cells were IF stained with γ-tubulin (green) and DAPI (blue). Centrosome number was counted about 50 cells (C). (D) pVHL status determines γ-tubulin expression. Indicated vectors transfected for 24 hours. Actin was used for loading control. (E) The expression of γ-tubulin was regulated through E3 ligase activity of pVHL. VHL-negative cells (C2 and A498) were transfected with WT or E3 ligase activity mutated VHL (L158S, C162F and R167W) vectors for 24 hours. (F) The effect of VHL mutants on Tax-resistance. A498 was not sensitized by mutant VHL transfection. After Tax (3 μM) treatment for 72 hours, the transfected cell's viability was measured by MTT assay. (G) The effect of mutant VHL on centrosome amplification. For IF staining, cells were fixed before stained with γ-tubulin (green) and DAPI (blue).
ER-α is Responsible for MTOC Amplification
To verify the target of pVHL, responding to MTOC amplification and Taxol-resistance, we first checked the effect of HIF-1α and HIF-2α on Taxol-resistance. Elimination or overexpression of HIFs did not change the sensitivity to Taxol (Figure 3A and Supplementary Figure S3A and B). So, we focused on ER-α, which has been recently identified as target of pVHL [13]. Si-ER-α could sensitize VHL-deficient cell lines to Taxol and also Colcemid (Figure 3B and Supplementary Figure S3C), whereas ER-α overexpression rendered the resistance to Taxol in VHL-intact ACHN (Figure 3C). However, pVHL and ER-α did not alter the sensitivity to Adriamycin (Figure 3D), suggesting that pVHL-ER-α network was not linked to general drug sensitivity, but to Taxol. To confirm the engagement of ER-α in Taxol-resistance, we examined the Taxol-induced cytotoxicity in C2, Caki-2 and MDA-MB-468 (ER-α negative breast cancer cell line) and found that Fulvestrant (FST), ER-α inhibitor and destabilizer, could sensitize these cell lines to Taxol, except MDA-MB-468 (Supplementary Figure S3D-F). Next, we stained MTOC using γ-tubulin in C2 cell and revealed that FST could block the MTOC amplification (Figure 3E and Supplementary Figure S3G). In addition, we observed the MTOC-increase by treatment of Estrogen (Est; Figure 3F). The inhibitory effect of FST on MTOC amplification and γ-tubulin expression were confirmed in A498, in which FST could reduce MTOC number (Figure 3, G–I). However, status of VHL, treatment of Est and FST did not alter γ-tubulin transcripts (Supplementary Figure 3H).
Figure 3.
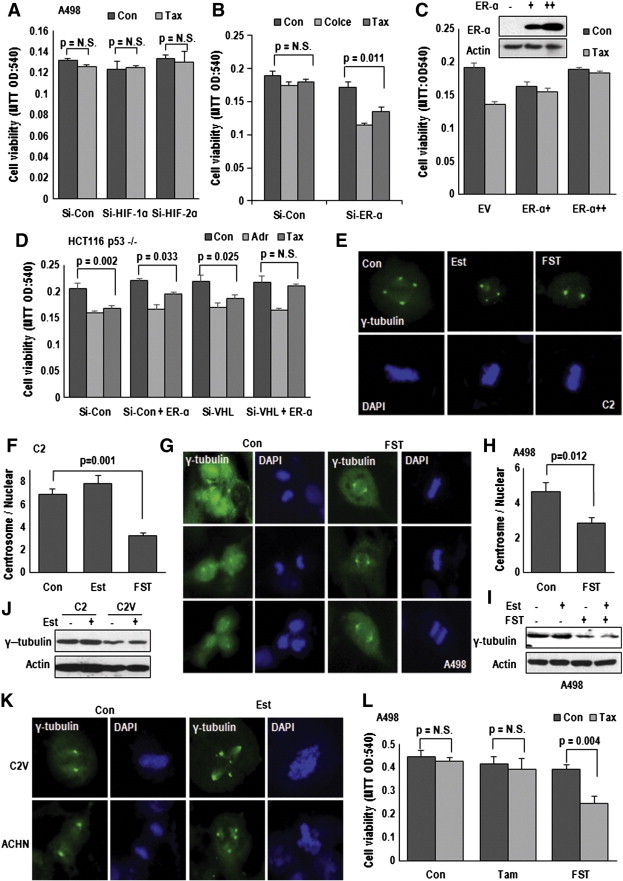
ER-α is responsible for centrosome amplification. (A) Tax sensitivity is not altered by HIFs. Si-HIF-1α or HIF-2α did not sensitize A498 cells to Tax-induced cell death. A498 cells were transfected indicated Si-RNA for 24 hours and incubated with Tax for 72 hours. Cell viability was determined by MTT assay. (B) Elimination of ER-α can restore the sensitivity to Tax and Colcemid (Colce). Si-ER-α sensitized Tax (3 μM) or Col (2 μM)-induced cell death in VHL-deficient A498 cell. A498 cells were transfected with Si-Con (Si-control) or Si-ER-α for 24 hours. After Tax or Col treatment for 72 hours, the viability of transfected A498 cells was monitored by MTT assay. (C) ER-α overexpression induces Tax-resistance. VHL-intact ACHN cells were transfected with ER-α expression vector (upper panel) for 24 hours. MTT assay was used for cell viability measurement (lower panel). (D) pVHL and ER-α are involved only in Tax-sensitivity but not Adriamycin-induced cell death (Adr; 2 μg/ml). Comparing to Tax, Adriamycin sensitivity was not affected pVHL or ER-α status. HCT116 p53 −/− cells were transfected with indicating vectors. After Taxol treatment, the cell viability was monitored by MTT assay. (E and F) Fulvestrant (FST) can block centrosome amplification in VHL-deficient C2 cell. C2 cells were incubated with FST (2 μM) or Estrogen (Est; 1 μg/ml) for 72 hours and IF stained with γ-tubulin (green) and DAPI (blue). Reduction of centrosome was detected in FST-treated cells (E). Centrosome number was counted from about 50 cells of each condition and presented as graph (F). (G and H) The effect of FST on centrosome in A498. Suppression effect of FST on centrosome amplification in A498 was confirmed by IF staining with γ-tubulin Ab (G) and counting (H). Experimental condition was identical to above. (I) Inhibitory effect of FST on γ-tubulin expression. FST reduced Est-induced γ-tubulin expression in A498 cells. (J) Est induces γ-tubulin expression in C2V. Treatment of Est for 72 hours could induce γ-tubulin. However, due to strong background expression, additional induction of γ-tubulin in C2 was not obviously detected. (K) Est promotes centrosome amplification in VHL-intact cell lines, C2V and ACHN. Cells were incubated with Est for 72 hours and IF stained with γ-tubulin Ab (green) and DAPI (blue). (L) The different effect of Tamoxifen (Tam) and FST on Tax-resistance. Tam (2 μM), FST and Tax were treated for 72 hours in VHL-negative A498 cells. The cell viability was estimated by MTT assay.
Estrogen Promotes MTOC Amplification
Since ER-α can induce Taxol-resistance and MTOC amplification, we tested that Est signaling can mimic VHL-deficient condition. To address this, we checked the MTOC number and expression of γ-tubulin in Est-treated C2V. The treatment of Est could induce γ-tubulin expression (Figure 3J) and MTOC numbers (Figure 3K and Supplementary Figure S4A and B). We could obtain the same results from Est-treated ACHN (Supplementary Figure S4C and D). In fact, MTOC amplification could be detected by Est-treatment in HEK293 (Supplementary Figure S4E and F). To determine that simple inhibition of Est-signaling is enough for restoration of MTOC and Taxol-sensitivity, we compared the effect of Tamoxifen (Tam; Estrogen antagonist) [24] and FST, ER-α inhibitor and destabilizer [25]. Tam did not compensate for VHL deficiency-induced Taxol resistance (Figure 3L), although both chemicals could block the Est-induced proliferation (Supplementary Figure S4G). This result indicates that ER-α itself but not transcription activity is important for MTOC amplification.
BRCA1 is Negative Regulator on ER-α-Mediated MTOC Amplification
To investigate how ER-α promotes MTOC amplification, we searched the physical binding partner of ER-α, and BRCA1 was raised as strong candidate. Indeed, BRCA1-ER-α binding has been published [3] and we also observed the interaction of them (Supplementary Figure S5A and B). In addition, involvement of BRCA1 in chromosome stability regulation has been reported by several group [4]. So, we examined the MTOC numbers in BRCA1 deficient HCC1937 [26] and found that FST could suppress the MTOC amplification in this cell (Figure 4A and B). Indeed, BRCA1-deficient cells showed elevated expression of γ-tubulin as well as its mitotic binding partner Rad51 (Figure 4C) and resistance to Taxol, which was recovered by FST (Figure 4D and Supplementary Figure S5C). To confirm the role of BRCA1 in MTOC amplification, we generated BRCA1 knock out ACHN using TALEN system. similarly with HCC1937, BRCA1-knock out ACHN, but not in BRCA2 knockout ACHN, showed the γ-tubulin incease and Rad51 reduction (Figure 4E) and MTOC amplification, which diminished by FST (Supplementary Figure S5D). To determine whether loss of VHL could be compensated by BRCA1 or conversely, we transfected VHL into HCC1937 or BRCA1 into A498. Interestingly, VHL transfection into HCC1937 or BRCA1 in A498 can induce Taxol-mediated cytotoxicity (Figure 4F and G). However, BRCA2 did not alter the sensitivity to Taxol in A498 (Figure 4G). These results indicate that VHL and BRCA1 possess replaceable and equivalent position on ER-α regulation. In addition, activity of Mitomycin C (MMC) was not altered by BRCA1, BRCA2 and VHL transfection in A498 (Figure 4G), indicating that BRCA1-pVHL system is not involved in DNA alkylation-induced cytotoxicity [27] and would be specifically linked to mitotic cell death. In fact, BRCA1 could suppress ER-α expression at translation level, although BRCA1 was also reduced by ER-α (Figure 4H).
Figure 4.
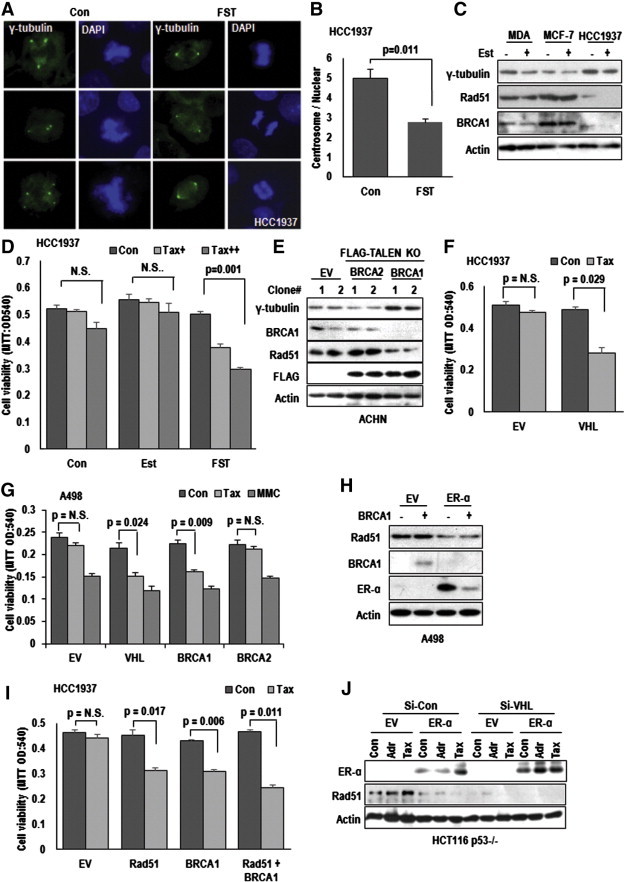
BRCA1 is a negative regulator on ER-α-mediated centrosome amplification. (A and B) Suppressive effect of FST on centrosome amplification in BRCA1-deficient HCC1937 cells. HCC1937 cells were treated with FST (2 μM) for 72 hours. After treatment, cells were IF stained with γ-tubulin (green) and DAPI (blue) (A). The number of centrosome was counted about 50 cells each as conditions (B). (C) The expression of γ-tubulin in human breast cancer cell lines. MDA-MB-468 (ER-α negative), MCF-7 (ER-α positive) and HCC1937 (BRCA1-deficient) cells were incubated with Est (1 μg/ml) for 72 hours. Protein expression was analyzed by western blot. Reduction of Rad51 was also detected in HCC1937. Actin was used for loading control. (D) Sensitization of BRCA1-deficient cells to Tax by FST. Tax-resistance of HCC1937 was restored by FST-treatment (2 μM). MTT assay was used for cell viability monitoring. (E) Deletion of BRCA1 induces γ-tubulin expression. BRCA1, but not BRCA1, knock out ACHN cells showed elevated γ-tubulin expression. ACHN cells were transfected with EV (Empty vector) or TALEN (TALEN2302/2303 for BRCA2 knock-out and TALEN2384/2385 for BRCA1 knock-out) vectors for 5 days in selection media. We performed the TALEN-transfection in replica and obtained the same result from each experiment (clone #). (F) pVHL can sensitize HCC1937 cells to Tax-induced cell death. After transfection with VHL, HCC1937 cells were incubated with Tax for 72 hours and monitored their viability through MTT assay. (G) BRCA1 induces Tax-sensitivity in VHL-negative cells. Consistent with VHL, BRCA1 could reduce Tax resistance. A498 cells were transfected with indicated vectors for 24 hours and treated with Tax or Mitomycin C (MMC; 1.5 μM) for 72 hours. (H) BRCA1 suppresses ER-α expression. A498 cells were transfected with indicated vectors for 24 hours. Protein expression was analyzed by western blot. (I) Rad51 sensitizes Tax-induced cell death. Overexpression of Rad51 could sensitize HCC1937 to Tax-induced cell death. However, Rad51 did not show synergic effect with BRCA1. HCC1937 cells were transfected with indicated vectors for 24 hours. Under same conditions to those described above, cell viability was monitored MTT assay. (J) ER-α overexpression or eliminated VHL suppress Rad51 expression. HCT116 p53 −/− cells were transfected with indicated vectors or si-RNAs for 24 hours. Adriamycin (Adr; 2 μg/ml) and Tax were treated for 24 hours.
Rad51 is a Novel Binding Target of ER-α
Since BRCA1 showed equivalent role with pVHL on ER-α, our next question is what is target of ER-α for regulating the MTOC duplication and Taxol resistance. Since the target should be related with MTOC regulation and BRCA1 pathway, we focused on Rad51. It has been reported to be associated with BRCA1 in mitosis and meiosis [28] and centrosome duplication [29]. In fact, Rad51, but not Rad50, could sensitize Taxol-induced cell death and γ-tubulin (Supplementary Figure S5E and F). So, we checked the effect of Rad51 in BRCA1- and VHL-deficient cell lines on Taxol-induced cell death and found that Rad51 overexpression could restore Taxol-response, similarly with BRCA1 transfection in HCC1937 and A498 (Figure 4I and Supplementary Figure S5E). Moreover, Rad51-induced Taxol sensitivity did not show synergic effect with FST (Supplementary Figure S5G) or BRCA1 co-transfection (Figure 4I), suggesting that Rad51 overexpression is enough for abolishing the ER-α-mediated Taxol resistance. Indeed, ER-α transfection or elimination of VHL could suppress Rad51 expression, even in Taxol-treated condition (Figure 4J).
Activated ER-α Disrupts BRCA1-Rad51 Interaction
To confirm the effect of ER-α on Rad51, we transfected ER-α and found that it could suppress Rad51 expression as well as induce γ-tubulin (Figure 5A and Supplementary Figure S6A). In addition, Rad51 could suppress γ-tubulin expression in VHL-null A498 (Supplementary Figure S6B), whereas si-Rad51 induced γ-tubulin expression (Figure 5B) and MTOC amplification in VHL-intact ACHN (Figure 5C and Supplementary Figure S6C). The same results were obtained from Rad51 inhibitor [28], B02-treated cells (Figure 5B and D). This feature is consistent with previous literatures that Rad51 Knock out cells show the defect in centrosome maintaining [29]. To address the reduction of Rad51 by ER-α, we first checked the interaction of them and found it by IP analysis (Supplementary Figure S6D). In our previous result, we showed the reduction of Rad51 in BRCA1-deficient condition (Figure 4C and E). In addition, it has been revealed that BRCA1 interacts with Rad51 during mitosis and meiosis [30]. Thus, we checked the involvement of ER-α in BRCA1-Rad51 interaction and found that Rad51 binding region of BRCA1 was overlapped with ER-α (Supplementary Figure S6E) and ER-α interrupted the BRCA1-Rad51 binding (Figure 5E and F). In addition, association of Rad51 with γ-tubulin was also blocked by ER-α transfection (Figure 5F). To confirm the interaction between Rad51 and γ-tubulin, we performed the GST-pull down assay using GST-γ-tubulin and confirmed the interaction of them (Supplementary Figure S6F), which was weaker in VHL-null cell lysate and reduced by Est-treatment (Supplementary Figure S6F). These results indicated that ER-α-mediated inhibition of BRCA1-Rad51-γ-tubulin binding was a cause for Taxol resistance and centrosome amplification in VHL- and BRCA1-deficient cells. To test this, we monitored the effect of B02 on FST-induced Taxol-effect. Restoration of Taxol toxicity in A498 was diminished by B02 (Figure 5G). We also obtained the similar results from C2 and C2V (Supplementary Figure S6G and H). Next, we examined the change of interaction between BRCA1-Rad51 in response to Est signaling and Taxol. In VHL-intact ACHN, Taxol could induce interaction between BRCA1-Rad51 that was reduced by Estrogen treatment (Figure 5H). In contrast, the interaction between BRCA1-Rad51 was very weak and induced by FST but not by Taxol in VHL-null A498 (Figure 5H). We also observed the increase of Rad51-γ-tubulin binding in A498 by FST (Supplementary Figure S6I). Indeed, inhibition of ER-α using FST could restore the interaction between BRCA1-Rad51 as well as Rad51-γ-tubulin in response to Taxol (Figure 5I). These results suggested that increase of BRCA1-Rad51 by Taxol might contribute to maintaining MTOC.
Figure 5.
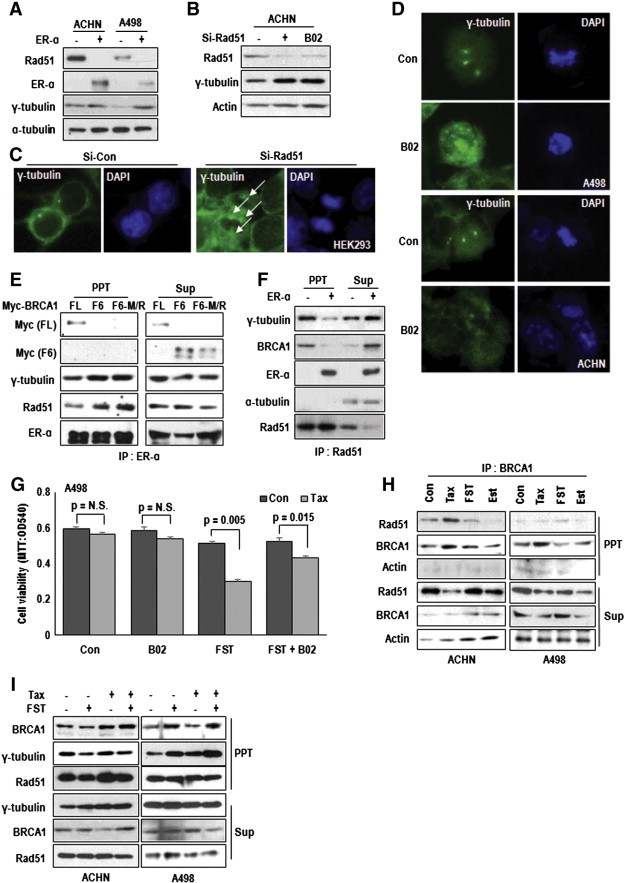
Activated ER-α disrupts BRCA1-Rad51 interaction. (A) The effect of ER-α on Rad51 expression. ER-α suppressed endogenous Rad51 expression as well as induced γ-tubulin. Cells were transfected with EV (Empty) or ER-α vectors for 24 hours. α-tubulin was used as loading control. (B) Rad51 inhibition induces γ-tubulin expression. Elimination of Rad51 or chemical inhibitor (B02) could induce γ-tubulin expression. ACHN cells were treated with B02 (5 μM) or transfected with Si-Rad51 for 24 hours. (C) si-Rad51 induces centrosome amplification. HEK293 cells were transfected with si-control (Si-Con) or si-Rad51 for 72 hours. cells were fixed and stained with γ-tubulin Ab (green) and DAPI (blue). White arrows indicates increased centrosome in si-Rd51-transfected cells. (D) Inhibition of Rad51 promoted centrosome amplification. A498 and ACHN were incubated with B02 for 72 hours and, after fixation, IF stained with γ-tubulin (green) and DAPI (blue). (E) The interaction between ER-α and wild-type BRCA1. ER-α did not bind with F6 fragment of BRCA1 or M1775R mutant of F6 fragment. Immunoprecipitation (IP) analysis was used for binding assay. Indicated vectors transfected HEK293 cell lysates were IPed by ER-α antibody. Under absence of intractable BRCA1, the binding between Rad51 and ER-α was increased (lane 2 and 3 in left panel). (F) ER-α disrupts the BRCA1-Rad51 binding. Interaction of between Rad51 and γ-tubulin was inhibited by ER-α. IP analysis was used for binding assay. EV (Empty vector) or ER-α vectors transfected HEK293 cell lysates were IPed by Rad51 antibody. (G) B02 blocks FST-mediated Tax-sensitization. Indicated chemicals were treated to A498 cells for 72 hours. The cell viability was measured by MTT assay. (H) Alteration of BRCA1-Rad51 binding in VHL status and chemical treatment. Tax (3 μM) induced BRCA1-Rad51 interaction in VHL positive ACHN cells but in VHL negative A498 cells did not. FST (2 μM) induced BRCA1-Rad51 binding in VHL-negative A498 cells. Est (1 μg/ml) reduced interaction of between BRCA1 and Rad51 in both A498 and ACHN cell. IP analysis was progressed using cell lysates of indicated chemical treated A498 and ACHN. (I) FST induces the binding of γ-tubulin and Rad51 in VHL-negative cell. Comparing to ACHN, A498 showed weak interaction of between γ-tubulin and Rad51. FST could induce the interaction. IP analysis was progressed using cell lysates of indicated chemical treated A498 and ACHN.
Increase of γ-Tubulin in VHL-Deficient Mice
To confirm the effect of VHL-ER-α signaling on MTOC regulation and BRCA1-Rad51 system, we examined the MTOC in VHL-deficient MEF cells, generated by Ubc-Cre-ERxpVHL flox/flox mouse. Increase of MTOC in cre-activated condition by treatment of 4-OHT was diminished by FST (Figure 6A and supplementary Figure S6J). Indeed, elimination of VHL in MEF could induce γ-tubulin expression (Figure 6B) and showed the resistance to Tax (Figure 6C). We could also observe the increase of γ-tubulin and reduction of Rad51 and BRCA1 in VHL-deficient kidney and liver tissues (Figure 6D and restoration of them by treatment of FST (Figure 6D). Our results are consistent with recently published literature, in which deletion of VHL in kidney can induce spindle mis-orientation and aneuploidy [31]. Furthermore, in this study, we showed more detailed mechanism for dysregulation of mitotic fidelity in VHL deficient condition. During cell cycle progression, VHL blocks ER-α-mediated cell cycle progression as well as MTOC duplication through direct interaction (Figure 6E). However, in VHL-null condition, elevated ER-α blocked the association of Rad51-BRCA1 and deregulated Rad51, which did not block the γ-tubulin, allowed γ-tubulin-induced multi-MTOC formation. It would be one of critical step for regulation of chromosome segregation, chromosome instability, and cancer initiation (Figure 6E).
Figure 6.
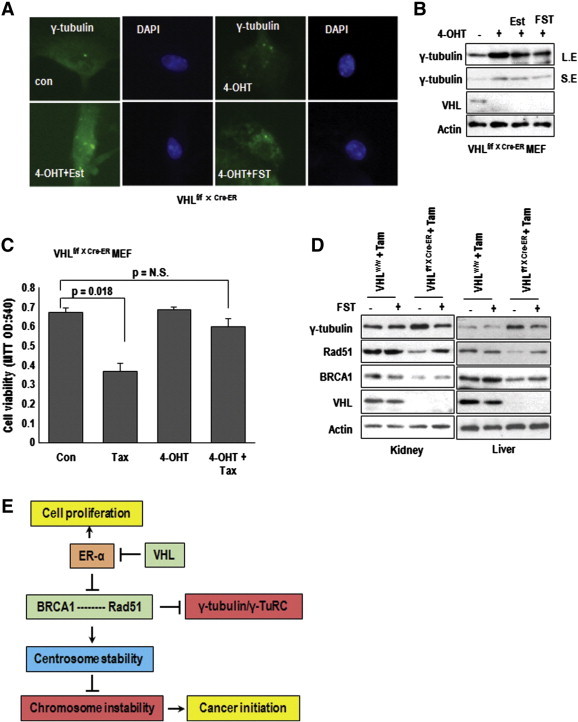
In vivo effect of VHL deficiency. (A) The centrosome number in VHL null MEF. MEF, obtained from VHLf/f xUbc-Cre-ER were incubated with 4-OHT to eliminate VHL (for 5 days) and Est or FST for 72 hours. Cells were stained with γ-tubulin Ab (green) and DAPI for DNA (Blue). (B) Increase of γ-tubulin by VHL elimination. Using the same samples, expression of VHL and γ-tubulin were determined by WB analysis. Actin was used for loading control. L.E and S.E indicate long exposed and short exposed data, respectively. (C) Tax-resistance in VHL deficient MEF. Cells were incubated with Tax for 72 hours and measured viability by MTT assay. VHL null MEF (treated by 4-OHT) showed the resistance to Tax. (D) The effect of VHL deletion in mouse tissues. 4 week old VHLf/f xUbc-Cre-ER mice were injected with 10 mg/kg Tam for 4 weeks and 5 mg/kg FST for 10 weeks. Cre-negative siblings (VHLf/f) were used for experimental control. Induction of γ-tubulin and reduction of BRCA1 and Rad51 by Tam injection in VHLf/f xUbc-Cre-ER mice were restored by FST treatment. (E) Diagram for summary. In VHL-deficient or ER-α activated condition, ER-α inhibits the association of Rad51-BRCA1 and Rad51 cannot regulate γ-tubulin. Thus, free γ-tubulin-induced multi-centrosome formation. Moreover, amplified centrosome causes chromosome instability and provides tumorigenic driving force.
Discussion
VHL is originally identified from von Hippel Lindau syndrome and has been revealed as tumor suppressor gene in clear cell renal carcinoma [5], [6]. Last two decades, molecular role of VHL has been reported to be E3 ligase and inhibit HIF-1α that induces VEGF, EPO and vascular neo-genesis related genes through its transcription ability [32], [33], [34]. Since neo-vasculogenesis is important step for angiogenesis in tumor progression, many researches have focused on pVHL-HIF-1α network. However, there is raveled problem why VHL should be mutated in early stage. In addition, there is no tumor initiating mutation that promotes mutation accumulation and cell cycle promotion in kidney cancer. Thus, we assumed that loss of VHL would contribute to cancer initiation, independently with HIF-1α. Concerning it, we previously showed that pVHL negatively regulate Estrogen signaling through direct interaction and through E3 ligase activity [13].
In this study, we showed additional role of pVHL in MTOC regulation. Since amplification of MTOC can induce chromosomal instability, it should be tightly regulated by tumor suppressor genes such as APC, Bub1, and Rad family [2], [35], [36]. Similarly with these tumor suppressor genes, loss of VHL can promote MTOC amplification (Figure 1F). In fact, while we were preparing this paper, the role of VHL on chromosome segregation was reported [31]. Although we and their observations were very similar, we showed more detailed network between VHL and ER-α-BRCA-Rad51 for MTOC duplication. Increased ER-α by VHL deletion blocks the BRCA1-Rad51 interaction and induces γ-tubulin expression. In addition, loss of VHL leads to increase of γ-tubulin expression that is closely linked to Taxol-resistance (Figure 1, Figure 2). In fact, increased γ-tubulin could generate multiple MTOC (Figure S1E). Thus, loss of VHL showed the resistance to relatively high concentration of Taxol (Figure S1C). However, in this study, we could not reveal the complete molecular mechanism about increase of γ-tubulin/duplication of MTOC in response to Estrogen and loss of VHL. Considering the different effect between Tamoxifen and FST (Figure 3L) and result of our RT-PCR for γ-tubulin (Supplementary Figure S3H), regulation of γ-tubulin would be achieved at post-translation level. So, next, we are trying to investigate γ-tubulin regulation and detailed molecular mechanism about amplification of MTOC under Rad51 inactivated condition. In particular, we are going to focusing on the role of Rad51 in inhibition of MTOC duplication and γ-tubulin increase at post-translation level.
In this study, we find very interesting feature that BRCA1 deficiency-induced MTOC amplification is also compensated by pVHL supplement or inhibition of ER-α (Figure 4). This result indicate that BRCA1 also have similar function with pVHL on regulation of MTOC and ER-α. However, it is not unraveled why inherit BRCA1 mutated patients do not produce renal cell carcinoma and rare breast cancer in VHL syndrome patients. Concerning this, there would unrevealed tissue specific factors that would protect MTOC amplification. Until now, we do not have any clue about this, but it should be investigated. Our results suggest that final effector or regulator of this pathway is Rad51 that has been revealed as essential factor of homologous recombination and chromosome stability. Indeed, Rad51 knock out cells produce multi-MTOC. In our system, elevated ER-α by VHL- or BRCA1 deficiency is cause for Rad51 inactivation. ER-α can bind and block the interaction with BRCA1 (Figure 5) and suppress expression. Moreover, ER-α blocks the interaction between Rad51 and γ-tubulin. Under normal condition, Rad51 would bind and block the γ-tubulin induced γ-tubulin ring complex (γ-TuRC). However, to clarify this, more intensive study should be performed.
In summary, elevated ER-α by loss of VHL or BRCA1 binds to Rad51 as well as BRCA1 and disrupt the BRCA1-Rad51 complex-mediated MTOC maintaining. Increased tubulin fibers will provide the resistance to microtubule-targeted drug such as Taxol. It would one of drug resistant mechanism in RCC as well as BRCA1-deficient cancers. In addition, MTOC deregulation in VHL/BRCA1 deficient cells is contributed to cancer initiation by increase of chromosomal instability.
Acknowledgment
We thanks to Dr. Joung JK (Massachusetts General Hospital) for TELAN vector system and Dr. Kaelin WG Jr. (Harvard Medical School) for VHL mutant vectors. This work was supported by National Research Foundation of Korea (NRF-2013R1A1A2060185).
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.neo.2014.09.013.
Appendix A. Supplementary Data
Supplementary Figures
References
- 1.Wang X, Fredericksen ZS, Vierkant RA, Kosel ML, Pankratz VS, Cerhan JR, Justenhoven C, Brauch H, GENICA, Olson JE. Association of genetic variation in mitotic kinases with breast cancer risk. Breast Cancer Res Treat. 2010;119:453–462. doi: 10.1007/s10549-009-0404-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bloom K. It's a kar9ochore to capture microtubules. Nat Cell Biol. 2000;2:E96–E98. doi: 10.1038/35014089. [DOI] [PubMed] [Google Scholar]
- 3.Ma YX, Tomita Y, Fan S, Wu K, Tong Y, Zhao Z, Song LN, Goldberg ID, Rosen EM. Structural determinants of the BRCA1: estrogen receptor interaction. Oncogene. 2005;24:1831–1846. doi: 10.1038/sj.onc.1208190. [DOI] [PubMed] [Google Scholar]
- 4.Kais Z, Chiba N, Ishioka C, Parvin JD. Functional differences among BRCA1 missense mutations in the control of centrosome duplication. Oncogene. 2012;31:799–804. doi: 10.1038/onc.2011.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arjumand W, Sultana S. Role of VHL gene mutation in human renal cell carcinoma. Tumour Biol. 2012;33:9–16. doi: 10.1007/s13277-011-0257-3. [DOI] [PubMed] [Google Scholar]
- 6.Peña-Llopis S, Christie A, Xie XJ, Brugarolas J. Cooperation and antagonism among cancer genes: the renal cancer paradigm. Cancer Res. 2013;73:4173–4179. doi: 10.1158/0008-5472.CAN-13-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maher ER, Kaelin WG., Jr. Von Hippel-Lindau disease. Medicine. 1997;76:381–391. doi: 10.1097/00005792-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 9.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 10.Thoma CR, Toso A, Gutbrodt KL, Reggi SP, Frew IJ, Schraml P, Hergovich A, Moch H, Meraldi P, Krek W. VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol. 2009;11:994–1001. doi: 10.1038/ncb1912. [DOI] [PubMed] [Google Scholar]
- 11.Young AP, Schlisio S, Minamishima YA, Zhang Q, Li L, Grisanzio C, Signoretti S, Kaelin WG., Jr. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat Cell Biol. 2008;10:361–369. doi: 10.1038/ncb1699. [DOI] [PubMed] [Google Scholar]
- 12.Jung YS, Lee SJ, Yoon MH, Ha NC, Park BJ. Estrogen receptor α is a novel target of the Von Hippel-Lindau protein and is responsible for the proliferation of VHL-deficient cells under hypoxic conditions. Cell Cycle. 2012;11:4462–4473. doi: 10.4161/cc.22794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thoma CR, Frew IJ, Hoerner CR, Montani M, Moch H, Krek W. pVHL and GSK3beta are components of a primary cilium-maintenance signaling network. Nat Cell Biol. 2007;9:588–595. doi: 10.1038/ncb1579. [DOI] [PubMed] [Google Scholar]
- 14.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;206:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 15.Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM. Mutation of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. 1994;7:85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 16.Jung YS, Lee SJ, Lee SH, Chung JY, Jung YJ, Hwang SH, Ha NC, Park BJ. Loss of VHL promotes progerin expression, leading to impaired p14/ARF function and suppression of p53 activity. Cell Cycle. 2013;12:2277–2290. doi: 10.4161/cc.25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci U S A. 2007;104:16633–16638. doi: 10.1073/pnas.0708043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lonergan KM, Iliopoulos O, Ohh M, Kamura T, Conaway RC, Conaway JW, Kaelin WG., Jr. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol. 1998;18:732–741. doi: 10.1128/mcb.18.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012;30:460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amato RJ. Chemotherapy for renal cell carcinoma. Semin Oncol. 2000;27:177–186. [PubMed] [Google Scholar]
- 21.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 22.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly (ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 23.Xiao H, Verdier-Pinard P, Fernandez-Fuentes N, Burd B, Angeletti R, Fiser A, Horwitz SB, Orr GA. Insights into the mechanism of microtubule stabilization by Taxol. Proc Natl Acad Sci U S A. 2006;103:10166–10173. doi: 10.1073/pnas.0603704103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chua ME, Escusa KG, Luna S, Tapia LC, Dofitas B, Morales M. Revisiting oestrogen antagonists (clomiphene or tamoxifen) as medical empiric therapy for idiopathic male infertility: a meta-analysis. Andrology. 2013;1:749–757. doi: 10.1111/j.2047-2927.2013.00107.x. [DOI] [PubMed] [Google Scholar]
- 25.Jiang D, Huang Y, Han N, Xu M, Xu L, Zhou L, Wang S, Fan W. Fulvestrant, a selective estrogen receptor down-regulator, sensitizes estrogen receptor negative breast tumors to chemotherapy. Cancer Lett. 2014;346:292–299. doi: 10.1016/j.canlet.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 26.DelloRusso C, Welcsh PL, Wang W, Garcia RL, King MC, Swisher EM. Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Mol Cancer Res. 2007;5:35–45. doi: 10.1158/1541-7786.MCR-06-0234. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz HS, Sodergren JE, Philips FS. Mitomycin C: chemical and biological studies on alkylation. Science. 1963;142:1181–1183. doi: 10.1126/science.142.3596.1181. [DOI] [PubMed] [Google Scholar]
- 28.Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol. 2011;6:628–635. doi: 10.1021/cb100428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daboussi F, Thacker J, Lopez BS. Genetic interactions between RAD51 and its paralogues for centrosome fragmentation and ploidy control, independently of the sensitivity to genotoxic stresses. Oncogene. 2005;24:3691–3696. doi: 10.1038/sj.onc.1208438. [DOI] [PubMed] [Google Scholar]
- 30.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 31.Hell MP, Duda M, Weber TC, Moch H, Krek W. Tumor suppressor VHL functions in the control of mitotic fidelity. Cancer Res. 2014;74:2422–2431. doi: 10.1158/0008-5472.CAN-13-2040. [DOI] [PubMed] [Google Scholar]
- 32.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 33.Yoon D, Pastore YD, Divoky V, Liu E, Mlodnicka AE, Rainey K, Ponka P, Semenza GL, Schumacher A, Prchal JT. Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J Biol Chem. 2006;281:25703–25711. doi: 10.1074/jbc.M602329200. [DOI] [PubMed] [Google Scholar]
- 34.Boutin AT, Weidemann A, Fu Z, Mesropian L, Gradin K, Jamora C, Wiesener M, Eckardt KU, Koch CJ, Ellies LG. Epidermal sensing of oxygen is essential for systemic hypoxic response. Cell. 2008;133:223–234. doi: 10.1016/j.cell.2008.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carr AM, Moudjou M, Bentley NJ, Hagan IM. The chk1 pathway is required to prevent mitosis following cell-cycle arrest at ‘start’. Curr Biol. 1995;5:1179–1190. doi: 10.1016/s0960-9822(95)00234-x. [DOI] [PubMed] [Google Scholar]
- 36.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures