

Abstract
With the aim of assuring more patient compliant pharmacotherapy for acquired immuno deficiency syndrome, a formulation of the first line anti-retroviral drug, nevirapine (NVP), has been developed by encapsulating it within niosomes. Biocompatible niosomes were fabricated using a biological surfactant, tyloxapol, with variable cholesterol concentrations. Formulation with surfactant/cholesterol molar ratio 1:0.1 exhibits maximum stability and optimum hydrophobicity. Thus, it is most suitable for the entrapment of NVP and has high entrapment efficiency of 94.3%. FTIR and DSC results indicate that NVP has sufficient compatibility with the excipients of the formulation. Photoluminescence quenching measurements were employed to elucidate the position of drug molecules in niosome bilayer along with the partition coefficient. Dissolution results indicate that the efflux of drug is sustained which creates a depot effect and decreases the fluctuations in drug release. Such a versatile and improved formulation of NVP is expected to increase its therapeutic index and alleviate toxic systemic side effects while improving the quality of life and duration of survival of the patients.
KEY WORDS: cholesterol, encapsulation, nevirapine, sustained release, tyloxapol niosomes
INTRODUCTION
Acquired immuno deficiency syndrome (AIDS) seems to be one of the most devastating epidemics that mankind has ever witnessed. According to the World Health Organization (WHO), more than 60 million people have been infected with the human immunodeficiency virus type 1 (HIV-1) and nearly 30 million people have died as a result of HIV infection till date. It is the sixth largest cause of death worldwide. Efficient management of AIDS includes highly active antiretroviral therapy (HAART) which comprises a combination of three or four anti-retroviral (ARV) drugs (1,2). Though the morbidity and mortality of AIDS patients have been reduced by 17% over the past few years and the survival time of the patient has been prolonged, the present treatments still fail to eradicate HIV and cure AIDS completely.
A relatively short half-life results in the fluctuation of drug levels in the body fluids which requires frequent dosing regimen leading to patient incompliance. Due to substantial first-pass metabolism and degradation in the gastrointestinal tract, these drugs have poor and erratic bioavailability. Furthermore, since the anti-HIV agents are unable to remove the provirus, a long-term treatment is required. Therefore, it is desirable to have a controlled or sustained release drug delivery system that could improve patient compliance, minimize side effects, and maximize therapeutic efficacy.
To cater to all these needs, nevirapine (NVP) has emerged as an excellent candidate for the treatment of AIDS as it inhibits HIV-1 reverse transcriptase at submicromolar concentration and shows benefits in HAART when used in combination with nucleoside analogs (3–5). However, over the years, drug-resistant forms of HIV both in vitro and in clinical trials have been witnessed. Often the use of NVP is accompanied by lethal side effects that include hepatotoxicity, central nervous system toxicity, insomnia, confusion, memory loss, depression, rashes, nausea, dizziness, Stevens–Johnson syndrome, toxic epidermal necrolysis and hyperlipidemia, etc. It has been issued a “black box label” by the US Food and Drug Administration owing to the severe hepatotoxicity caused within the first few weeks of treatment (6). As a result, its use is confined to the cases where the benefit to the patient exceeds the risk. A promising substitute to enhance the clinical potential of NVP is the fabrication of a drug delivery module that can assist in its sustained and targeted release.
NVP exhibits low aqueous solubility and high permeability and has been classified as class II drug, according to Biopharmaceutics Classification System (BCS). Complexity in the formulation of dosage forms (7,8), poses a challenge in the achievement of optimal dissolution kinetics (9) which results in a decrease in bioavailability. Given the paradoxical content of NVP, it is apparent that there is a need for a better formulation which is less toxic and more efficacious.
Of the numerous systems available, no drug delivery system accomplishes the site-specific delivery with sustained release kinetics of drugs in an expected manner. Supramolecular systems such as micelles, liposomes, and other nanoparticles are used in chemical and pharmaceutical fields (10–12). However, they exhibit limited utility in terms of their partial degradation that occurs before they reach a desired target in the body (13). Non-ionic surfactant vesicles or niosomes surmount these drawbacks due to their structural similarity to the cell wall and thus are of high pharmacological interest (14). Niosomes are microscopic lamellar structures obtained on the hydration of synthetic non-ionic surfactants, with or without the incorporation of cholesterol or other lipids. They fulfill a majority of the prerequisites of a delivery system. Being non-ionic, they are less toxic and improve the therapeutic index of a drug by restricting their action to target cells. They possess a microstructure consisting of both hydrophilic and hydrophobic moieties together and thus can accommodate drug molecules with wide range of solubilities. In addition to conventional, oral, and parenteral routes, they are acquiescent to be delivered by ocular, transdermal, vaginal, and inhalation routes (15).
Tyloxapol has been described as a liquid polymer of 4-(1,1,3,3-tetramethylbutyl) phenol with ethylene oxide and formaldehyde in the official monographs of the US Pharmacopeia (16). It is a non-ionic biological surfactant of the alkyl aryl polyether alcohol type with a HLB value of 12.9 (17). It is popularly employed as an excipient (e.g., as emulsifier) and is mostly used in marketed ophthalmic products and as a mucolytic agent for treating pulmonary diseases (18–22). It has been used for the development of new sustained release drug administration systems (21). Tyloxapol molecules assemble in aqueous solution to form vesicles.
Considering the advantages offered by niosomes and biological significance of Tyloxapol, the chief focus of the current study is to gain insight into the potential of tyloxapol-based niosomes in improving the bioavailability of NVP.
MATERIALS AND METHODS
Materials
Tyloxapol was procured from Sigma Aldrich. Cholesterol was obtained from Merck (India). NVP was obtained as a gift sample from Panacea Biotec (Mohali, India). All the chemicals were used as received without further purification. HPLC-grade water was used to prepare the samples for electron microscopic images. Deionized water was used for the other experiments. The molecular structures of the materials used are represented in Fig. 1.
Fig. 1.
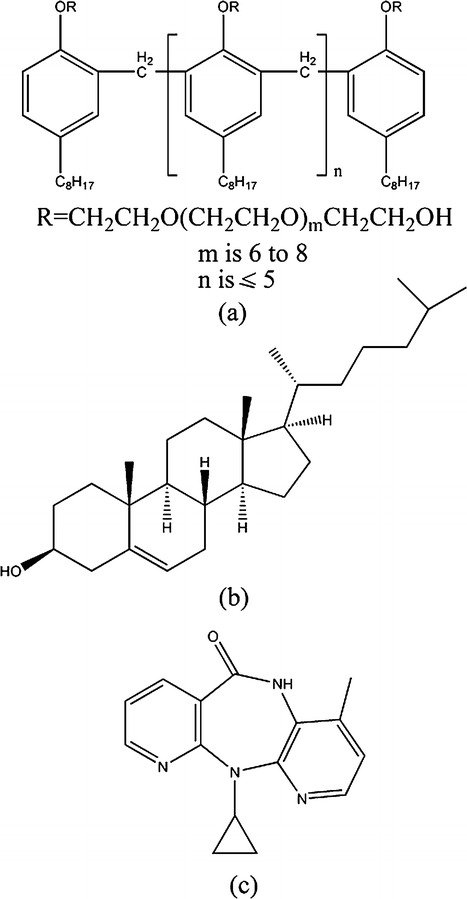
Molecular structures of a tyloxapol, b cholesterol, and c nevirapine
Methods
Standard Curve for Nevirapine
Standard curve for NVP has been prepared using absorption spectroscopy. Absorbance of known concentrations (standards) of drug was measured. From the linear regression of the graph of absorbance at λmax as a function of drug concentration, the extinction coefficient was determined (Fig. 2). It was then used to calculate the unknown concentration of NVP. The method was found to be 99 ± 0.5% accurate.
Fig. 2.
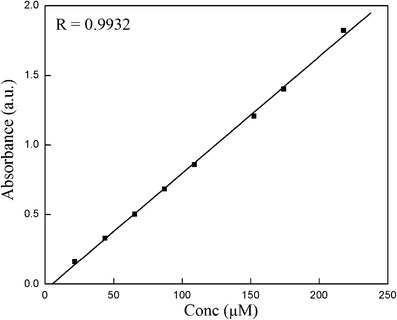
Standard curve of NVP
Fabrication of Blank- and Drug-Loaded Tyloxapol Niosomes
Tyloxapol niosomes were prepared by thin-film hydration method. Precisely weighed quantities of NVP, tyloxapol, and cholesterol were dissolved in chloroform in a flask. Tyloxapol/cholesterol molar ratio was varied as 1:0.1 (formulation F1), 1:0.25 (formulation F2), and 1:0.5 (formulation F3). The chloroform was evaporated under reduced pressure using a rotary flash evaporator (Stuart RE 300DB, Germany). The thin films were hydrated with water while the flask was rotated at 60°C for 20 min. Formulations were then sonicated for 15 min in a bath sonicator (Powersonic 405, Hwashin technology Co., Seoul, South Korea).
Electron Microscopy
The size and morphology of niosomes were obtained using transmission electron microscopy (TEM). Niosomes were diluted using HPLC-grade water (1:10) and deposited on carbon-coated copper grid, followed by the removal of excess formulation using a fine filter paper. The samples were then analyzed at 200 kV using JEOL 2100F TEM (Jeol Ltd. Japan).
Shape and topography of the formulation was confirmed using JEOL JSM-6610LV scanning electron microscopy (SEM) (JEOL USA Inc.). The filtration of formulation was avoided so as to view the morphology of bigger niosomes in the formulation, if there are any.
Particle Size Analysis (PSA)
The size analysis of fabricated formulations was done using Malvern zetasizer nano series (Nano-S90) instrument based on photon correlation spectroscopy. Two milliliters of prepared niosomes was taken in a polystyrene cuvette and exposed to laser light diffraction at an angle of 90°.
Compatibility of NVP with Excipients
The most significant task in a pre-formulation study is the analysis of compatibility of drug with the excipients of the formulation. Differential scanning calorimeter (DSC) was anticipated to be a speedy and reliable technique for appraising physico-chemical interactions between components of the formulation. Pure substances and physical mixtures of drug and excipients were analyzed using DSC (Q20 from TA instruments, USA) under dynamic nitrogen atmosphere in the temperature range of 0–300°C. A heating rate of 10°C per min was used, and the thermograms were examined for evidence of any kind of interaction.
The DSC results were further confirmed using Fourier transform infrared spectroscopy (FTIR). FTIR spectra of pure and physical mixture of drug and excipients were recorded on PerkinElmer (RX1; MA, USA) using AgCl plates in the frequency range of 4,000–350 cm−1 with a 4-cm−1 spectral resolution.
Determination of Percentage Entrapment
Encapsulation efficiency of the NVP-loaded niosomes (0.1% w/w) was determined by dialysis method. Free NVP was separated from the entrapped drug using dialysis (23). Pre-weighed amount of NVP-loaded niosomes was dialyzed using a Sigma dialysis bag against phosphate buffer saline (PBS) (pH 7.4). It was maintained at 25 ± 0.05°C, using Lauda Ecoline RE 320 thermostat. The absorbance of the dialysate was measured against PBS at the characteristic wavelength of drug using UV–vis spectrophotometer (Jasco V 530, Japan). The concentration of free drug was obtained using standard free curve, and the entrapment efficiency was calculated as follows:
| 1 |
where Ctotal is the concentration of total drug and Cout is the concentration of unentrapped drug.
Stability Studies
Calorimetric studies were performed to study the effect of cholesterol concentration on tyloxapol niosomes. Formulations with varied cholesterol concentrations were monitored using DSC in the temperature range 0–150°C with a uniform heating rate of 10°C per min. The DSC thermograms of all the formulations were explored for structural changes in the niosomes.
Distribution Coefficient
Distribution coefficient (24) of NVP was calculated using photoluminescence quenching measurements of the pyrene-labeled blank and drug-loaded niosomes. Spectra were recorded using PerkinElmer LS 55 fluorescence spectrophotometer from USA. The experiments were performed in triplicate to verify the reproducibility of results.
Location of NVP
Steady-state fluorescence quenching of pyrene was used to investigate the location of NVP (25,26) within tyloxapol niosomes. Blank niosomes labeled with the probe were titrated against drug-incorporated niosomes. To comprehend the influence of drug on the niosome membrane, the microenvironment of the probe was explored using the ratio (I1/I3) of the first peak to the third peak.
Microviscosity Studies
The microviscosity of niosomal membrane with varying cholesterol content was calculated by steady-state fluorescence polarization (P) spectroscopy (27). The extent of depolarization expressed as a polarization value was calculated according to the following equation:
| 2 |
where Ip and Iv were the fluorescence intensities of the emitted light polarized parallel and vertical to the exciting light, respectively, and G is the grating correction factor. The fluorescence intensities Ip and Iv were measured at various temperatures using Hitachi F 7000 fluorescence spectrofluorophotometer, Japan.
Diffusion Kinetics of NVP
In vitro release of the formulation was accessed to get an idea of in vivo performance (28). NVP-loaded niosomes 0.1% w/w were put into a dialysis tube which was dialyzed against 50 ml PBS (pH-7.4) at 37°C and 100 rpm. At pre-determined time intervals (5, 10, 15, 20, 25, 30, 40, 50, 60, 90, 120, 150, 180, 210, 240, and 300 min after starting the experiment), samples were withdrawn from the outside of the dialysis tube to estimate the percentage of drug released and replenished with fresh buffer solution kept at same temperature. Concentration of NVP at different intervals was examined spectrophotometrically at 277 nm after necessary dilution. The kinetics of diffusion was studied by using various kinetic equations (29–31), and the order of the release has been determined by comparing parameters of the linear regression line for different rate equations.
RESULTS AND DISCUSSION
Tyloxapol is a non-ionic surfactant having detergent properties. It has been widely used not only as an additive in pharmacy but has also been introduced as a pharmacologically active substance (22). PEGylated tyloxapol niosomes for the controlled release of first-line anti-tuberculosis drugs have already been reported by our group (32). However, these niosomes exhibit poor entrapment efficiency and non-sustained release in the case of NVP (data not shown). Considering this, an effort has been made in the present study to formulate niosomes composed of tyloxapol with variable concentrations of cholesterol that could successfully control the release of NVP in addition to high entrapment efficiency. Cholesterol is used as a membrane-stabilizing material since it alters the fluidity of chains in bilayers and, when present in sufficient concentration, abolishes the gel-to-liquid phase transition endotherm of surfactant bilayers. In addition, it also averts the niosomes from aggregation (33) and markedly decreases the efflux of entrapped solute. The decreased permeability is attributed to the filling of voids created by the assembly of surfactant monomers.
Size and Morphology of Niosomes
It is evident from TEM photomicrographs that in the formulations F1 and F2, the niosomes are spherical in shape with a size range of 60–70 nm (Fig. 3a, b). However, in formulation F3 (Fig. 3c), the niosomes are predominantly elongated which perhaps may be due to high cholesterol concentration which disrupts the regular linear structure of niosomes (34). Morphology of the formulation F1 was further confirmed using SEM analysis (Fig. 3d). In SEM, the smaller niosomes could not be observed due to the limitation of instrument. The scale has not been provided in the SEM image (Fig. 3d) since it is only for the representation of morphology.
Fig. 3.

TEM images of formulation a F1, b F2, c F3, and d SEM image of formulation F1
Figure 4 depicts the size distribution graph by intensity. The particle sizes and polydispersity index (PDI) for the different formulations of tyloxapol as measured by PSA have been found to be 52.1 (0.362), 63.3 (0.276), and 68.3 nm (0.331), respectively, which are in good conformity with the TEM measurements. The high PDI (>0.1) points toward the heterogeneity of the formulation which is perhaps due to the multicomponent system.
Fig. 4.
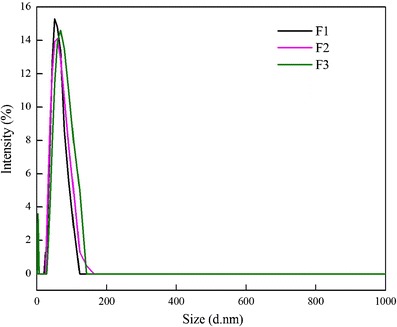
Particle size distribution by intensity of different formulations of tyloxapol
Compatibility Study with Excipients
FTIR spectra of pure drug, surfactant, and physical mixture of drug and surfactant have been recorded (Fig. 5) to explore any kind of interactions or incompatibility between the drugs and components of niosomes. The IR spectra of the mixture of tyloxapol and NVP shows disappearance of peak at 3,480 corresponding to the –OH band of tyloxapol. This could be due to hydrogen bond formation with NVP. Physical mixture of cholesterol and NVP gives IR spectrum which is the superposition of individual ones without absence, shift, or broadening in the vibration bands. This confirms the absence of any major physical or chemical interactions between the two components. Therefore, no major drug–excipient interactions were detected in FTIR spectra, signifying that NVP is compatible with the components of formulation.
Fig. 5.
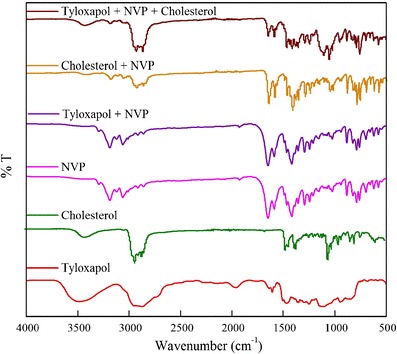
FTIR spectra of physical mixture of NVP with the excipients to check the compatibility of formulations
In pre-formulation studies, DSC is a significant tool to quickly obtain information about possible interactions among the formulation components, based upon the appearance, shifting, or disappearance of endothermic or exothermic peaks in thermal curves of drug–excipient mixtures. DSC curves for pure NVP and physical mixtures with excipients are shown in Fig. 6. The sharp melting point peak of pure NVP appeared at 245°C whereas no such peak has been observed in the case of physical mixture of NVP and cholesterol, signifying the amorphization of drug (35). However, in the case of tyloxapol–NVP mixture, a smaller peak is observed for NVP, suggesting that some crystalline drug still remains. In the case of physical mixture of NVP with both tyloxapol and cholesterol, no peak is seen corresponding to the pure drug, suggesting complete amorphization or solubilization of drug. However, a small shift in the peak of cholesterol is seen which may be considered as the presence of some interactions in the physical mixture. However, FTIR spectrum of the three components together exhibits all the peaks corresponding to cholesterol which indicates absence of any sort of interactions. The thermal curves and the FTIR spectra of the physical mixtures can be considered practically as an evidence for the absence of incompatibility between NVP and the excipients.
Fig. 6.
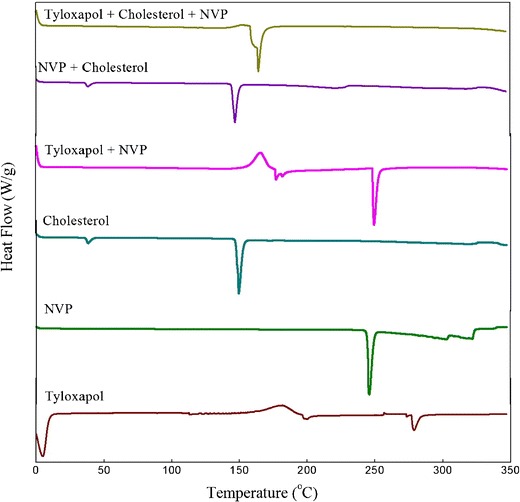
DSC thermogram of physical mixture of NVP with the excipients
Microviscosity
Fluorescence polarization correlates to the microviscosity of the probe environment. Higher value of fluorescence polarization designates greater microviscosity and thus more rigidity. Figure 7 depicts the variation of microviscosity as a function of temperature measured using photoluminescence. As expected, an increase of cholesterol ratio from 0.1 to 0.5 resulted in an increase of microviscosity of the membrane; however, there was not much significant difference observed.
Fig. 7.
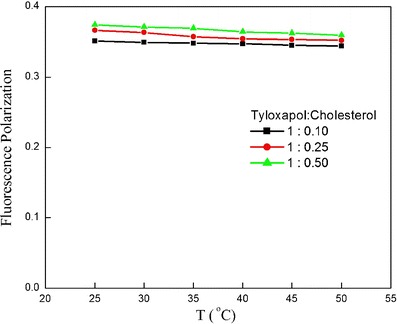
Microviscosity studies of different formulations of tyloxapol having different cholesterol concentrations
Stability Studies
The stability of the formulations has been measured as a function of temperature using calorimetric studies. DSC thermograms of the blank formulations have been depicted in Fig. 8. An increase in cholesterol concentration is known to abolish the gel-to-liquid phase transition resulting in less leaky niosomes (34). However, peak area for F1 has been found to be at the minimum as compared to F2 and F3. It may seem that the DSC results oppose the microviscosity studies; however, since the later shows only trivial enhancement in the microviscosity with an increase of cholesterol ratio, it can be ignored. The contradictory behavior in the present system can be attributed to the fact that an increase in cholesterol beyond certain concentration may disrupt the regular linear structure of niosomes (34). It can thus be concluded that cholesterol at molar ratio of 0.1 provides optimum hydrophobicity which diminishes the membrane fluidity. This is due to the reduction in formation of the transient hydrophilic holes which are responsible for leakage of drug (34).
Fig. 8.
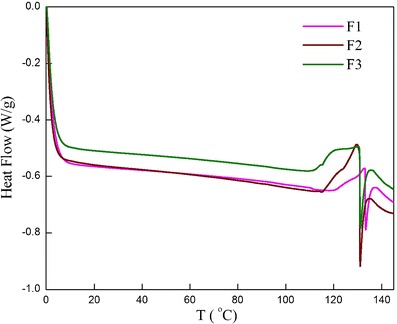
DSC heating thermograms of tyloxapol niosomes in the presence of increasing concentration of cholesterol
Entrapment Efficiency
Sufficient entrapment of therapeutic agent is the most desirable property for successful and practical usage of any drug delivery module since it will assist in solving the problem of dose-related systemic side effects and thus enhancing patient compliance. An increase in cholesterol content in the niosomal system elevates the drug-loading capacity since the incorporation of cholesterol influences vesicle stability and permeability (36). Therefore, in the present study, an attempt has been made to explore encapsulation efficiency as a function of tyloxapol to cholesterol molar ratio. High encapsulation efficiency has been observed for NVP; however, variation of cholesterol concentration was found to have little effect on the entrapment of NVP into the tyloxapol niosomes (Table I). There was a trivial increase of 0.2% in the entrapment efficiency as the cholesterol ratio is increased from 0.1 to 0.25 followed by a decrease when the cholesterol ratio is further increased to 0.5. The decrease can be attributed to the fact that higher amounts of cholesterol may compete with the drug for hydrophobic packing space within the amphiphile bilayers, hence barring the drug entrapment into vesicles.
Table I.
Characteristics of Niosomes Fabricated Using Tyloxapol Having Different Cholesterol Concentrations
| Formulations | Tyloxapol/cholesterol molar ratio | Entrapment efficiency (%) |
|---|---|---|
| F1 | 1:0.10 | 94.3 |
| F2 | 1:0.25 | 94.5 |
| F3 | 1:0.50 | 91.6 |
From the above studies, it can be concluded that the niosomal formulation composed of tyloxapol/cholesterol molar ratio of 1:0.1 is advantageous for further investigation.
Photoluminescence Studies
Micropolarity Measurements
The sites occupied by NVP in micro heterogeneous atmosphere of formulated niosomes can be assessed by monitoring the polarity of probe. Pyrene, a hydrophobic fluorescent probe, is preferentially distributed in the hydrophobic domains of niosomes, if such sites are present (25). Local polarity around pyrene in biomimetic environments like that of niosomes can be anticipated by the ratio of the first to the third vibronic band (I1/I3) that represents the solubilization sites occupied by the ensemble of pyrene molecules (26). Figure 9a portrays the I1/I3 as a function of NVP concentration. As the concentration of NVP increases, the ratio of I1/I3 decreases and hence the polarity decreases which is an indication of its solubilization in the niosome bilayer. The decrease can be credited to the fact that hydrophobicity of NVP makes the probe shift gradually into the film phase thus decreasing the micropolarity.
Fig. 9.
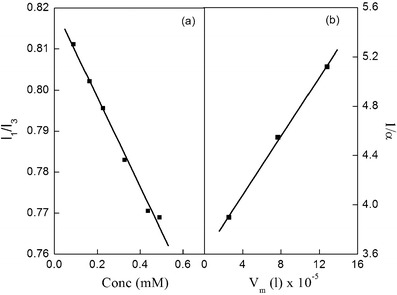
Plot of a pyrene micropolarity index (I 1/I 3) vs conc to determine the location of NVP in the formulation and b 1/α vs V m to determine the distribution coefficient for NVP in formulation F1
Distribution Coefficient
Distribution coefficient is one of the vital parameters to resolve the extent of incorporation of drugs into the niosomes. The higher the extent of penetration, the more will be the entrapment efficiency. Distribution coefficient for NVP in the selected formulation was determined by measuring the quenching of fluorescence of pyrene by non-fluorescent NVP.
According to the Stern–Volmer (SV) model, quenching of pyrene occurs due to the collision of a NVP molecule with a pyrene molecule in excited state (24). This is a dynamic process and can be described by the following equation:
| 3 |
where Io, I, and [Q] represent the initial fluorescence intensity, intensity after quenching, and the total concentration of the quencher, respectively. KSV is the SV constant which explains the interactions between donor and acceptor molecules. Absence of any upward or downward curvature in the plot signifies that SV model is applicable for the determination of interactions of the drug and niosome membrane.
By performing a series of experiments with various surfactant concentrations (Fig. 9b), the distribution coefficient of NVP was calculated (37). Large value of partition coefficient (KD = 4.26) indicates that NVP exists more in the niosome membrane than in the ambient bulk phase.
The molar Gibbs free energy change (∆Gm) for the distribution of NVP in niosome/water continuum was calculated using Gibbs equation (Eq. (4)) (26).
| 4 |
The estimated ∆Gm value for the solubilization of NVP is highly negative (−24.31 kJ/mol) indicating spontaneous hydrotrope solubilization action of niosomes toward NVP (26).
In Vitro Release Studies
The in vitro release of NVP from different formulations was explored over a period of 5 h at physiological conditions using dialysis bag method. The output obtained can provide a good correlation with the in vivo performance. Figure 10 displays the release behavior of NVP from the fabricated niosomes. During the first hour, 16.1% of NVP is released from formulations F1. Speedy drug release initially can be explained by the amorphization of NVP, which is more soluble than the crystalline form of the drug (35). Later, slow or sustained drug release is attributed to the expulsion of drug from the internal lamellae of niosomes. 43.7% of NVP is released from F1 in 5 h. It is apparent from the release profile that the efflux of the drug occurs in a controlled manner. This can be attributed to the fact that the cholesterol insertion fills the pores in vesicular bilayer systems resulting in niosomes that are less leaky. This substantiates the fact that cholesterol acts as a membrane-stabilizing agent that helps to sustain the drug release. It can be inferred that the fabricated niosomal delivery will decrease the drug fluctuations and permit longer dosing intervals thus increasing patient compliance. Higher amount of NVP is released in F2 and F3 as compared to F1 as expected because an increase in cholesterol concentration leads to slight disruption in the regular linear structure of niosomes (34).
Fig. 10.
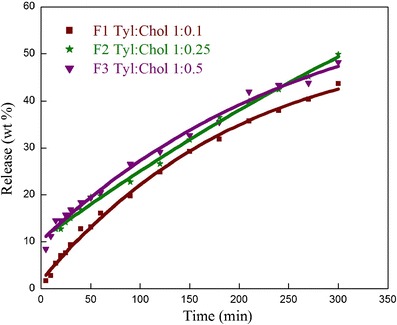
Effect of cholesterol on the in vitro release of NVP from the tyloxapol niosomes having different tyloxapol/cholesterol molar ratio
Mode of release of NVP from F1 has been assessed or analyzed using five different kinetic models (Table II). High correlation coefficients were observed in the case of Higuchi plot. Thus, the drug release is proportional to the square root of time, signifying that the efflux is diffusion controlled. The value of n in KP model should be equal to 0.5 for diffusional (Fickian) release, 1 for zero-order kinetics, and 0.5 < n < 1 for anomalous (non-Fickian) release. n value of 0.7636 (Table II) indicates that the release of NVP is anomalous or non-Fickian (31).
Table II.
Rate Constants and Regression Correlations Using Rate Equations for the Release of NVP from Formulation F1 (29–31)
| Zero-order kinetics Q = k o t |
First-order kinetics ln Q = ln Q o − k 1 t |
Higuchi kinetics Q = k H t 1/2 |
KP model |
Exponential first order Q = Q o − be−kt |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| k o | R | k 1 | R | k H | R | k p | n | R | k | R |
| 0.1659 | 0.9855 | 0.0083 | 0.8588 | 2.3278 | 0.9989 | 0.0063 | 0.7636 | 0.9920 | 0.0078 | 0.9943 |
KP model Korsmeyer-Pappas model
CONCLUSIONS
One of the tangible goals in finding the perfect cure for HIV/AIDS is reducing the gap between state-of-the-art and “ideal” formulations, thus assuring a more compliant pharmacotherapy. Keeping this in view, tyloxapol niosomes have been explored for changes in the stability, microviscosity, and entrapment of NVP as a function of cholesterol concentration. There is insignificant enhancement in the entrapment efficiency with an increase in cholesterol ratio from 0.1 to 0.5, followed by a slight decrease in the encapsulation of NVP, when the cholesterol ratio is further increased from 0.5. The fall in the entrapment efficiency is due to the competition of cholesterol with the drug for hydrophobic voids within the amphiphile bilayers. DSC thermograms also indicate that cholesterol at molar ratio of 0.1 provides the most favorable hydrophobicity and thus maximum stability. High partition coefficient in niosome water continuum and more negative molar Gibbs free energy change specifies the spontaneous solubilization of NVP in the niosome membrane than in the ambient bulk phase. The fabricated niosomes thus act as good reservoir for the sustained release of NVP which can provide the desired requirements of higher bioavailability and consequently lower dosage.
Acknowledgments
SKM and NJ are thankful to the Council of Scientific and Industrial Research, India (CSIR), for the financial assistance.
References
- 1.Lin JH. Role of pharmacokinetics in the discovery and development of indinavir. Adv Drug Deliv Rev. 1999;39:33–49. doi: 10.1016/S0169-409X(99)00018-6. [DOI] [PubMed] [Google Scholar]
- 2.Shah CA. Adherence to high activity antiretroviral therapy (HAART) in pediatric patients infected with HIV: issues and intervention in India. Pediatrics. 2007;74:55–60. doi: 10.1007/s12098-007-0028-8. [DOI] [PubMed] [Google Scholar]
- 3.Merluzzi VJ, Hargrave KD, Labadia M, Grozinger K, Skoog M, Wu JC, et al. Inhibition of HIV-1 replication by a non-nucleoside reverse transcriptase inhibitor. Science. 1990;250:1411–1413. doi: 10.1126/science.1701568. [DOI] [PubMed] [Google Scholar]
- 4.Larder BA. Interactions between drug resistance mutations in human immunodeficiency virus type 1 reverse transcriptase. J Gen Virol. 1994;75:951–957. doi: 10.1099/0022-1317-75-5-951. [DOI] [PubMed] [Google Scholar]
- 5.Balzarini J, Pelemans H, Karlsson A, DeClercq E, Kleim J-P. Concomitant combination therapy for HIV infection preferable over sequential therapy with 3TC and non-nucleoside reverse transcriptase inhibitors. Proc Natl Acad Sci U S A. 1996;93:13152–13157. doi: 10.1073/pnas.93.23.13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kontorinis N, Dieterich D. Hepatotoxicity of antiretroviral therapy. AIDS Rev. 2003;5:36–43. [PubMed] [Google Scholar]
- 7.Hawi A, Bell G. Preformulation studies of nevirapine, a reverse transcriptase inhibitor. Pharm Res. 1994;11(Suppl):36. [Google Scholar]
- 8.Lamson MJ, Sabo JP, Macgregor TR, Pav TW, Rowland L, Hawi A. Single dose pharmacokinetics and bioavailability of nevirapine in healthy volunteers. Biopharm Drug Dispos. 1995;20:285–291. doi: 10.1002/(SICI)1099-081X(199909)20:6<285::AID-BDD187>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 9.Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennernas H, Hussain AS, et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol Pharm. 1995;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 10.Ristsehel WA. Microemulsions for improved peptide absorption from the gastrointestinal tract. Methods Find Exp Clin Pharmacol. 1991;13:205–220. [PubMed] [Google Scholar]
- 11.Gabizon A, Goren D, Cohen R, Barenholz Y. Development of liposomal anthracyclines: from basics to clinical applications. J Control Release. 1998;53:275–279. doi: 10.1016/S0168-3659(97)00261-7. [DOI] [PubMed] [Google Scholar]
- 12.Navalakhe RM, Nandedkar TD. Application of nanotechnology in biomedicine. Indian J Exp Biol. 2007;45:160–165. [PubMed] [Google Scholar]
- 13.Liu X, Huang G. Formation strategies, mechanism of intracellular delivery and potential clinical applications of pH-sensitive liposomes. Asian J Pharm Sci. 2013;8:319–328. doi: 10.1016/j.ajps.2013.11.002. [DOI] [Google Scholar]
- 14.Girigoswami A, Susmita D, Swati D. Fluorescence and dynamic light scattering studies of niosomes-membrane mimetic systems. Spectrochim Acta A. 2006;64:859–866. doi: 10.1016/j.saa.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 15.Marianecci C, Marzio LD, Rinaldi F, Celia C, Paolino D, Alhaique F, et al. Niosomes from 80s to present: the state of the art. Adv Colloid Interf Sci. 2014;205:187–206. doi: 10.1016/j.cis.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 16.US Pharmacopeia, US Pharmacopeial Convention, Rockville, 22nd Revision (USP XXII), 1990, 1434.
- 17.Kulshreshtha AK, Singh ON, Wall GM. Pharmaceutical suspensions: from formulation development to manufacturing. New York: Springer; 2010. [Google Scholar]
- 18.Scott H. Comparing the surface chemical properties and the effect of salts on the cloud point of a conventional nonionic surfactant, octoxynol 9 (Triton X-100), and of its oligomer, tyloxapol (Triton WR-1339) J Colloid Interface Sci. 1998;205:496–502. doi: 10.1006/jcis.1998.5721. [DOI] [PubMed] [Google Scholar]
- 19.Regev O, Zana R. Aggregation behavior of tyloxapol, a nonionic surfactant oligomer, in aqueous solution. J Colloid Interface Sci. 1999;210:8–17. doi: 10.1006/jcis.1998.5776. [DOI] [PubMed] [Google Scholar]
- 20.Westesen K. Phase diagram of tyloxapol and water—I. Int J Pharm. 1994;102:91–100. doi: 10.1016/0378-5173(94)90043-4. [DOI] [Google Scholar]
- 21.Westesen K, Koch MHJ. Phase diagram of tyloxapol and water—II. Int J Pharm. 1994;103:225–236. doi: 10.1016/0378-5173(94)90172-4. [DOI] [Google Scholar]
- 22.Tainter ML, Nachod FC, Bird JG. Alevaire as a mucolytic agent. N Engl J Med. 1955;253:764–767. doi: 10.1056/NEJM195511032531804. [DOI] [PubMed] [Google Scholar]
- 23.Hao Y, Zhao F, Li N, Yang Y, Li K. Studies on a high encapsulation of colchicine by a niosome system. Int J Pharm. 2002;244:73–80. doi: 10.1016/S0378-5173(02)00301-0. [DOI] [PubMed] [Google Scholar]
- 24.Vostrikov VV, Selishcheva AA, Sorokoumova GM, Shakina YN, Shvets VI, Savel’ev OY, et al. Distribution coefficient of rifabutin in liposome/water system as measured by different methods. Eur J Pharm Biopharm. 2008;68:400–405. doi: 10.1016/j.ejpb.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 25.Kalynasundaram K, Thomas JK. Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J Am Chem Soc. 1977;99:2039–2044. doi: 10.1021/ja00449a004. [DOI] [Google Scholar]
- 26.Liu T, Guo R. Preparation of a highly stable niosome and its hydrotrope-solubilization action to drugs. Langmuir. 2005;21:11034–11039. doi: 10.1021/la051868b. [DOI] [PubMed] [Google Scholar]
- 27.Marangoni AG. Steady-state fluorescence polarization spectroscopy as a tool to determine microviscosity and structural order in food systems. Food Res Int. 1992;25:67–80. doi: 10.1016/0963-9969(92)90027-3. [DOI] [Google Scholar]
- 28.Zhang JA, Anyarambhatla G, Ma L, Ugwu S, Xuan T, Sardone T, et al. Development and characterization of a novel Cremophor® EL free liposome based paclitaxel (LEP-ETU) formulation. Eur J Pharm Biopharm. 2005;59:177–187. doi: 10.1016/j.ejpb.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, Sun X, Zhang ZR. An investigation on liver-targeting microemulsions of norcantharidin. Drug Deliv. 2005;12:289–295. doi: 10.1080/10717540500176829. [DOI] [PubMed] [Google Scholar]
- 30.Dai H, Chen Q, Qin H, Guan Y, Shen D, Hua Y, et al. A temperature-responsive copolymer hydrogel in controlled drug delivery. Macromolecules. 2006;39:6584–6589. doi: 10.1021/ma060486p. [DOI] [Google Scholar]
- 31.Washington C. Drug release from microdisperse systems: a critical review. Int J Pharm. 1990;58:1–12. doi: 10.1016/0378-5173(90)90280-H. [DOI] [Google Scholar]
- 32.Mehta SK, Jindal N. Formulation of tyloxapol niosomes for encapsulation, stabilization and dissolution of anti-tubercular drugs. Colloids Surf B: Biointerfaces. 2013;101:434–441. doi: 10.1016/j.colsurfb.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 33.van Hal DA, Bouwstra JA, van Rensen A, Jeremiasse E, de Vringer T, Junginger HE. Preparation and characterization of nonionic surfactant vesicles. J Colloid Interface Sci. 1996;178:263–273. doi: 10.1006/jcis.1996.0114. [DOI] [Google Scholar]
- 34.EL-Samaligy MS, Afifi NN, Mahmoud EA. Increasing bioavailability of silymarin using a buccal liposomal delivery system: preparation and experimental design investigation. Int J Pharm. 2006;308:140–148. doi: 10.1016/j.ijpharm.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 35.Zerrin SB, Yuksel N. Investigation of formulation variables and excipient interaction on the production of niosomes. AAPS PharmSciTech. 2012;13:826–835. doi: 10.1208/s12249-012-9805-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gregoriadis G. Liposome technology. 2. Boca Raton: CRC Press; 1993. [Google Scholar]
- 37.Lurie E, Kaplun AP, Dubovskii PV, Shvets VI. Interaction of N-(2-hydroxybenzyl)-ω-amino carbonic acids, novel amphipathic fatty acid derivatives, with membrane: partition coefficients. Biochim Biophys Acta. 1995;1235:256–262. doi: 10.1016/0005-2736(95)80012-5. [DOI] [PubMed] [Google Scholar]