

Abstract
Glycoside hydrolases are important enzymes that support bacterial growth by enabling the degradation of polysaccharides (e.g., starch, cellulose, xylan, and chitin) in the environment. Presently, little is known about the overall phylogenetic distribution of the genomic potential to degrade these polysaccharides in bacteria. However, knowing the phylogenetic breadth of these traits may help us predict the overall polysaccharide processing in environmental microbial communities. In order to address this, we identified and analyzed the distribution of 392,166 enzyme genes derived from 53 glycoside hydrolase families in 8,133 sequenced bacterial genomes. Enzymes for oligosaccharides and starch/glycogen were observed in most taxonomic groups, whereas glycoside hydrolases for structural polymers (i.e., cellulose, xylan, and chitin) were observed in clusters of relatives at taxonomic levels ranging from species to genus as determined by consenTRAIT. The potential for starch and glycogen processing, as well as oligosaccharide processing, was observed in 85% of the strains, whereas 65% possessed enzymes to degrade some structural polysaccharides (i.e., cellulose, xylan, or chitin). Potential degraders targeting one, two, and three structural polysaccharides accounted for 22.6, 32.9, and 9.3% of genomes analyzed, respectively. Finally, potential degraders targeting multiple structural polysaccharides displayed increased potential for oligosaccharide deconstruction. This study provides a framework for linking the potential for polymer deconstruction with phylogeny in complex microbial assemblages.
INTRODUCTION
Together, carbohydrates account for ∼75% of the earth's biomass (1), and therefore, our understanding of how this material is processed by microorganisms is a key question for global biogeochemistry. Of these carbohydrates, the more abundant are cellulose, xylan, chitin, starch, and glycogen. Cellulose, the most abundant polysaccharide on earth, is produced by a variety of organisms, including plants and bacteria. Xylan and several other polysaccharides (e.g., glucomannan) together form hemicellulose, associated with cellulose, to constitute plant cell wall material. Chitin, produced by many arthropods (e.g., crustaceans and insects) and fungi, is the second most abundant structural polysaccharide. On the other hand, starch and glycogen, found in many organisms, are used as a means to store energy inside the cell. Additional important polysaccharides are dextrans and fructans (e.g., inulin). These polysaccharides represent the major source of energy for most heterotrophs, including many microbes (e.g., bacteria and fungi). These heterotrophs produce glycoside hydrolases (GHs) to degrade polysaccharides and to release monosaccharides (e.g., glucose, fructose, N-acetylglucosamine, and xylose) that are key resources for microbial growth (2–4). However, not all microorganisms are directly involved in polysaccharide deconstruction. Active degraders express enzymes (e.g., endo-/exocellulases and endo-/beta-xylanase) that act synergistically in the deconstruction of complex polymers (e.g., cellulose and xylan) and enzymes for processing small degradation products (5, 6). Many degraders are likely to be involved in the degradation of multiple substrates since, in nature, polysaccharides form complex mixed materials (e.g., plant material) (7).
Some other lineages (i.e., opportunists) are solely capable of processing the smaller substrates (2). In addition, many members of the Tenericutes, Spirochaetes, or Cyanobacteria are unable to process cellulose or its deconstruction product (8).
Understanding the distribution of genes for polysaccharide utilization in bacteria is key to understanding how microbial communities, including bacteria and fungi, are assembled and evolve and, more importantly, how many ecosystems function (2, 9–11).
However, to date, little is known about the distribution and the cooccurrence of the bacterial potentials for deconstruction of the variety of polysaccharides encountered in nature.
The GH superfamily encompasses proteins involved in glucan hydrolysis, including cellulose, hemicellulose, xylan, chitin, starch, and glycogen (12). The classification of glycoside hydrolases is primarily based on protein structures that common allow for Pfam (i.e., probabilistic models for statistical inference of homology) identification (13). Generally, some enzymes are active on polymers and generate oligosaccharides, which are subsequently processed by a variety of enzymes with activity on oligosaccharides (i.e., the “-osidases”) that are found in a few GH families (e.g., GH1 to -3). Considering their central function in carbon metabolism (14), their impact on nutrient cycling (15), and their biotechnological potential (16), the biochemistry of these enzymes has been widely investigated. In some cases, glycoside hydrolase families with different folds display similar activities (e.g., endocellulases from GH5 and -9), while, in some families, minor modifications of the active site/substrate-binding cleft result in different specificities/activities while conserving the overall fold (e.g., exocellulase and endocellulase from GH6). However, despite examples where minor modifications of the active site lead to distinct substrate specificities (e.g., cellulases and chitosanases from GH8), the substrate specificities of many GH families are remarkably conserved (see Table S1 in the supplemental material). This suggests a strong selective pressure on key enzymes for polysaccharide deconstruction (8, 17–19).
In microorganisms, the phylogenetic distribution of enzymes is not random (8, 20), and the presence of glycoside hydrolases is restricted at different taxonomic levels. For example, as described in CAZy (12), cellulases from glycoside hydrolase family 7 are exclusively observed in members of the Fungi, suggesting an ancestral specialization event. In contrast, the distribution of various traits targeting cellulose and chitin has been shown to be broad and the corresponding GHs to be phylogenetically conserved in clusters at the genus or species level (8, 17). Functional status (e.g., potential degrader or opportunist) has been proposed as a concept to discriminate lineages according to their potential for cellulose deconstruction (8). However, cellulose in plant material is commonly associated with other polymers. Thus, an important question is whether or not potential cellulose degraders are also involved in hydrolysis of other plant polymers or are reliant on the properties of other organisms. This information, in association with taxonomic identification of microbial assemblages, is key in order to predict and understand how changes in microbial community composition in response to environmental perturbation (e.g., global climate change) would affect polysaccharide processing. Thus, to improve our understanding of polysaccharide processing by bacteria, we asked the following questions: (i) what is the phylogenetic distribution of enzyme potentials for polysaccharide deconstruction in bacteria, and (ii) how are the enzyme potentials for deconstruction of different polysaccharides linked? To answer these questions, we analyzed the distribution of orthologs of 53 glycoside hydrolase families targeting various plant, fungal, bacterial, and animal polysaccharides in 8,133 sequenced bacterial genomes. Our results provide a basic framework for determining the potential for polysaccharide processing in taxonomically resolved bacterial assemblages.
MATERIALS AND METHODS
Glycoside hydrolase mapping.
To prevent the loss of potential GHs, we developed a custom bioinformatics pipeline based on a well-defined hidden Markov model (HMM) to detect potential GHs with low similarity to known enzymes. Linking this annotation with the genomic context, as provided by the FIGfam annotation, allowed us to further discriminate the functions of proteins belonging to the same GH family, as follows. (i) We extracted protein sequences for all SEED-annotated genes in fully sequenced bacterial genomes retrieved from the PATRIC database (21) using the SEED API (22, 23). (ii) A custom Pfam database for glycoside hydrolase families with Pfam accession numbers was created (see Table S1 in the supplemental material) (12, 24). (iii) Sequences with more than 150 amino acids were tested for glycoside hydrolase domains against the custom Pfam library using HMMscan (e < 10−10) (25). (iv) Positive hits were reciprocally analyzed against the entire Pfam-A database (e < 10−10). Only sequences returning positive hits for some GHs upon the reciprocal analysis and covering ≥70% of the length shown in the Pfam database were considered potential GHs. (v) FIGfams from positive hits were retrieved using the SEED API. The specificity of GH combinations in each individual FIGfam was defined when >80% of genes for a specific FIGfam returned this GH combination. The abundant FIG 00001088 returned multiple GH combinations (GH1 or GH20). Thus, protein sequences affiliated with the FIG 00001088 gene were further analyzed using HMMscan (with previously described cutoffs) and manually added to the final GH count.
When possible, broadly defined substrates were assigned to glycoside hydrolase families according to the substrate specificities of characterized enzymes, as stated in the CAZy database (see Table S1 in the supplemental material) (8, 12, 19, 26). Biosynthetic cellulases from GH8 were separated from hydrolytic enzymes (8).
Functional groups.
GH families targeting the same substrate were classified in functional groups targeting oligosaccharides, starch and glycogen, cellulose, xylan, chitin, dextran, fructan, or other animal or plant polysaccharides. In addition, we introduced a group of GH families targeting structural polysaccharides (i.e., cellulose, chitin, and xylan). Next, genomes were classified according to their potential for oligosaccharide and polysaccharide processing. Potential degraders of these substrates were defined as bacteria having at least one gene targeting one of these specific substrates.
Taxonomy and phylogenetics.
The complete taxonomy of each individual genome was retrieved from the NCBI taxonomy server (from phylum to genus), and corresponding prealigned/high-quality 16S rRNA sequences were retrieved from the Silva database (27). The alignment was trimmed for conserved positions detected in >95% of analyzed sequences using trimAl (28). The resulting DNA alignment (1,275 bp) was analyzed and used for tree construction using Phylip (F84 DNA distance matrix and neighbor joining) (29). The clustering of glycoside hydrolases was investigated using the previously described indices phylogenetic signal (D) (30) and trait depth (τD) (20). Briefly, D was computed using the R package CAPER, with 1,000 permutations. A D value of ≥1 represented an overdispersed distribution, D ≅ 1 a random distribution, 0 < D < 1 a Brownian motion-like evolutionary distribution, and D = 0 a strongly clumped trait distribution (30). τD was estimated as the average 16S rRNA sequence distance between members of a clade where at least 90% of the strains carry a trait and the root node of this clade (determined using consenTRAIT) (20). For abundant traits (i.e., glycoside hydrolase families 1, 2, 3, 4, 5, 13, 15, 18, 20, 31, 32, 38, 65, 70, 78, and 88), we computed D and τD for strains with or without these traits.
Statistical analyses.
The glycoside hydrolase distribution in sequenced bacterial genomes was analyzed using functions within the Vegan and Stats packages in the R software environment (31, 32). Trait redundancy (σ) was defined as the ratio of genomes with multiple copies of a trait (>1) over the number of genomes with the trait (≥1).
RESULTS
Glycoside hydrolase mapping.
We identified 392,166 potential glycoside hydrolases (GHs) across 8,133 sequenced bacterial genomes (see Table S2 in the supplemental material). The most frequent GHs belonged to families targeting oligosaccharides (i.e., GH1 to -4, -20, and -31), starch and glycogen (i.e., GH13 to -15, -57, and -65), and chitin (e.g., GH18, -19, and -85) (Fig. 1). These GHs were observed in most sequenced bacterial genomes. Besides these abundant functions, some individual GH families were also frequently detected, including GH5, GH70, GH32, GH78, and GH38, targeting cellulose, dextrans, fructans, rhamnose, and mannose, respectively.
FIG 1.

Glycoside hydrolase (GH) contents in all sequenced bacterial genomes, including members of the Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, and Cyanobacteria. The horizontal bars represent medians, the boxes represent the 25th to 75th percentiles, and the vertical bars represent the 2.5th to 97.5th percentiles.
Phylogenetic distribution of glycoside hydrolases.
The GH content was extremely variable across phyla. For example, both Actinobacteria and Bacteroidetes displayed abundant and diverse GHs with high potential for structural polysaccharide processing, while, perhaps not surprisingly given that most are photoautotrophic, Cyanobacteria harbored reduced potentials for polysaccharide processing (Fig. 1). In phyla with few sequenced genomes, such as Verrucomicrobia and Acidobacteria (16 and 5 analyzed genomes, respectively), the GH content was generally abundant but highly variable. At a finer taxonomic level, the distribution of GHs across lineages from the same phylum was also highly variable, as suggested by the comparison of GH distribution between the mostly pathogenic Mycobacterium strains and the mostly environmental Streptomyces strains (phylum Actinobacteria) (see Fig. S1 in the supplemental material). When present, genes for GH families targeting starch and oligosaccharides displayed high intragenomic redundancy (σ > 0.80) compared to that of genes for GHs targeting structural polymers. Among these, some GH families displayed extremely low redundancy (e.g., GH8 [σ < 0.10]) (see Table S1). Thus, many strains had important potential for oligosaccharides and starch or glycogen processing. Conversely, GHs for structural polysaccharide deconstruction were less frequent or, when present, less abundant.
Next, we investigated the phylogenetic distribution of each individual GH family in the bacterial genomes using metrics for phylogenetic signal (D) and trait depth (τD). For most of the GH families, the phylogenetic signal D, either positive or negative, was close to zero, revealing clumped (Brownian) distributions (Fig. 2A). Conversely, GH families 71, 75, 44, and 45 displayed D values corresponding to more dispersed traits. On average, most of the GH families were observed in bacteria forming clusters of relatives with a clade depth τD of less than 0.03 for 16S rRNA gene distance (i.e., sharing of traits was more common in organisms sharing 94% or more 16S rRNA sequence similarity). Few GHs, mostly the abundant ones, like GH1 and 13, displayed higher τD values. D and τD were also computed for functional groups. As described for individual GH families, the phylogenetic signal D for functional groups targeting starch and glycogen, cellulose, fructose, and dextran was positive and close to 0 (D ≤ 0.1). In contrast, the potential to target oligosaccharides and xylan displayed a more dispersed distribution (D = 0.24 and D = 0.17, respectively). The corresponding τD values were also estimated. On average, the potential to target xylan, dextran, or fructan was observed in bacteria forming clusters of relatives with a clade depth τD of below 0.025 for 16S rRNA gene distance. The potential to target other substrates was observed in groups of relatives forming clusters with τD values of >0.80. For these functional potentials, we computed the τD of strains lacking these functional groups. On average, strains having no detected potential for processing of oligosaccharides, starch, cellulose, and chitin formed clusters of bacteria with average τD values equal to 0.01, 0.01, 0.02, and 0.02, respectively.
FIG 2.

Phylogenetic distribution of glycoside hydrolases in sequenced bacterial genomes. Phylogenetic signal D (A) and trait depth τD (B) for glycoside hydrolases and lack of glycoside hydrolases (*) in sequenced bacterial genomes.
Since strains that are closely related according to their 16S rRNA sequences are assumed to share increased numbers of functional genes, we then investigated how phylogeny and the overall genome-specific glycoside hydrolase content were linked (Fig. 3; see also Table S3 in the supplemental material). Strains forming operational taxonomic units with 100, ≥99, ≥97.5, and ≥95% 16S rRNA gene similarities displayed median functional dissimilarities of 0.062, 0.100, 0.183, and 0.308, respectively. This suggested that even very closely related strains may differ regarding their GH composition and, thus, may have distinct potential regarding polysaccharide deconstruction. Strains with lower 16S rRNA gene sequence similarity displayed further increases in functional dissimilarity (median overall functional dissimilarity of 0.733).
FIG 3.
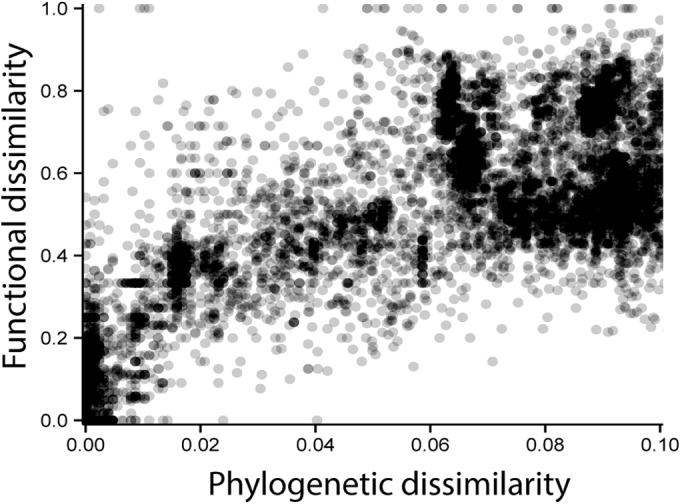
Relationship between phylogeny and functional dissimilarity. Dependence of F84-corrected pairwise dissimilarity for 16S rRNA gene sequences and pairwise Bray-Curtis dissimilarity for 53 GH families across 8,133 sequenced bacterial genomes.
Cooccurrence of potential for polysaccharide processing.
Next, we examined the cooccurrence of potentials for polysaccharide hydrolysis across organisms (Fig. 4). First, 3.1% of the strains analyzed had genes for oligosaccharide processing only (i.e., -osidases), whereas 88.3% displayed the combined potential to process oligosaccharides and starch/glycogen. The ability to target at least one structural polysaccharide (i.e., cellulose, xylan, or chitin) was observed in 66.7% of the genomes (Fig. 4A), and the potential for cellulose, xylan, and chitin deconstruction was detected in 40.8, 24.6, and 53.1% of sequenced bacterial genomes, respectively. Most of the strains classified as potential degraders contained genes for the hydrolysis of multiple targets, with 32.9 and 9.4% having the potential to target 2 or 3 structural polysaccharides, respectively, and only 24.4% of the genomes having the potential to target a single structural polysaccharide. Most of the potential chitin degraders (74.8%) also displayed the potential to target plant structural polysaccharides. Of the potential cellulose degraders, 29.5 and 76.9% also targeted xylan and chitin, while 48.9 and 71.8% of the potential xylan degraders could also target cellulose and chitin, respectively. A small percentage of the strains, 5.1%, had no detected enzymes for oligosaccharide hydrolysis but had enzymes for starch/glycogen, structural polysaccharides, or other substrates (i.e., dextran, fructan, or other plant or animal polymers) or multiple substrates (i.e., mixed). Cellulases from GH8 for cellulose biosynthesis (BcsZ) were observed in 1,686 genomes (mostly Proteobacteria); among these, 476 genomes also possessed the potential for cellulose deconstruction.
FIG 4.
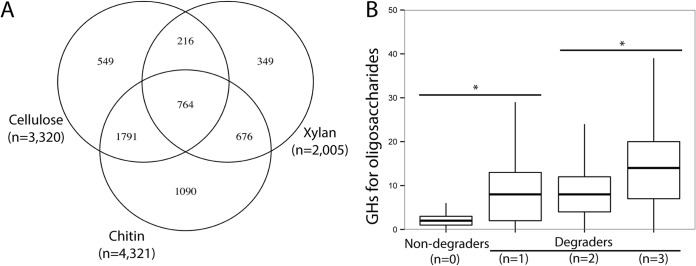
Cooccurrence of potential to target different substrates in sequenced bacterial genomes. (A) Venn diagram for distribution of potentials to target different structural polysaccharides (n = number of genomes with potential for polysaccharide deconstruction). (B) Relationship between potentials for structural polysaccharide hydrolysis and abundance of genes encoding enzymes for oligosaccharide processing (-osidases) *, P < 0.001 (t test; n = number of targeted structural polysaccharides).
Next, we investigated the link between the potential for structural polysaccharide and oligosaccharide processing. Strains with the potential to target multiple structural polysaccharides displayed significantly more enzymes for oligosaccharides than strains with reduced potential (Fig. 4B). Finally, we investigated the correlation between potentials for polysaccharide deconstruction (see Table S4 in the supplemental material). The abundance of genes for enzymes targeting cellulose was positively correlated with the abundance of genes for most of the other functions, including the potential for xylan and chitin deconstruction. So were most of the potentials for polysaccharides hydrolysis. However, cellulose biosynthesis and dextran deconstruction were negatively correlated with the potential for cellulose deconstruction.
DISCUSSION
Linking phylogeny to function is a recurrent question in microbiology and is key to understanding how changes in microbial communities in response to environmental perturbation would affect ecosystem functioning (e.g., carbon cycling). The increasing number of sequenced bacterial genomes annotated using consistent techniques is enabling a more thorough assessment of the patterns of distribution of functions across the microbial tree of life. Recently, a systematic investigation of sequenced bacterial genomes for functional traits related to cellulose (8) or chitin (17) deconstruction has improved our understanding of the evolutionary forces shaping the present distribution of potentials for cellulose and chitin deconstruction in sequenced bacterial genomes (33). Understanding the distribution of cellulases in bacteria allows the discrimination of potential cellulose degraders, opportunists, and bacteria not involved in cellulose deconstruction. This information can be used as a basic framework to identify and investigate the distribution of functional groups, e.g., cellulose degraders, in environmental microbial assemblages (34–36). In nature, cellulose is associated with various polymers (e.g., xylan) in most plant materials (5). Starch and glycogen are found in many organisms and are also important carbon sources for many bacteria (37), whereas chitin produced by fungi and arthropods is present in many environments (38). Finally, dextran and fructan (i.e., inulin) are also frequently observed polymers. Thus, understanding how bacteria access these resources is a key prerequisite to understanding the functioning of many ecosystems.
Here, we present an integrated phylogenomic analysis of the distribution of 392,166 sequences from the major GH families targeting the most abundant polysaccharides in 8,133 sequenced bacterial genomes. On average, there are 48.2 GHs/genome. Considering an average bacterial genome of ∼3 Mbp and an average gene size of ∼1.5 kbp (39), this represents 2.4% of the genes in bacteria. This value is in good agreement with previously assumed glycoside hydrolase frequency in bacterial genomes (40). However, GHs display broad distribution and most glycoside hydrolase families, being nonrandomly distributed, are found in smaller clusters of relatives at the genus or species level. The functional dissimilarity between strains increases with the phylogenetic distance, although closely related strains can display substantial functional divergences. This suggests that some recent events may have affected some genomes (e.g., gene duplication and horizontal gene transfer). Nevertheless, most of the traits analyzed are predominantly inherited from parent strains.
Knowing the phylogenetic distribution of the GHs, with some information regarding their substrate specificity, allowed us to expand the terminology introduced for cellulose degradation (i.e., opportunists and potential degraders) to other polysaccharides (8). First, the vast majority of sequenced bacteria (89%) are potential starch and glycogen utilizers. This supports the idea that enzymes for starch and glycogen processing are frequent in bacteria, either to access plant or animal material or to process endogenous storage material (37, 41). The lack of potential for starch and glycogen processing is observed in smaller clusters of relatives that occur from species to genus level. Thus, the loss of potential for starch and glycogen processing is observed in multiple clusters (e.g., in some members of the Mycoplasma, Neisseria, and Mycobacterium genera) and corresponds to genome modifications that are sometimes associated with specific ecological lifestyles (e.g., parasitism or symbiosis) (37). The potential for chitin deconstruction is also abundant in the sequenced bacterial genomes. Most of the chitinases detected are from GH18 and, to a lesser extent, GH19. Across all strains, chitinases are observed in deep clusters, including members of Cytophaga, Flavobacterium, Actinomyces, and groups of Proteobacteria and Firmicutes. Also, the prevalence of genes for GH20, mostly encoding β-1,6-N-acetylglucosaminidase, suggests that, as described for cellulose (8), many lineages may opportunistically compete with chitin degraders for the degradation products (e.g., chitobiose) (38). The high prevalence of strains (e.g., Vibrio [42] and Serratia [43] strains) with the potential for chitin degradation highlights chitin as a key resource across ecosystems (i.e., marine and soil ecosystems) (44, 45). However, chitinolytic activity is known to be involved in other ecologically relevant pathways, such as pathogenicity (i.e., Listeria strains [46]) and antifungal activity (i.e., Collimonas strains [47]). On average, the lack of potential for chitin processing is observed in clusters of relatives with an average 16S rRNA similarity of 96.1%. Thus, in large groups of bacteria, the potential for chitin degradation is absent.
Potential xylan degraders mostly express enzymes from GH10 and -30. A few lineages, like the Firmicutes, also possess enzymes from GH11. Distinct distributions of genes for similar substrates suggest that the potential to target similar substrates appeared independently in different lineages and that the exchange of genetic material between lineages is somehow constrained. This supports our result showing that most glycoside hydrolases are inherited from parent strains.
Most potential degraders have the potential to target multiple substrates. This highlights the tight connection between polysaccharides (e.g., cellulose and xylan in plant cell wall), as well as bacterial potential to degrade these substrates in nature. As described for cellulose deconstruction (8), the distribution of GHs in sequenced bacterial genomes reveals that lineages having increased potential for structural polysaccharide deconstruction harbor increased potential for oligosaccharide processing. This is consistent with the idea that an increased number of oligosaccharide types produced by the degraders requires an increased diversity of -osidases for processing small substrates. This increased potential for oligosaccharide processing is assumed to prevent the inhibition of polysaccharides hydrolysis (48).
Together, the sequenced bacterial genomes, although largely skewed toward pathogenic organisms, are a valuable resource to investigate the distribution of functional genes for polysaccharide degradation in regard to niche specialization in microbial lineages. For example, autotrophs (e.g., Cyanobacteria strains) consistently harbor reduced glycoside hydrolase diversity and frequency. Conversely, heterotrophs (e.g., Streptomyces strains) display increased potential for polysaccharide deconstruction in order to cope with changing environments and fluctuating carbon sources. However, pathogens (e.g., Mycobacterium strains), relying on their hosts to provide resources, tend to have reduced potential for polysaccharide deconstruction.
Globally, we provide herein an integrated framework for potential polysaccharide deconstruction that connects glycoside hydrolases to functional status (e.g., potential xylan degrader) and to the phylogeny of sequenced bacterial lineages. This information is key in order to estimate the potential for polysaccharide deconstruction in microbial populations dominated by bacteria (49) and to parameterize simulated microbial guilds for ecosystem modeling (50). More broadly, this information will be useful to investigate how fluctuations of environmental bacterial communities, together with fungal populations (9), in response to natural or anthropogenic changes may affect the polysaccharide deconstruction in the environment and, more globally, the carbon cycling.
Supplementary Material
ACKNOWLEDGMENTS
We thank Eoin L. Brodie for many helpful comments on the manuscript.
This material is based upon work supported by the NSF Dimensions of Biodiversity program (OCE-1046297) and the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research (BER), under award number DE-PS02-09ER09-25.
The authors declare no conflict of interest.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03718-14.
REFERENCES
- 1.Thomas F, Hehemann J-H, Rebuffet E, Czjzek M, Michel G. 2011. Environmental and gut bacteroidetes: the food connection. Front Microbiol 2:93. doi: 10.3389/fmicb.2011.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldfarb KC, Karaoz U, Hanson CA, Santee CA, Bradford MA, Treseder KK, Wallenstein MD, Brodie EL. 2011. Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front Microbiol 2:94. doi: 10.3389/fmicb.2011.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson DR, Goldschmidt F, Lilja EE, Ackermann M. 2012. Metabolic specialization and the assembly of microbial communities. ISME J 6:1985–1991. doi: 10.1038/ismej.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haichar FEZ, Achouak W, Christen R, Heulin T, Marol C, Marais M-F, Mougel C, Ranjard L, Balesdent J, Berge O. 2007. Identification of cellulolytic bacteria in soil by stable isotope probing. Environ Microbiol 9:625–634. doi: 10.1111/j.1462-2920.2006.01182.x. [DOI] [PubMed] [Google Scholar]
- 5.Wilson DB. 2011. Microbial diversity of cellulose hydrolysis. Curr Opin Microbiol 14:259–263. doi: 10.1016/j.mib.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Anderson I, Abt B, Lykidis A, Klenk HP, Kyrpides N, Ivanova N. 2012. Genomics of aerobic cellulose utilization systems in actinobacteria. PLoS One 7:e39331. doi: 10.1371/journal.pone.0039331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hou Bin P, Li YZ, Wu BH, Yan ZC, Yan BX, Gao PJ. 2006. Cellulolytic complex exists in cellulolytic myxobacterium Sorangium. Enzyme Microb Technol 38:273–278. doi: 10.1016/j.enzmictec.2004.08.044. [DOI] [Google Scholar]
- 8.Berlemont R, Martiny AC. 2013. Phylogenetic distribution of potential cellulases in bacteria. Appl Environ Microbiol 79:1545–1554. doi: 10.1128/AEM.03305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eichlerová I, Homolka L, Žifčáková L, Lisá L, Dobiášová P, Baldrian P. 2015. Enzymatic systems involved in decomposition reflects the ecology and taxonomy of saprotrophic fungi. Fungal Ecol 13:10–22. doi: 10.1016/j.funeco.2014.08.002. [DOI] [Google Scholar]
- 10.Stursová M, Zifčáková L, Leigh MB, Burgess R, Baldrian P. 2012. Cellulose utilization in forest litter and soil: identification of bacterial and fungal decomposers. FEMS Microbiol Ecol 80:735–746. doi: 10.1111/j.1574-6941.2012.01343.x. [DOI] [PubMed] [Google Scholar]
- 11.Allison SD, Lu Y, Weihe C, Goulden ML, Martiny AC, Treseder KK, Martiny JBH. 2013. Microbial abundance and composition influence litter decomposition response to environmental change. Ecology 94:714–725. doi: 10.1890/12-1243.1. [DOI] [PubMed] [Google Scholar]
- 12.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. 2014. Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeBoy RT, Mongodin EF, Fouts DE, Tailford LE, Khouri H, Emerson JB, Mohamoud Y, Watkins K, Henrissat B, Gilbert HJ, Nelson KE. 2008. Insights into plant cell wall degradation from the genome sequence of the soil bacterium Cellvibrio japonicus. J Bacteriol 190:5455–5463. doi: 10.1128/JB.01701-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trivedi P, Anderson IC, Singh BK. 2013. Microbial modulators of soil carbon storage: integrating genomic and metabolic knowledge for global prediction. Trends Microbiol 21:641–651. doi: 10.1016/j.tim.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Lynd LR, Weimer PJ, Van Zyl WH, Pretorius IS. 2002. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev 66:506–577. doi: 10.1128/MMBR.66.3.506-577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zimmerman AE, Martiny AC, Allison SD. 2013. Microdiversity of extracellular enzyme genes among sequenced prokaryotic genomes. ISME J 7:1187–1199. doi: 10.1038/ismej.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantarel BL, Lombard V, Henrissat B. 2012. Complex carbohydrate utilization by the healthy human microbiome. PLoS One 7:e28742. doi: 10.1371/journal.pone.0028742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martiny AC, Treseder K, Pusch G. 2013. Phylogenetic conservatism of functional traits in microorganisms. ISME J 7:830–838. doi: 10.1038/ismej.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gillespie JJ, Wattam AR, Cammer SA, Gabbard JL, Shukla MP, Dalay O, Driscoll T, Hix D, Mane SP, Mao C, Nordberg EK, Scott M, Schulman JR, Snyder EE, Sullivan DE, Wang C, Warren A, Williams KP, Xue T, Yoo HS, Zhang C, Zhang Y, Will R, Kenyon RW, Sobral BW. 2011. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect Immun 79:4286–4298. doi: 10.1128/IAI.00207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Disz T, Akhter S, Cuevas D, Olson R, Overbeek R, Vonstein V, Stevens R, Edwards RA. 2010. Accessing the SEED genome databases via Web services API: tools for programmers. BMC Bioinformatics 11:319. doi: 10.1186/1471-2105-11-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang H-Y, Cohoon M, de Crécy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Rückert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. 2005. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res 40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput Biol 7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen W, Xie T, Shao Y, Chen F. 2012. Phylogenomic relationships between amylolytic enzymes from 85 strains of fungi. PLoS One 7:e49679. doi: 10.1371/journal.pone.0049679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felsenstein J. 1989. PHYLIP—Phylogeny Inference Package (version 3.2). Cladistics 5:164–166. [Google Scholar]
- 30.Fritz SA, Purvis A. 2010. Selectivity in mammalian extinction risk and threat types: a new measure of phylogenetic signal strength in binary traits. Conserv Biol 24:1042–1051. doi: 10.1111/j.1523-1739.2010.01455.x. [DOI] [PubMed] [Google Scholar]
- 31.Oksanen J, Blanchet FG, Kindt R, Legendre P, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2012. vegan: Community Ecology Package. R Package version 2.0-8. http://vegan.r-forge.r-project.org/. [Google Scholar]
- 32.R Development Core Team. 2012. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 33.Baldrian P, López-Mondéjar R. 2014. Microbial genomics, transcriptomics and proteomics: new discoveries in decomposition research using complementary methods. Appl Microbiol Biotechnol 98:1531–1537. doi: 10.1007/s00253-013-5457-x. [DOI] [PubMed] [Google Scholar]
- 34.Nyyssönen M, Tran HM, Karaoz U, Weihe C, Hadi MZ, Martiny JBH, Martiny AC, Brodie EL. 2013. Coupled high-throughput functional screening and next generation sequencing for identification of plant polymer decomposing enzymes in metagenomic libraries. Front Microbiol 4:282. doi: 10.3389/fmicb.2013.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo HL, Hazen TC, Simmons BA, DeAngelis KM. 2014. Enzyme activities of aerobic lignocellulolytic bacteria isolated from wet tropical forest soils. Syst Appl Microbiol 37:60–67. doi: 10.1016/j.syapm.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Whitaker J, Ostle N, Nottingham AT, Ccahuana A, Salinas N, Bardgett RD, Meir P, McNamara NP. 2014. Microbial community composition explains soil respiration responses to changing carbon inputs along an Andes-to-Amazon elevation gradient. J Ecol 102:1058–1071. doi: 10.1111/1365-2745.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henrissat B, Deleury E, Coutinho PM. 2002. Glycogen metabolism loss: a common marker of parasitic behaviour in bacteria? Trends Genet 18:437–440. doi: 10.1016/S0168-9525(02)02734-8. [DOI] [PubMed] [Google Scholar]
- 38.Beier S, Bertilsson S. 2013. Bacterial chitin degradation—mechanisms and ecophysiological strategies. Front Microbiol 4:149. doi: 10.3389/fmicb.2013.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilmes P, Bond PL. 2006. Metaproteomics: studying functional gene expression in microbial ecosystems. Trends Microbiol 14:92–97. doi: 10.1016/j.tim.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 40.Davies GJ, Gloster TM, Henrissat B. 2005. Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr Opin Struct Biol 15:637–645. doi: 10.1016/j.sbi.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 41.Goh YJ, Klaenhammer TR. 2013. A functional glycogen biosynthesis pathway in Lactobacillus acidophilus: expression and analysis of the glg operon. Mol Microbiol 89:1187–1200. doi: 10.1111/mmi.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keyhani NO, Roseman S. 1999. Physiological aspects of chitin catabolism in marine bacteria. Biochim Biophys Acta 1473:108–122. doi: 10.1016/S0304-4165(99)00172-5. [DOI] [PubMed] [Google Scholar]
- 43.Vaaje-Kolstad G, Horn SJ, Sørlie M, Eijsink VGH. 2013. The chitinolytic machinery of Serratia marcescens—a model system for enzymatic degradation of recalcitrant polysaccharides. FEBS J 280:3028–3049. doi: 10.1111/febs.12181. [DOI] [PubMed] [Google Scholar]
- 44.Hjort K, Bergström M, Adesina MF, Jansson JK, Smalla K, Sjöling S. 2010. Chitinase genes revealed and compared in bacterial isolates, DNA extracts and a metagenomic library from a phytopathogen-suppressive soil. FEMS Microbiol Ecol 71:197–207. doi: 10.1111/j.1574-6941.2009.00801.x. [DOI] [PubMed] [Google Scholar]
- 45.Souza CP, Almeida BC, Colwell RR, Rivera ING. 2011. The importance of chitin in the marine environment. Mar Biotechnol (NY) 13:823–830. doi: 10.1007/s10126-011-9388-1. [DOI] [PubMed] [Google Scholar]
- 46.Larsen MH, Leisner JJ, Ingmer H. 2010. The chitinolytic activity of Listeria monocytogenes EGD is regulated by carbohydrates but also by the virulence regulator PrfA. Appl Environ Microbiol 76:6470–6476. doi: 10.1128/AEM.00297-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Boer W, Leveau JHJ, Kowalchuk GA, Klein Gunnewiek PJA, Abeln ECA, Figge MJ, Sjollema K, Janse JD, van Veen JA. 2004. Collimonas fungivorans gen. nov., sp. nov., a chitinolytic soil bacterium with the ability to grow on living fungal hyphae. Int J Syst Evol Microbiol 54:857–864. doi: 10.1099/ijs.0.02920-0. [DOI] [PubMed] [Google Scholar]
- 48.Gefen G, Anbar M, Morag E, Lamed R, Bayer EA. 2012. Enhanced cellulose degradation by targeted integration of a cohesin-fused β-glucosidase into the Clostridium thermocellum cellulosome. Proc Natl Acad Sci U S A 109:10298–10303. doi: 10.1073/pnas.1202747109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berlemont R, Allison SD, Weihe C, Lu Y, Brodie EL, Martiny JBH, Martiny AC. 2014. Cellulolytic potential under environmental changes in microbial communities from grassland litter. Aquat Microbiol 5:639. doi: 10.3389/fmicb.2014.00639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Allison SD. 2012. A trait-based approach for modelling microbial litter decomposition. Ecol Lett 15:1058–1070. doi: 10.1111/j.1461-0248.2012.01807.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.