

Abstract
Curcumin is a natural product with several thousand years of heritage. Its traditional Asian application to human ailments has been subjected in recent decades to worldwide pharmacological, biochemical and clinical investigations. Curcumin’s Achilles heel lies in its poor aqueous solubility and rapid degradation at pH ~ 7.4. Researchers have sought to unlock curcumin’s assets by chemical manipulation. One class of molecules under scrutiny are the monocarbonyl analogs of curcumin (MACs). A thousand plus such agents have been created and tested primarily against cancer and inflammation. The outcome is clear. In vitro, MACs furnish a 10–20 fold potency gain vs. curcumin for numerous cancer cell lines and cellular proteins. Similarly, MACs have successfully demonstrated better pharmacokinetic (PK) profiles in mice and greater tumor regression in cancer xenografts in vivo than curcumin. The compounds reveal limited toxicity as measured by murine weight gain and histopathological assessment. To our knowledge, MAC members have not yet been monitored in larger animals or humans. However, Phase 1 clinical trials are certainly on the horizon. The present review focuses on the large and evolving body of work in cancer and inflammation, but also covers MAC structural diversity and early discovery for treatment of bacteria, tuberculosis, Alzheimer’s disease and malaria.
Keywords: monocarbonyl analogs of curcumin, MACs, inflammation, cancer, infectious disease, anti-angiogenesis, NF-κB, TNF-α
1. Introduction
Curcumin (1, diferuloylmethane, Figure 1) is an ancient and tantalizing molecule characterized by nicknames such as Indian Saffron, The Spice of Life and Indian Solid Gold. Extracted from fresh dried roots of Curcuma Longa and related species in the ginger family, it is distributed annually in over million ton quantities world-wide as the rough and heterogeneous extract “turmeric”, which contains over two hundred other natural small molecules. The mixture with 2%–8% curcumin can be refined to deliver both pure 1 and isomeric mixtures of the agent dominated almost entirely by the enol isomers (Figure 1). Many varieties of the natural product are popular primarily as food coloring and flavoring agents, spices, cosmetics, botanical supplements and medicines [1]. The internet is rich with the range of products available.
Figure 1.

Curcumin and its demethoxy isomers isolated from turmeric.
The medical history of turmeric and curcumin, particularly in Asia, is extensive and stretching from centuries-old traditional ayurvedic practice to modern times. In the current environment that combines medicinal chemistry, pharmacology, biochemistry and molecular biology, cucumin has surfaced as a pleiotropic agent able to interact directly or indirectly with a multitude of cellular proteins while appearing to exert a whole organism effect on an extensive range of human disorders. The literature includes claims that the molecule can serve as an antioxidant, antimicrobial, antifungal, antiinflammatory and wide-ranging anticancer agent. In the latter category, it has been reported to elicit benefits in connection with drug-resistance and metastasis. The extended list includes protection for heart ailments, arthritis, wound healing, depression and Alzheimer’s disease among many others. It is not surprising, then, that considerable health care research has been devoted to testing the efficacy of curcumin as a pure agent, in various formulations and in combination with other proven drugs. In the 2013–2014 time frame, the NIH reported over 90 clinical trials with curcumin integral to the therapy under investigation [2]. Yet no single curcumin-containing agent has been approved by the FDA. One possible reason could be the limited opportunity for protection of such a compound in an aggressive marketplace and a historical geographical context. In 1995, two researchers at the University of Mississippi (UM) sought and won a patent for curcumin’s ability to heal wounds. They also garnered the exclusive right to market turmeric. Within two years the Indian government’s Council of Scientific and Industrial Research protested the patent as biopiracy and challenged its novelty by showing that wound-healing is an ancient practice supported by equally ancient Sanskrit documents. Needless to say, the patent was revoked and India’s “national molecule” was rescued from exploitation by UM and its faculty [3].
In parallel with recent research on parent curcumin, many laboratories around the globe went in search of easily prepared novel agents with biological properties similar or superior to those of curcumin. A major chemical class, the monocarbonyl analogs of curcumin (MACs) evolved and is the focus of this review.
One might conclude that the driving force for this curcumin re-direction arose from the patent conflict between UM and India. However, a number of other crucial factors have been at work. That most often quoted is the meager bioavailability of the drug in humans resulting from aqueous insolubility, low absorption, rapid metabolism, poor chemical stability and fast systemic elimination [4] These considerations noted in the overwhelming majority of MAC papers cited herein imply the molecule to be less tantalizing as a drug candidate than its ancient legacy might otherwise suggest. Influential structural modifications of curcumin that improve stability and solubility involve elimination of the hydrolysis-prone keto-enol functionality in 1–3 [5,6,7,8] and incorporate a range of alternative substituents on the terminal phenyl rings. Two such replacements involve dialkyl substitution of the hydrogens on the carbon between the two carbonyl groups in the diketo tautomer (the FLLL family, 4, Figure 2) [9] or installation of a single carbonyl group either as an acyclic agent or embedded in a small ring (the MAC family) (5, Figure 2). Both avoid the extraordinarily rapid decomposition of curcumin at pH 6.5 and above in aqueous medium [10] and deliver improved pharmacokinetic profiles in mouse models [11,12,13,14]. Enhancement of solubility is likewise readily achieved by appropriate substituent modification of MAC structures, the acyclic GO-series represented by GO-Y030 (Figure 2) [15,16,17] and combination of pyridines and piperidines such as EF24 (6) [18] and UBS109 (7b) [19] providing excellent examples. Accordingly, such molecular structures have attracted interest as models for development of novel curcumin mimics. On the other hand, not all of the natural product’s liabilities are bypassed by structural modification. MACs such as 6 and 7b like curcumin [4,20,21], experience rapid reductive metabolism and generate metabolites that carry only a fraction of the activity of the fully unsaturated parent compounds [19].
Figure 2.

Curcumin mimics. FLLL series (4), GO-series (GO-Y030), MACs as acycle or ring (5), EF24 (6), EF31 (R = H, 7a), UBS109 (R = Me, 7b).
Other reasons for turning from 1 to MACs are ease of synthesis [22,23] selectivity [9,24,25] and recognition that the pleiotropic nature [26] of the curcumin-like architecture permits rapid evaluation of drug potential for currently troublesome disorders such as highly resistant bacteria, Alzheimer’s syndrome, HIV, tuberculosis, malaria and diabetes [22,27]. Several excellent reviews detailing the diversity, applications and biological foundations of MACs for utility in human disease have appeared in the recent past [10,22,27,28,29,30,31,32].
2. Structural Diversity
2.1. 2D Diversity
A casual survey of both reviews of the monocarbonyl curcumin literature and the vast collection of supporting peer-reviewed research papers reveals hundreds of variations on the theme created by elimination or restructuring of the keto-enol moiety in curcumin. Nonetheless, almost all of the individual compounds can be clustered into two core templates in the diarylpentanoid class of molecules, namely the acyclic form 8 and the cyclic variation 9 (Figure 3).
Figure 3.

Core structures representing the diversity of individual MAC derivatives.
The majority of analogs are symmetrical consistent with ease of synthesis, however, many asymmetric versions have been prepared including chalcone variants [28,33]. The terminal aromatic rings may contain up to three different substituents and one or more nitrogen atoms in the ring to deliver heteroaromatic pyridine analogs (8, 9, Y = Z = N) with N located at o, m and p sites. The most popular phenyl ring substituents are OR and OH followed by halogen atoms. However, N- and C-linked substituents have been probed as well. The terminal phenyls have also been replaced with heteroaromatic rings such as thiophene in a few cases. The central ring in 9 most often appears as a 5- or 6-membered ring, although a number of 7-membered ring congeners are known. The central 6-ring is often accompanied by X = C, NR, O, S and SO2. Surprisingly, no B-R or Se derivatives have appeared to date. Finally, the central carbonyl group is the overwhelming favorite functionality at its position, although C=NOH, C=C(CN)2 and related substances have been prepared. A sampling of individual structures representing MAC diversity is presented along with their biology below.
2.2. 3D Diversity
Apart from the topological and structural variations described above, each of the mono-carbonyl analogs adopts a unique 3D structure in the solid state and a corresponding conformational profile in solution. The implication is that the delicate requirements for a molecule to bind and influence the behavior of a target protein will be dependent on the 3D geometry of the ligand. Thus, 2D representations such as 10–16 imply an undeserved similarity in terms of their complementarity to a chiral protein pocket. This is illustrated with the X-ray crystal structures of a small subset of MACs, several of which are substituted with fluorine as a rather diminutive replacement for hydrogen (Figure 4).
Figure 4.

Five X-ray crystal structure geometries of selected diarylpentanoid MACs. In the structures shown, the aromatic rings are each substituted with fluorine at the ortho-postions C2 and C6 or without substituents; (a) Acyclic series exhibiting planar geometry; (b) Central 6-membered ring with an approximate plane of symmetry bisecting an envelope conformation; (c) Central 5-membered ring with a twist ring conformation; (d) 7-membered ring derivatives presenting two different conformations of the central ring.
The structures depicted in Figure 4 reveal four separate 3D motifs. Acyclic fluorinated 10 [34] and the unfluorinated analog 13 [35,36,37] are both fully planar. Nonetheless, a polymorphic form of 13 [38] and a number of substituted analogs [38,39,40,41] adopt a twisted shape. Many MAC entries in the Cambridge Structural Database (CSD) show distortions from planarity, but the great majority involve metal complexation to one or both of the C=C double bonds. In the uncomplexed acyclic cases, ortho-fluoro substitution of the terminal phenyl rings does not perturb planarity. However, larger ortho-groups, e.g., CF3 or i-Pr, accompanied by steric hindrance will certainly induce non-planarity. Introduction of a 6-membered central ring, on the other hand, produces a butterfly shape as in 14 regardless of the nature of X (C, N, O or S) [42,43,44,45,46,47,48,49,50,51]. Interestingly, a variety of structural modifications including the formation of a nitrogen heterocycle, neutral or charged (Figure 4b, variations of X and Y), results in a very similar conformation verified by molecular superposition of the structures. One apparent exception to this observation is the cationic N-dimethyl analog 14 (X = NMe2+, terminal phenyl rings carry p-NMe2) [52]. The six-membered ring is a distorted half-chair, while the distal phenyl rings are twisted away from the butterfly shape. The 5-membered variant 12 (Figure 4c) adopts a highly unsymmetrical structure resulting from the adoption of a twist conformation by the central ring [42]. The X-ray structures of a family of nearly 20 analogs of the corresponding unfluorinated analog 15 (R = H) lacking ortho-substituents on the phenyl rings are fully planar similar to 10 and 13 [53]. Thus, it appears that internal steric effects occasioned by the four o-fluorine atoms in 12 is the basis for the observed asymmetric non-planarity. Other more bulky ortho-substituents can be expected to enhance the effect. A complement is the structure of 15 (R = Me) [54]. This molecule likewise exhibits a twisted 5-membered ring conveying both modest non-planarity and asymmetry to the overall molecular shape. Within the same MAC family, 15 (R = H) has recently been isolated as two different but nearly superimposable conformations representing a second polymorph of the compound. The origin of the two conformers and the new polymorph has been ascribed to C-H---O, π-π and C-H---π interactions [55]. Two 7-membered ring analogs (16a,b, Y = CH, X = CH2-CH2, Figure 4d) reveal yet other geometrical options in the form of two different conformers for the central ring and novel positioning of the terminal phenyl groups [56].
The message of this analysis is that 2D representations of MACs decorated with a range of substituents provide an incomplete picture of the fundamental nature of the interactions between potential drugs and their protein targets. Furthermore, the X-ray structures discussed above still reveal only the “tip of the iceberg”. The molecular shapes captured by small molecule crystal structures do not necessarily represent those for the same molecule bound to a protein. In solution, ligand molecules are properly described by an ensemble of conformations, one of which is likely to match the conformer within a protein pocket. It has been shown that low population conformers (<20%) are often the favored structure [57,58,59]. In the MAC context, head-to-head comparisons of the bio-potencies of 5- and 6-membered ring analogs occasionally reveal sharp differences most likely due to a combination of substituent effects, overall molecular shape and the conformation that is selected for binding to specific targets (Figure 3). Unfortunately, this makes SAR development complicated and reasoned molecular design difficult.
One recent study concerned with the interaction of pleiotropic MACs with kinases has nonetheless attempted to illustrate that the binding of similar core structures is tempered by both molecular shape and specific substitution pattern [26]. EF31 (7a and 14, Y = N, X = NH) was used to screen a 50-member kinase library, and the top 12 enzymes were then subjected to IC50 inhibition measurements with five MACs. AKT1 and AKT2 delivered the lowest range of values (IC50 0.02 to >100 μM) followed by a kinetic analysis to strongly suggest that the dominant mechanism for inhibition is competitive displacement of ATP. Accordingly, a structure-based analysis performed with AKT2. The top hit, N-protonated EF31 (IC50 0.02 μM) was subsequently docked into the ATP binding and shown to make hydrogen bonds from its carbonyl group (CO---HN(CH2)) and one of the pyridine nitrogens, a salt bridge to Glu236 (N-H---OC(O)) and several attractive hydrophobic contacts. Comparison with protonated EF24 (7a and 14, Y = C-F, X = NH2) with 40-fold lower potency (IC50 0.8 μM) demonstrated loss of a key hydrogen bond and introduction of weaker ligand-protein associations. Protonated UBS109 (7b and 14, Y = N, X = NMe), lowering potency still further (IC50 1.9 μM), was predicted to relocate somewhat in the binding site to accommodate the slightly bulky equatorial N-methyl group. This movement not only created a pair of close steric contacts, but also eliminated the electrostatically enhanced C=O-HN(Lys181) hydrogen bond. The largest structure activity perturbation takes place with 7b/14 (X = S, Y = N; IC50 > 100 μM) relative to EF31 (7a) in which the central NH is replaced by sulfur, and the AKT2 potency drops by several thousand fold. Molecular modeling suggests a major relocation of the molecule in the binding pocket and the loss of two key electrostatically enhanced H-bonds (i.e., from C=O and N-H). The relatively large sulfur atom and the expanded volume of the molecule due to its long S-C bonds are contributing factors. The reader is referred to the original literature [26] for additional details. One concludes that relatively small changes in drug-ligand structure, not necessarily apparent from comparison of flat textual structures (e.g., Figure 4), can have a drastic effect on the degree of binding with target proteins when they are known. In such circumstances, structure-based models can prove useful for understanding quantitative structure activity relationships (QSARs) and generating ideas for further synthesis and bioassay. In such cases, a ligand-based analysis can sometimes compensate for the lack of detailed structure. Unfortunately, in the sequel, most of the cases discussed do not yet allow a consideration of specific ligand-protein interactions. However, recognition that structural variation as illustrated in Figure 4 and the accompanying discussion, is operating beneath the cover of superficial structural comparison can alert one to expect both unintended frustration and surprises.
In this section, we have provided a consolidated structural introduction to be followed by a therapeutic organization, which brings together structure, mechanism and biology under specific disease/biology subsections. We have made an attempt to track disease indications associated with the range of structural modifications in the hope that this overview may serve as a useful basis for further analog development.
3. Inflammation Control in Vitro and in Vivo by MACs
In general, the anti-inflammatory activity of curcumin analogs results primarily from inhibition of nuclear factor kappa-B (NF-κB), tumor necrosis factor (TNF)-α, and interleukin (IL)-6, NF-κB being a key transcriptional factor in the inflammatory signaling pathway. Many studies have reported that MACs may target both inflammation and tumors by inhibiting the activation of NF-κB [8,23,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74]. The anti-inflammatory properties and the ability to inhibit the immune response by MACs, at least in part, result from inhibition of the activation of the latter multi-protein complex, since many of the genes that are implicated in the immune/inflammatory response are upregulated by NF-κB. MACs have also been shown to be a direct inhibitor of enzymes that are important in the inflammatory response, including lipoxygenase (5-LOX) and cyclooxygenase (COX-2) [75]. With few exceptions, most of the curcumin analogs with good anti-inflammatory action incorporate the diarylpentanoid linker instead of the β-diketone moiety and incorporate heteroatom and halogen moieties (17–25, Figure 5). Parallel studies have also confirmed that these analogs exhibit better anti-tumor, anti-inflammatory and anti-oxidant activity relative to curcumin (17–19, 26–29, Figure 5).
Figure 5.

Structures of anti-inflammatory MACs 17–29.
3.1. NF-κB/TNF-α
Our group has synthesized two structurally similar analogs, 3,5-bis-(2-fluorobenzylidene)-4-piperidone (6, EF24) and 3,5-bis-(2-pyridinylmethylidene)-4-piperidone (7a, EF31) and compared their NF-κB inhibition activities in mouse RAW264.7 macrophages [61] Results showed that 7a (IC50 ~ 5 μM) exhibits significantly more potent inhibition of lipopolysaccharide (LPS)-induced NF-κB DNA binding compared to both 6 (IC50 ~ 35 μM) and curcumin (IC50 > 50 μM). Compound 7a also effectively blocks NF-κB nuclear translocation and the induction of downstream inflammatory mediators including pro-inflammatory cytokine mRNA and protein (TNF-α, IL-1β and IL-6). Furthermore, 7a (IC50 1.9 μM) shows significantly greater inhibition of IkB kinase β compared to 6 (IC50 ~ 131 μM). In addition to these efforts, Vileker et al. conducted a series of studies demonstrating the effectiveness of 6 to block mRNA synthesis of NF-κB dependent inflammatory factors [64].
Liang et al. reported a series of MACs (30–40, Figure 6) with the ability to inhibit LPS-inducing macrophages that release inflammatory cytokines TNF-α and IL-6 via in vitro cell experiments [65,66]. Systematic structure-activity relationship studies on these compounds showed that multiple analogs can block expression of the inflammatory factors. Cyclohexanone-containing derivatives are somewhat more effective than acetone or cyclopentanone-derived compounds [65]. Installation of a long chain substituent such as 3-(dimethylamino) propoxyl (compound 40) shows an inhibitory effect on LPS-induced TNF-α expression similar to curcumin, but a more potent inhibitory effect on LPS-induced IL-6 expression than curcumin. However, the dimethylamino-analogs 41–43 (Figure 7) exhibit either similar or reduced inhibitory effects on LPS-induced TNF-α or IL-6 expression relative to curcumin, indicating that nitrogenous substitution by itself does not enhance anti-inflammatory activity. On the other hand, 33 and 44 (Figure 6 and Figure 7) with a long chain allyloxyl moiety shows a stronger inhibitory effect on LPS-induced TNF-α indicating that the length and flexibility of the distal substituents may be favorable to the anti-inflammatory activity.
Figure 6.

Structures of anti-inflammatory MACs 30–40.
Figure 7.

Structures of anti-inflammatory MACs 41–58.
Among curcumin-like compounds, 30, 31, and 45 (Figure 6 and Figure 7) deliver the best inhibition activities while 46, 47, and 48 (Figure 7) are essentially inactive, suggesting that the presence of a 3-methoxy group in combination with the 4-OH group is critical to activity. The electronegative property of a substituent in the 4’-position plays an important role in anti-inflammatory activities [66]. Compounds without a para substituent in the phenyl rings show little inhibitory activity, whereas the presence of electron-withdrawing chloro substituents removes the anti-inflammatory activity completely (49–52, Figure 7). By comparison, tetra-methoxy 53 or tetra-methyl 54 (Figure 7) with 4’-substitution showed significant inhibitory activities against LPS-induced TNF-α and IL-6. These results indicate that the anti-inflammatory activity induced by LPS may be associated with the electronegativity of 4’-substituents. Electron-donating capacity from this position may increase the anti-inflammatory abilities, whereas a neutral and electron-withdrawing moiety may reduce or remove such bioactivity. Among all the compounds studied, 40 and 44 showed the highest potential as anti-inflammatory agents. However, the underlying molecular mechanisms of N-substituted long-chain substituents at the transcriptional or post-transcriptional levels are yet to be determined. One possible origin of the substituent-influenced inflammatory variation may be the enhanced resonance interaction of electron-donating functionalities with the enone group so as to attenuate its electrophilic properties. Since thiol conjugation appears to be a critical feature for biologically active enones [76,77] perturbation of the Michael addition for MACs by para-substituents may have a decisive influence on the degree of anti-inflammatory action. In mouse primary peritoneal macrophages, 55 (Figure 7) potentially inhibited the production of pro-inflammatory gene expression including TNF-a, IL-1b, IL-6, iNOS, COX-2 and PGE synthase. This activity was ascribed in part to the inhibition of ERK/JNK phosphorylation and NF-κB activation. Compound 55 likewise shows significant in vivo effects on pro-inflammatory cytokine production in plasma and liver; namely, attenuated lung histopathology and reduced mortality in endotoxemic mice [67]. In LPS-challenged mice, pretreatment by 56 (Figure 7) attenuated the increase of plasma levels of NO, TNF-α and IL-6, while significantly reducing the hepatic inflammatory gene transcription by the inhibition of various inflammatory mediators [68]. Compounds 57 and 58 (Figure 7) significantly alleviate renal and cardiac injuries in diabetes mellitus by means of an anti-inflammatory mechanism [69,70]. Further studies revealed that these anti-inflammatory actions are mediated by inhibiting the JNK/NF-κB signal pathway [67,69,71]. The Liang group has reported that, in general, the six-membered cyclohexanone ring system (IC50 values from 4 to 180 µmol) is superior to the five-membered cyclopentanone system (IC50, 1 to 222 µmol) for inhibitory activity [8]. The difference is often modest, but can be significant depending on the cell line used for the comparison. Some of analogs screened by Liang et al. have undergone preclinical study for the treatment of arthritis, pyemia (multiple abscesses caused by pus-forming microorganisms) and nephritis (kidney inflammation).
Weber et al. studied the inhibition of TNFα-induced activation of NF-κB by dienone MACs (17, 19, 27, 28, 59, Figure 5 and Figure 8) by using the Panomics’ NF-κB Reporter Stable Cell Line [72]. The enones tested included analogs with both a 5-carbon spacer (17, 26, 30, 60–62, Figure 5, Figure 6 and Figure 8) and a 3-carbon spacer (63, Figure 8) separating the aromatic rings. The former are highly active and include cases with heterocyclic rings such as 17 (IC50 = 3.4 µM), the most active agent among the tested enones. While these compounds retain the enone functionality, their NF-κB inhibition does not correlate with their anti-oxidant activity. The study revealed that prevention of stress-induced activation of NF-κB by MAC analogs was achieved by inhibition of specific targets rather than by an overall anti-oxidation process. The latter action by MACs depends on the ability to quench free radical reactions, but can be complemented by inhibition or inactivation of specific targets. Researchers have tested the abilities of MACs to quench the pre-formed radical monocation of 2,2'-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid), known as the total radical-trapping anti-oxidant parameter (TRAP) assay, and their ability to reduce the ferric tripyridyltriazine complex, namely the ferric reducing/antioxidant power (FRAP) assay [78]. Most active analogs probed in this study (17, 19, 28) show no activity in the TRAP or FRAP assay, which led to the conclusion that there is no correlation between anti-oxidant activity and inhibition of the TNFα-induced activation of NF-κB. This lack of correlation suggests that MACs inhibit specific targets rather than operate through redox chemistry.
Figure 8.

Structures of anti-inflammatory MACs 59–73.
Yadav et al. synthesized the series of heterocyclic cyclohexanone analogues 22, 20–25, 65–67 (Figure 5 and Figure 8) which were screened for inhibition of NF-κB transactivation in non-adherent leukemia cells [73]. The pyridine heteroaromatic substituted bis-methylene cyclohexanones 22 and 23 were the most active heteroaromatic analogues (IC50 = 1.0 µM and 0.8 µM), while the corresponding five-membered heteroaromatic agents exhibit considerably less activity (e.g., 36, IC50 = 34 µM). The polymethoxyphenylmethylene N-methylpiperidone derivatives 66 and 67 (Figure 8) reveal good activity (IC50 = 0.9 and 4.0 µM, respectively) in the luciferase assay compared to the cyclohexanone core derivatives (~30 fold less activity), which had no activity. These results suggest that in addition to the electronic effects of substituents on the terminal aromatic rings described above, a nitrogen heteroatom in the aromatic rings or heteroatoms in the core cyclic ketone enhance potency. Cao et al. have reported pyridinyl analogs of dibenzylideneacetone (68, 69, Figure 8), and these MACS were evaluated for their anti-inflammatory activity by the NF-κB inhibition assay in colorectal carcinoma cells [74]. Almost all synthesized analogs exhibited better cytotoxicity than curcumin, and 68 in particular delivered the highest anti-NF-κB (IC50 = 0.52 µM) potency in suppressing growth of colon cancer cells (IC50 = 1.6 µM).
Recently, Liu et al. synthesized a series of allylated or prenylated MACs and evaluated their anti-inflammatory effects in RAW 264.7 macrophages [79]. A majority of the compounds effectively inhibited the LPS-induced expression of TNF-α and IL-6. The preliminary and quantitative SAR analyses showed that the asymmetric MACs possess higher anti-inflammatory activity than symmetric analogs, and suggested that the electronegativity and molecular polarizability of the MAC structures are important for the inhibition of LPS-induced IL-6 expression. Among the tested compounds, 82–85 (Figure 9) exhibited stronger inhibition of both TNF-α and IL-6 than curcumin. In particular, 83 delivered the most potent effects with inhibitory rates reaching 68% and 91%, respectively, and exhibited significant protection against LPS-induced death in both septic mice models and primary peritoneal macrophages. A related and recent report described the synthesis and evaluation of various asymmetric MACs as anti-inflammatory agents by inhibiting the LPS-induced secretion of TNF-α and IL-6. Among the test subjects, 86–90 exhibited dose-dependent inhibition. The anti-inflammatory activities of analogs 86 and 87 were associated with inhibition of the phosphorylation of the extracellular signal-regulated kinase ERK and the activation of NF-κB [28].
Figure 9.

Structures of anti-inflammatory MACs 74–94.
3.2. 5-LOX/COX-2
Cyclovalone (91) and three analogs (92–94) incorporating the diarylpentanoid linker between the terminal phenyl rings show anti-COX activity. The dimethylated analogs 92 and 94 are more potent than 95 and 97 that are, in turn, more potent than curcumin, suggesting the addition of methyl groups on the phenyl rings enhances anti-COX activity [80].
A series of MACs 70–79 (Figure 8 and Figure 9) containing a variety of heterocyclic rings (the indolyl, imidazolyl and thienyl rings) was synthesized by Katsori et al [75] and investigated for their ability to inhibit the inflammation related enzymes 5-LOX and aldose reductase (ALR2). Results of these studies revealed that 71 and 79 are more potent inhibitors of inflammatory enzymes 5-LOX and ALR2 than curcumin. Compounds 75 and 76 containing bromobenzene or heterocycles exhibit a high in vivo anti-inflammatory activity assessed by using the functional model of carrageenin-induced rat paw edema (expressed as percent inhibition of carrageenin-induced inflammation), while 75 showed much higher efficacy than indomethacin.
Gafner et al. designed and synthesized MACs (59, 80) for inhibition of COX-1 and COX-2 and tested them in murine macrophages. Fluoro substitution in the MACs cyclohexanone analog 80 (Figure 9) enhanced anti-inflammatory activity, while nitro and tert-butyl substitution decreased it [81]. Based on COX-2 inhibition, 59 (IC50 = 5.5 µM) is more effective than other derivatives as well as curcumin (IC50 = 15.9 µM). The data suggests that structural elements responsible for COX-1 and COX-2 inhibition do not correlate well with those responsible for inhibiting COX-2 and iNOS gene expression. However the same elements do contribute to inhibition of 12-O-tetradecanoyl-13-acetate (TPA)-induced ornithine decarboxylase (ODC) activity. TPA-induced ODC is a rate-limiting enzyme process in the polyamine biosynthetic pathway. Certain polyamines are known to be important for cell growth and differentiation and have been implicated in the early phase of tumor promotion. Thus, diminishing ODC activity has been used frequently as a marker for inhibition of tumor promotion. Weber et al. reports highlight MAC analogs 17 (IC50 4.1 μM) and 81 (IC50 3.8 µM) as potential COX-2 inhibitors, which imply an important role in pro-inflammatory stimulation via TPA-induced activation of AP-1 [82,83].
In general, the position and electronegativity of substituents on the terminal aromatic rings and the length of the spacer between these rings determine the anti-inflammatory activities of MAC’s. For example, bromo substitution at the 2-position (compound 58) shows little activity, whereas substitution at the 3-position results in superior anti-inflammatory activity compared to curcumin (IC50 10–20 µM). MACs incorporating the cyclohexanone moiety are reported to be slightly more effective than those with acetone and cyclopentanone as the central core of the molecules [84]. Analogs bearing a long chain allyloxy substituent (40, 55, 56) exhibit enhanced activity, while analogues with dimethylamino or trifluoromethane substituents reveal diminished activity. Analogs possessing 3-methoxy (30, 31, 45) or trimethoxy (66) substitution display higher activity than curcumin. Heteroaromatic ring-substituted MACs (17, 18, 21–25) also show moderate anti-inflammatory activity [65,72,83,84]. All of these effects have their counterparts in the action of MACs in vitro in tumor cells and in vivo in murine models. The complementary actions of ablating both inflammation and cancer have their origin in blocking the same set of cell signaling pathways. This phenomenon is explored in detail in the next section addressing the action of MACs in the cancer environment.
The astute reader may note that several reports cited in this section highlight the influence of electronegative substituents at the para-positions of the MAC terminal aromatic rings as a mediator of anti-inflammatory activity. This point arises from the intuitive observation that electron-donating substituents appear to decrease inflammation, while electron-withdrawing groups are less effective or eliminate it [28,66,84]. The productive use of electronegativity descriptors in QSAR correlation treatments is consistent [79]. These works do not, however, provide a qualitative rationale for how differential electronegativity might be exerting the proposed effect. Prompted by a Reviewer, we propose the phenomenon to be complex and composed of several reinforcing forces. The following Figure 10 suggests the interplay of some of them.
Figure 10.

Electrostatic potentials for a series of piperidinone analogs with para-substituents H, CH3, OCH3, OH, F and CF3 situated at the purple centers; charges: green/neutral, red/negative and blue/positive; surfaces generated with the semiempirical PM3 method in Student Spartan [85].
For substituents H and CH3, apart from the C=O moiety, the molecular surface is neutral, but the methyl groups of OCH3 begin to exhibit slight positive charge in the molecular view shown. This is enhanced by OH substitution and further magnified across the entire molecular surface for F and CF3. These changes in electrostatic potential contribute to the binding of these molecules to the protein targets in the body’s cells. This is clearly not the whole, picture, however, since the protein binding pockets also need to accommodate the steric bulk of the p-substituents. In addition the OCH3, OH, F and CF3 groups must be compatible with any H-bonding or complementary electrostatic interactions of the MACs within reach at the binding center. Consequently, the charge distributions induced by the different electronegative atoms or groups partner with atomic size and non-bonded contacts to provide the maximum binding arrangement and, thereby, result in the associated ligand-protein affinities. These, in turn, undoubtedly modulate the strength of the ultimate anti-inflammatory output at the terminus of a long train of linked intra- and intercellular physiological events. For now, the latter remain a mystery yet to be unraveled.
4. Cancer Mediation in Vitro and in Vivo by MACs
4.1. In Vitro Probes of Cancer Cell Lines and Signaling Factors
The major actions of MACs reported in the anticancer literature are mainly anti-angiogenic and cytotoxic/anti-proliferative in the context of in vitro assays. These two phenomena correlate well, suggesting that the same signaling pathways or proteins are involved in the inhibitory processes. Not surprisingly, the same mechanistic factors may also be involved in the inflammatory events described in the previous section. However, unlike studies in inflammation research, investigators in cancer have most frequently evaluated MAC curcumin analogs in phenotypic assays, such as proliferation and angiogenesis, rather than mapping signaling pathways or identifying specific protein targets. Curcumin is, of course, the exception. A recent review on breast cancer by Cridge, Larsen and Rosengren summarizes associated molecular targets and points out the intervention of cytokines, growth factors, apoptosis and cell cycle proteins in addition to transcription factors and enzymes for a subset of MACs [32]. A recent modest kinase screen demonstrated that one MAC, EF31 (7a = 14, X = NH, Y = N, Figure 2), is able to block 22 of 50 cancer-related kinases and suggests a mechanism dominated by competition with ATP [26]. Clearly more work needs to be done in this area, but it is highly likely that a majority of intracellular pathways established for curcumin will be followed by the MAC class of compounds, but with at least 10–20 fold greater potency.
For example, Dimmock and colleagues synthesized a series of symmetric piperidones (95–118, Figure 11 and Figure 12) and used murine P388 and L1210 cells as well as human Molt4/C8 and CEM T lymphocytes to evaluate cytotoxic effects [86,87]. The average IC50 values for the N-acryloyl analogs 104–110 for the four cell lines was 1.8 μM, while the N-unsubstituted compounds 111–117 delivered a considerably higher average of 44 μM. Thus, within this series of substances, the N-substituted analogs furnish considerably greater cytotoxicity, and the SAR correlates positively with the size of the aryl substituents. However, in the three clusters of agents investigated, the SARs are specific to each series. Thus, for 111–117, electronic parameters were deemed the most important factor influencing cytotoxicity.
Figure 11.

Structures of anti-cancer MACs 95–103.
Figure 12.

Structures of anti-cancer MACs 104–126.
Adams et al. synthesized a separate series of MACs and screened them for anti-proliferation and anti-angiogenic activity. Analogs 6 (EF24), 119, 120 and 121, among others, exhibited excellent cytotoxicity superior to that for cisplatin [23]. Those analogs effective in the anti-proliferation assay were also efficacious in anti-angiogenisis assays. For example, 6 is almost as potent as TNP-470, which has undergone clinical evaluation as an anti-angiogenic drug [23]. These data suggest: (1) The symmetrical α,β-unsaturated ketone moiety installed in the analogs shows increased anti-cancer and anti-angiogenesis activity compared with the β-diketone structure of curcumin; (2) Ortho-substitution on aromatic rings (119–121, Figure 12) in some cases enhances the activity for symmetrical analogs, the meta- or para-substitutions (124, 125, Figure 12) being somewhat less active possibly due to alterations in molecular geometry (Figure 4); and (3) Introduction of a heteroatom in the cyclic ketone (120, 121 and 126, Figure 12) generally yields improved anti-cancer and anti-angiogenic activity.
Compound 6 (EF24) was further studied by Thomas et al. and found to decrease cell viability of lung cancer cells via upregulated mitogen-activated protein kinases (MAPK) as evidenced by increased ERK1/2, c-Jun N-terminal kinase (JNK) and p38 (stress-activated protein kinase) phosphorylation [62]. A synergistic effect between the P38 inhibitor and 6 with respect to clonogenic activity of A549 lung cancer cells and apoptosis induction was also reported. Another interesting observation for 6 was revealed in relation to its anti-hypoxia inducible factor (HIF)-1 activity compared to curcumin. While curcumin inhibited HIF-1α gene transcription, 6 inhibited HIF-1α post-transcriptionally. The inhibition phenomenon occurred in a von Hippel Lindau (VHL)-dependent, but proteasome-independent manner. An additional difference was that curcumin induces microtubule stabilization in cells while 6 has no effect [88].
To evaluate MAC effects on colon cancer cells, Ohori et al. synthesized and screened 127–131 (Figure 13) against cell growth. The analogs are symmetrical 1,5-diarylpentadienones, the aromatic rings of which possess alkoxy substituents at meta- or meta/para-positions. Compound 128 was found to exhibit four-times higher potency than curcumin (IC50 2 µM vs. 8 µM) [89].
Figure 13.

Structures of anti-cancer MACs 127–148.
Chandru and colleagues prepared the dienone cyclopropoxy curcumin analogs 132–135 (Figure 13) and evaluated the quartet by anti-proliferation and anti-angiogenic assays employing an in vivo Ehrlich ascites tumor mouse model. The agents significantly reduced ascite volumes accompanied by increased apoptosis. Anti-angiogenic activity was demonstrated by the significant reduction of microvessel density in the peritoneum wall sections. The study was interpreted to imply that the two aromatic regions might be critical for potential drug-protein interactions [90].
Aromatic enone and dienone analogues (136–144, Figure 13) were prepared by Robinson et al. and screened in an in vitro anti-angiogenic assay [91]. The compounds inhibited cell proliferation, 140 and 143 being particularly potent, suggesting the importance of heterocyclic substitution. The same group subsequently generated derivatives differing in either the substitution pattern of the benzene rings or the fusion characteristics of the aromatic rings. The most notable compounds 145–148 (Figure 13) are tetralones which introduce a measure of rigidity by tethering the central enone moiety. The 2-naphthyl analog 146 exhibits the best anti-angiogenic activity, 85% at 1 µg/mL in the following sequence: 146 > 147 > 145 > 148 [92].
Woo et al. conceived a series of asymmetric MAC chalcones by pairing substituted phenyl amides with the terminal curcumin styrene unit carrying m-OMe and p-OH; i.e., 149–162 (Figure 14). The in vitro growth inhibition of human umbilical vein endothelial cells (HUVEC) caused by 149, 158, 161 and 162 reflects potent anti-angiogenic activity and suggests it may be particularly important for asymmetric phenyl alkyl amides coupled with heteroaromatic moieties [93].
Figure 14.

Structures of anti-cancer MACs 149–162.
Jha et al. synthesized a series of highly polar MACs 163–179 (Figure 15) by replacing the unstable keto-enol moiety of curcumin with a substituted piperidone and testing the series against tumor inhibition activity [94]. In human Molt 4/C8 cells and CEM T-lymphocytes, compounds 171–179 were significantly more potent than the control agent melphalan [95] in inhibiting leukemia and colon cancer cell lines. However, compounds 163–170 lost activity. The change in potency is undoubtedly due to the geometric disposition of the double-bond configuration within the (O=C)-CH=CH-(C=O) moiety of the piperidone substituent; i.e., E vs. Z stereochemistry. The most likely planar E form is expected to be twisted out of planarity in the Z isomer by steric effects encountered from the syn olefin orientation. The corresponding X-ray structures would offer deeper insights into this hypothesis, but in the spirit of the message of Figure 4, we presume that co-planarity of the compounds plays a key role in exerting cytotoxicity in the target proteins.
Figure 15.

Structures of anti-cancer MACs 163–179.
Fuchs et al. synthesized a collection of largely acyclic MACs 180–192 (Figure 16) and tested their anti-tumor properties by blocking the proliferation of prostate and breast cancer cells [96]. Compound 188, decorated with three methoxy groups at ortho and para terminal ring sites, is particularly attractive with an IC50 within the 10−6 M range corresponding to an inhibitory potency of more than 50-fold higher than curcumin. Suarez and colleagues prepared a similar subset of compounds 61, 180, 181, 191 and 192 (Figure 8 and Figure 16) and tested them against several tumor cell lines [97]. All the compounds exhibited different degrees of inhibitory activity against colon cancer cells HT-29, but analogs 61, 180 and 181 furnished superior potency (IC50 < 2.3 μM).
Figure 16.

Structures of anti-cancer MACs 180–192.
Yamakoshi et al. reported the cytotoxicity of MACs 193–205 (Figure 17) to the human colon cancer cell line HCT-116 [98]. SAR analysis complemented other studies by highlighting the structural motifs for bis-(arylmethylidene)acetone and 3-oxo-1,4-pentadiene and the degree of substitution as being important for maintaining high levels of cell cytotoxicity. Interestingly, the compound structures studied in this work suggests that the symmetry of the compounds is relatively insignificant for cytotoxicity. Zhang and co-workers prepared 26 asymmetric monocarbonyl analogs and demonstrated that five of them strongly inhibit the release of tumor necrosis factor-α and interleukin-6, while also showing much higher chemical stability than curcumin itself [28]. Thus, while the compounds are certainly suitable for use as agents against acute inflammatory disease, it remains to be seen whether the additional synthetic steps and the low-to-modest yields in some cases justifies the asymmetry. Liang’s team likewise tested a series of MACs for the anti-tumor activity and presented an SAR that implied compounds such as 206 and 207 (Figure 17) can induce tumor cell apoptosis by activating the stress mechanism of endoplasmic reticulum [99,100]. The two compounds have been reported to be under preclinical study for non-small cell lung cancer.
Figure 17.

Structures of anti-cancer MACs 193–210.
Other curcumin analogs, such as 30 (Figure 6) and 208 (Figure 17) also inhibited phosphorylation of STAT3 in breast and prostate cancer cells. In addition, these analogs exhibited more potent activities than curcumin on the down-regulation of signal transducer and activator of transcription (STAT3), AKT, and HER-2/neu, as well as the inhibition of cancer cell growth and migration [101,102].
Malhotra and Rawat et al. reported a series of novel 3,5-bis(arylidene)-4-piperidone-based symmetrical MACs and screened them for their potential anticancer activity. Among reported compounds, 209 and 210 (Figure 17) showed significant inhibition against various human tumor cell lines. Mechanism studies with the COLO 205 cell line suggests that compound 209 activates both caspase 8 and 9 and moderately activates effector caspase 3, which combined with the DNA fragmentation event suggests an apoptotic mechanism. Compound 210 delivers the characteristic annexin positive result, DNA fragmentation and caspase 3 activation. The activation of caspase 8, but not caspase 9 however, suggests an apoptosis extrinsic mechanism [103].
More recently, compound 211 (Figure 18) was subjected to HUVEC in vitro assay and shown to elicit anti-angiogenic activity by suppressing the downstream protein kinase activation of vascular endothelial growth factor (VEGF) via decreasing phosphorylation of AKT and p38 [104]. The same group reported that 212 displays an anti-tumor effect in the MTT cell proliferation assay using a H460 non-small cell lung cancer cell line [105].
Figure 18.

Structures of anti-cancer MACs 211–222.
A Guangzhou-New Jersey collaboration has reported in vitro activity for two series of analogs, the thiopyran-4-ones 213–216 [106,107] and the benzyl piperidones 217–219 [108] (Figure 18). The thiopyranones were tested in an MTT proliferation assay against prostate PC-3, HT-29 colon and Panc-1 cancer cell lines and shown to deliver suppressive IC50 values < 1 μM. All block transcriptional activity of NF-κB, modulate phosphorylation of ERK1/2 and reveal potent stimulation of apoptosis. The compounds are also uniformly 10–40 fold superior to curcumin in growth inhibition assays. In the parallel study with the benzyl piperidones 217–219, PC-3, pancreas BxPC-2, HT-29, and H1299 lung cancer cell lines were probed with growth inhibition, MTT and trypan blue exclusion assays. The compounds are active with IC50 values < 2 μM and, similar to the thiopyranones, cause apoptosis in PC-3 cells by reduction of phosphorylation of ERK1/2 and AKT. Utilization of the benzyl moiety as a linker, the introduction of F and OCH3 substitution on the benzyl aromatic ring and increase of steric bulk appears to enhance the cytotoxicity. A final investigation by Samaan et al. [109] presented a 32-compound series of nitrogen-containing heterocyclic derivatives represented by 220–222 (Figure 18) as a challenge against the h-androgen independent prostate cancer cell lines PC-3 and DU-145. The analogs are reported to furnish anti-prostate cytotoxicity IC50 values of 50–390 nM, but no toxicity against MCF-10A normal mammary epithelial cells. The terminal 5-membered ring heterocyclic molecular class (220–221) would appear to serve as an effective bio-isostere of the now well-recognized potency and solubility enhancing pyridine series (222 and others).
4.2. In Vivo Cancer Models, Tumor Growth and Regression: MACs
It is often the case that an apparently exciting in vitro profile proves to be ineffective in an animal model. As a result, the cell-based outcomes described in the previous two sections and those below require in vivo complementation. To date, a number of rodent models have been reported. One of the earliest by Shoji et al. involved treatment of athymic nude mice carrying MDA-MB-231 breast cancer solid tumors with EF24 (6) [23]. Significant anti-tumor effects were observed at 20 mg/kg with a 30% reduction of tumor weight to control. At 100 mg/kg, the tumor weight dropped to 55% without harmful side effects; namely liver, kidney and spleen toxicities were absent, and the mice experienced normal weight gain. The maximum tolerated dose (MTD) of 200 mg/kg i.v. indicated that in this setting 6 appears to be considerably more effective and substantially safer than the clinical drug cisplatin (MTD = 10 mg/kg i.p.). Perturbation of the same cell line with 6 demonstrated cell cycle arrest in the G2/M phase, an increase in intracellular ROS levels [67] post-transcriptional inhibition of the pro-angiogenic transcription factor HIF-1α, unlike curcumin, induction of microtubule stabilization in cells [87]. In a parallel study by the same group [110] coagulation factor VIIa (fVIIa) was employed as a carrier to deliver EF24 to tissue factor (TF) on the surface of the cancer cells, significantly decreasing the viability of TF-expressing MDA-MB-231 and HUVEC cells. Subsequent i.v. administration of the EF24-FFRck-fVIIa conjugate to human breast cancer xenografts in athymic nude mice leads to complexation of the conjugate to TF, endocytosis of the complex and presumed hydrolysis of the drug within the cytoplasm. The internally sequestered EF24 induces apoptosis and significantly reduces tumor size by comparison with systemically distributed and unconjugated EF24. By conjugating potent and toxic drugs to fVIIa, this targeted drug delivery system has the potential to enhance therapeutic efficacy in breast cancer and possibly other cells, while reducing toxic side effects by a combination of anti-angiogenic and anti-cancer actions.
Similar encouraging results for EF24 (6) have been observed in the recent past for both subcutaneous and orthotopic hepatocellular carcinoma (HCC) models [111]. DU145 prostate cancer xenografts in immunocompromised mice [112] energy metabolism and ovarian cancer metastasis models [113] ovarian carcinoma xenografts [114] and potent growth inhibition (GI50) of a HCT-116 human colon tumor xenograft in mice with little apparent toxicity [115,116]. As a companion to these murine studies, comprehensive pre-clinical mouse pharmacokinetics and metabolism have been performed for EF24 (6) by application of an LC/MS/MS assay [18]. The compound is stable in human and rat plasma at 37° for over two days and over a month at −20 °C. Protein binding in human, dog, rat and mouse plasma was >94% for all concentrations, albumin in human plasma being the primary target. EF24 pharmacokinetics in male CD2F1 mice were carried out at 10 mg/kg by i.v., i.p. and oral administration and best fit to a 3-compartment model. Terminal elimination half-life was 74 min accompanied by rapid absorption after oral administration, while i.v. dosing delivered a peak plasma concentration of 2.5 μM well above the in vitro GI50 value for sensitive cell lines in the NCI 60 cell line screen. Metabolism was monitored with liver microsome preparations and shown to be dominated primarily by NADPH-dependent reductive metabolism with a contribution from aromatic ring hydroxylation in the human fractions. A related liver microsome study for 7 (UBS109) reveals that reductive metabolism is also the primary deactivating mechanism, and that the primary metabolite with a single reduced C=C bond retains ~20% of the activity of 7 [19]. The high oral bioavailability of 6 (35 and 60% after i.p. and oral administration, respectively) contrasts markedly with the extremely low value for curcumin in both rodents and humans. These features, along with the potent cytotoxic activity, were interpreted to suggest that EF24 (6) is a promising candidate for cancer drug development. Closely related analogs of EF24 (6) in which the terminal aromatic rings are replaced by pyridine rings, namely EF31 (7a) and UBS109 (7b) (Figure 2), both supplement and confirm the present observations for a range of other cancer types and for inflammation as well. A comparative study examined the effects of the two analogs on human head and neck squamous cell carcinoma (SCC) Tu212 xenograft tumors established in athymic nude mice. The more soluble and more effective EF31 provided a dose response at 15 and 25 mg/kg i.p. and average AUC(0–∞) values of 3386 and 7769 h × ng/mL, respectively [12]. UBS-109 likewise delivers a dose response at 50, 100, and 150 mg/kg p.o. on SCC Tu212 xenografts, and 10 and 20 mg/kg i.p. on human pancreatic carcinoma Mia-Paca xenografts in athymic nude mice [14].
Separate investigations have examined bone mass loss in connection with metastatic breast cancer. Initially, curcumin, EF31 and UBS109 were compared for their relative effects on osteoblastogenesis and osteoclastogenesis in vitro. EF31 and UBS109 revealed a suppressive effect on osteoclastogenesis relative to curcumin, while UBS109 (7b) has a unique stimulatory effect on osteoblastic differentiation and mineralization associated with Smad signaling. 7b also potently inhibits NF-κB, which plays a pivotal role in osteoclastogenesis [117]. In a follow-up, UBS109 (7b) was examined for preventive effects on bone loss induced by breast cancer cell bone metastasis [118]. Nude mice were inoculated with the corresponding cells into the head of the right and left tibia. Within a week, the mice were treated with control and UBS109 under both oral (50 or 150 mg/kg) and i.p. (10 or 20 mg/kg) administration once daily for 5 days/week for 7 weeks. X-ray diagnosis was used to ascertain remarkable bone loss in the untreated femurs and tibias. On the contrary, bone loss was prevented by both p.o. and i.p. treatment in the subject animals presumably by stimulating osteoblastic mineralization and suppressing osteoclastogenesis. The potential for human intervention resides in the possible control of breast cancer accompanied by parallel bone growth and restructuring.
A particularly challenging condition without effective remedy is pancreatic cancer. An important mechanistic feature is aberrant DNA methylation, driven mainly by DNA methyltransferases. Accordingly EF31 (7a) and UBS109 (7b) have been probed for their ability to inhibit methylation in both in vitro and in vivo settings [14]. It is known that DNMT-1 is dependent on Hsp90 for activation and stability. Treatment of two pancreatic cell lines, Mia-PaCa-2 and PANC-1, with 7a and 7b resulted in inhibition of Hsp90 activity through its decreased binding to DNMT-1, decreased levels of Hsp90 expression and the overexpression of three pivotal silenced genes commonly silenced in the spontaneous promotion of pancreatic cancer. Under in vivo conditions, Mia-Paca-2 xenografts were established in 5-week old nude mice and treated with 25 mg/kg doses of 7a and 7b i.v. three times weekly for 30 days. A panel of biomarkers was extracted from the tumors to show, among other things, that DNMT-1 is clearly downregulated and associated with re-expression of the silenced genes. In addition, EF31 (7a) and UBS109 (7b) potentiate the inhibition of cell proliferation by oxaliplatin and 5-fluorouracil (5FU), two agents in common anti-cancer use in the clinic. A follow-up study reveals that these two compounds also exert an increased antiangiogenic effect on pancreatic cancer both in vitro and in vivo. [119]. The unique properties of 7a and 7b appear to make them promising therapeutic agents for development in pancreatic cancer as single agents or as part of chemosensitized chemotherapy cocktails.
Finally, two piperidines 22 (Figure 5) and 223 (Figure 19) assayed by Yadav and colleagues display the well-known inhibition of NF-κB activation complemented by substantial cytotoxicity towards MBA-MB-231, MDA-MB-468 and SkBr3 breast cancer cell lines; models for triple negative breast cancer disease [73]. Against the MBA-MB-231 cell line, the EC50 values are submicromolar at 0.8 and 0.3, respectively, while the two agents also cause apoptosis in 40%–45% of the SKBr3 and MDA-MB-231 cells, respectively.
Figure 19.

MACs 22 and 223 are effective in vitro against triple negative breast cancer.
In a subsequent investigation, 22 suppressed the growth of MDA-MB-468 xenograft tumors by 48% compared to control following 10 weeks of daily oral dosing (8.5 mg/kg) [120]. The compound also evidenced anti-angiogenic properties in vitro by inhibiting 46% of HUVEC cell migration and the induction of these cells to form tubular networks. Thus, 22 presents potent proapoptotic and anti-angiogenic assets in vitro and in vivo, suggesting it to be a potential and promising remedy for treatment of ER-negative breast cancer. Other in vivo rodent studies not specifically reviewed here are likewise encouraging that the wide-ranging MAC family of anti-inflammatory and anti-cancer agents may well be able to address the corresponding debilitating conditions in humans sometime in the future [8,15,16,17,67,68,90,97,110,111,114,118].
5. Bacterial Growth
Curcumin has received far less attention as inspiration for a potential antibacterial agent by comparison with its influence on the development of cancer blockers. This is surprising given that each year in the U.S. at least 2 million people become infected with bacteria resistant to antibiotics causing an annual death toll of approximately 23,000 arising directly from the infections [121]. The situation has become sufficiently dire that during the fourth week of September, 2014, President Obama has directed the CDC to create a national plan to control the situation [122]. Importantly, since inflammation follows infection as described in the previous section, were curcumin and mimics to become effective antibiotic agents, they offer an advantage in the ability to mediate simultaneously both bacterial titer and the inflammatory response. Early reports catalog curcumin’s blockade of various bacteria [123,124]. One recent study applied the molecule and its more soluble conjugate, curcumin glucoside, to two Gram-positive (Bacillus cereus, Staphylococcus aureus) and two Gram-negative (Escherichia coli and Yersinia enterocolitica) pathogenic bacteria. All four exhibited moderate growth inhibition in response to treatment by both molecules [125]. In a related study [126], it was noted that curcumin induces filamentation in Gram-positive Bacillus subtilis (strain 168), suggesting the molecule to be able to modulate bacterial cytokinesis (cell division to daughter cells). At the molecular level, the process is regulated by the assembly dynamics of FtsZ protein protofilaments, a protein aggregate that assembles into a ring at the future midpoint of bacterial cell division [127]. FtsZ is a prokaryotic homologue of the eukaryotic protein tubulin that regulates the function and trafficking of microtubules. Curcumin was shown to inhibit assembly in vitro, increase GTPase action and bind to the protein accompanied by perturbation of secondary structure. Such actions, lethal to the bacteria, provide a provocative mechanistic hypothesis for the action of the drug. These observations were followed by a docking study exploring the interaction of curcumin with the catalytic cores of the E. coli and B. subtilis FtsZ proteins [128] The tentative location of the binding site, supported by bacterial mutagenesis, was proposed as an opportunity to access novel inhibitors by structure-based molecular design [129,130,131].
Three interesting reports have explored MACs as candidates for bacterial control. An early exploration presented 40 compounds in the a, b and c classes shown in Figure 4 and compared their actions as monitored by zones of inhibition against seven multi-drug resistant bacteria containing both Gram-positive and Gram-negative examples [132]. A few members of each inhibitor class are very similar to the action of curcumin, but all are less potent than the β-lactam antibiotic ampicillin. Encouraging, however, most were found to be active against Enterobacter cloacae, an organism that is clinically resistant to ampicillin. On average, the acyclic a-class was claimed to be overall most effective, though the differences would appear to be small. Only one substitution offered a sizeable ortho-substituent (Br). This member of each of the three classes proved to be inactive, perhaps due to steric distortion (cf. Figure 4).
The second study prepared 15 piperidones and screened them against Salmonella typhi, Vibrio cholerea, Escherichia coli and Staphylococcus aureus [133] Several compounds produced significant zones of inhibition (e.g., 224, Figure 20) for the first two bacteria superior to the action of both curcumin and penicillin. More modest activity was observed for the other analogs against all four bacteria.
Figure 20.

MACs 224 and 225 block the growth of multiple bacterial strains in vitro.
Finally, a very recent investigation explored the effects of EF24 (6 and 14, Y = C-F, X = NH) and its dimer 225 (EF2DTPA) against Gram-negative (Escherichia coli) and Gram-positive (Enterococcus faecalis, Staphylococcus aureus) bacteria [134]. Both classes experience direct bacterial growth suppression. Additional experiments demonstrated that the compounds do not influence bacterial uptake or localization in dendritic cells. Although the antibacterial potencies of the synthetics are considerably lower than that of traditional antibiotics, the authors suggest that such substances might be useful in combination with other chemopreventive agents in appropriate situations.
Although the literature supporting the results discussed in this section have not explored the FtsZ connection, they clearly illustrate that MACs carry antibacterial activity and, in some cases, are effective against highly resistant bacteria. Considering the structural insights derived from examining the FtsZ-curcumin complexes [124]. it is likely that a similar evaluation of modeled FtsZ-MAC complexes would lead to suggestions for compound modification and drive investigations to confirm the prospective binding and lead to novel antibacterial candidates.
6. Tuberculosis
Tuberculosis (TB) is an airborne disease caused by the contagious and ancient bacterium Mycobacterium tuberculosis (M. tuberculosis). TB is considered latent when the M. tuberculosis infection is present but the immune system is able to prevent active multiplication. If the immune system is unable to prevent bacterial growth, TB becomes active and can be fatal, if untreated. About one third of the world’s population is infected with the microorganism. In 2012 there were nine million new cases and 1.3 million TB-related deaths around the world [135]. Not surprisingly, TB is the leading killer of people infected with immune compromised HIV, making TB one of the most deadly diseases in the world. Currently, ten drugs are approved by the FDA to treat active TB [136] with a combined success rate reported to be 87% in 2011 [137]. While current drugs are highly effective against the TB bacteria, drug-resistant strains are becoming more prevalent especially in less developed countries. Contemporary treatment methods for drug-resistant TB are less effective and 50 to 200 times more expensive than treatment for classic cases of TB [138]. As a consequence, discovery of alternative drugs is of high importance.
Curcumin was reported to inhibit M. tuberculosis growth as early as the mid-twentieth century, making it an underexplored but promising drug candidate against TB. In 1949 Schraufstatter and colleagues reported that curcumin is able to completely block M. tuberculosis growth in vitro at a dilution of 1:40,000 [139] A recent review by Marathe and co-workers argues that curcumin may serve as an inhibitor against efflux pumps in TB bacteria. Efflux pumps mediate the efflux of antibiotics, drugs and other classes of compounds. Several studies indicate that efflux pumps are behind the survival mechanism for resistant M. tuberculosis [140,141,142]. Another investigation by Dube and colleagues used computational models to focus on a specific domain of an essential enzyme for M. tuberculosis (the BRCT domain) known as M. tuberculosis enzyme (MtuLigA) [143]. MtuLigA is essential and unique to TB bacteria, making it an intriguing drug target. Computational models suggested that a curcumin derivative, monoacetylcurcumin, binds to the BRCT domain, but no experiments were performed to confirm inhibition of MtuLigA by curcumin or its analogs. Takeuchi and co-workers conducted in vitro experiments to study the inhibitory activity of curcumin derivatives against DNA polymerase λ [144], which also contains the BRCT domain as part of its active site. Curcumin itself demonstrated strong inhibition of DNA polymerase λ (IC50 7.0 μM), while its monoacetyl derivative proved to be nearly twice as effective (IC50 3.9). Agrawal et al. reported that demethoxycurcumin (2) can block M. tuberculosis H37Rv strain with a minimum inhibitory concentration (MIC) of 200 μg/mL (5.8 × 10−4 M) [145]. Of four synthetic derivatives of 2, two analogs exhibited values of 125 and 8 μg/mL MICs. This compares with a rifampicin control at 2 μg/mL. The latter most active compound carries an R = OCH2-CH=CH-CO2C2H5 p-substitution, while the former lacking a conjugated ester (R = OCH2CO2C2H5) is 16-fold weaker. Michael addition may therefore play an important role in the mechanism of such compounds. Bairwa et al. have recently reviewed a broader set of similarly substituted keto-enol curumin derivatives and drawn a similar conclusion [27]. Finally, while curcumin shows promising anti-TB activity in vitro, it fails to show similar effectiveness in vivo [146]. One wonders if the curcumin derivatives mentioned here would show the same fate.
As previously stated, replacement of curcumin’s keto-enol functionality with a single carbonyl group may serve as a way to achieve greater stability and perhaps enhanced performance. MACs and related analogs have not been thoroughly explored at the time of writing, implying a potentially important direction for this class of molecules as anti-tubercular agents. Dimmock and co-workers have addressed this problem by combining the MACs architecture with a Mannich base and evaluating the compounds in vitro for antimycobacterial properties against M. tuberculosis H37Rv [147]. At 12.5 ug/mL, the Mannich bases achieved 99% inhibition, while a related series of unsaturated ketones blocked microorganism growth by only 21%–66%. Two of the bases were both exceptionally active with MIC values of 0.39 ug/mL (226, second structure not shown), which compares very favorably with rifampin (0.16 uM/mL). A follow-up evaluation on 226 sought to understand the origin of the high activity by preparing the partial structures 227 and 228 (Figure 21) [148]. Compound 227 proved to be essentially inactive, but 228 gave a moderate MIC value of 12.5 uM obviously highlighting the N(CH3)2 moiety. A probe of toxicity against Vero cells showed selectivity indices (IC50/MIC) for 226 and 228 to be 21 and 0.004, the former considered to signal a lead. Thus, enone 226 was evaluated against seven strains of M. tuberculosis including multidrug resistant (MDR) and extensively drug-resistant (XDR) strains to deliver MIC values across the board very similar to that of M. tuberculosis H37Rv. Accordingly, the drug-resistant strains do not evidence cross resistance to Mannich base 226, which appears to exert a different mode of antimycobacterial action relative to that by a panel of other TB drugs.
Figure 21.

Structures of various MACs serving as in vitro antimycobacterial agents against TB (226–230).
A separate investigation focused on the preparation of a series of piperidones, which showed excellent in vitro antimycobacterial properties against M. tuberculosis H37Rv [149]. Among others, 229 and 230 are 100% inhibitory at 6.3 μg/mL with MICs at 0.2 and 0.8 μg/mL, respectively. In addition, the compounds are generally well tolerated in mice as judged by toxicity and neurotoxicity screens. Oral and ip dosing of a selection of the piperidones in rats likewise failed to reveal neurotoxicity, although some swelling and stimulated respiration in rat liver mitochondria was observed. The two effects seem to be disconnected. Missing from this report is a study of the effects of the synthetic agents in animals infected by M. tuberculosis.
More recently, EF24 (6), EF31 (7a) and UBS109 (7b) were examined for in vitro growth inhibition of both M. marinum and M. tuberculosis, including rifampicin-resistant strains [150]. The compounds deliver IC50 values in the 4-25 μM range, considerably lower than curcumin. An important mechanistic finding of this work is that the anti-mycobacterial action of the MACs appears to be dependent on Michael addition chemistry.
We finish with a caveat. It has recently been shown that in a Drosophila model host system, host autophagy activation is essential for antimicrobial action against M. tuberculosis in adult flies [151]. Such activation would appear to be required for homeostatic control during chemotherapy against the TB microorganism. Blocking autophagy with antibiotics, i.e., with curcumin derivatives or MACs, might well compromise the host autophagy in the successful delivery of drug to M. tuberculosis during treatment. One of our MACs, EF25 (231, Figure 22), has recently been shown to block autophagy and contribute to cell death in a xenograft-based hepatocellular carcinoma study [152]. It remains to be seen whether the inhibition of autophagy by MACs in a cancer model would translate adversely to a TB model in which the drug would eliminate M. tuberculosis but simultaneously counter the effect by providing a safe haven for the microorganism during chemotherapy.
Figure 22.
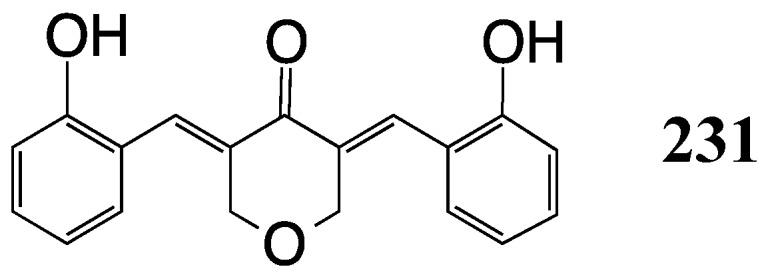
A heterocyclic MAC that blocks autophagy (231).
7. Alzheimer’s Disease
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder that most often progresses to dementia for the aging population. It is estimated that 36 million people are afflicted by dementia worldwide with eight million new cases annually. The most common cause of dementia is AD accounting for 60%–70% of all such cases. While this obviously takes an irreversible toll on the recipient, AD also exacts a devastating price for families and caregivers [153]. The condition can be characterized in part by amyloid β (Aβ) aggregation, inflammation and oxidative damage in the brain. Unfortunately, not only are the pathological mechanisms not fully understood, no chemotherapeutic agents yet exists to either ameliorate or eliminate the illness. Curcumin, with its anti-oxidative and anti-inflammatory properties, is an under-explored but promising candidate for AD treatment.
Recent studies indicate that curcumin or its analogs may be able to effectively address a wide array of AD symptoms [154]. In an early study conducted in 2004, curcumin was shown to bind to and inhibit the formation of Aβ oligomers and fibrils. In vitro experiments showed that curcumin inhibits Aβ aggregation more effectively than the over-the-counter NSAIDs ibuprofen and naproxen, anti-inflammatory drugs known to reduce amyloid formation [155]. In vivo experiments from the same study disclosed that curcumin injected peripherally crosses the blood-brain barrier, binds to plaques and reduces levels of amyloid aggregation in mice with advanced amyloid accumulation. The outcome is consistent with the hypothesis that fibrils and plaques are the cause of AD, and that they can be diminished in vivo. Accordingly, the authors suggested that curcumin should be taken to the clinic. A number of trials have been performed, and at the time of this writing (October, 2014) seven are underway [156,157]. Unfortunately, none have revealed a decisive counter to AD. In another subsequent study, a seven-day curcumin treatment regimen reduced levels of plaque by 30% in a mouse model of AD. The regimen also significantly reversed structural dendritic abnormalities. Curcumin was found to have limited effectiveness in crossing the blood-brain barrier, but results indicate a high affinity for Aβ that may contribute to the inhibition of amyloid formation [158]. A molecular approach to understanding how small molecules interact with amyloid-β has generated X-ray structures of four compounds complexed with amyloid-like segments, the small molecules lying within cylindrical cavities running along β-spines of the fibers [159]. The electron density for curcumin at low resolution does not permit modeling in atomic detail, but it strongly suggests that the long axis of the molecule lies parallel to the fiber axis as it does for the other ligands examined. The work proposes a general pharmacophore for Aβ aggregates suggesting that mixtures of compounds may be necessary in any future treatment to address the collection of polymorphic aggregates that appear to characterize the amyloid fibrils. A separate analysis sought to determine the tautomeric form of curcumin binding to Aβ aggregates [160]. It was concluded that the keto-enol form is the dominant form associated with the latter and that targeting the enolization process might offer a target for design of novel amyloid-binding agents. However, observation of the enol form in the amyloid context is entirely consistent with detailed examination of curcumin itself demonstrating the overwhelming presence of the keto-enol form [161], apparently eliminating the possibility to target tautomerization.
While curcumin shows tantalizing effectiveness against AD, its low bioavailability and stability as discussed in the Introduction poses problems for its potential as a drug. As emphasized in previous sections, one way to bypass such characteristics is pursuit of curcumin analogs with more suitable biophysical and physicochemical properties.
Orlando and co-workers [162] recently screened a library of curcumin derivatives and MACs with a novel ELISA-based assay that quantifies oligomeric Aβ peptide and distinguishes the oligomeric aggregate from fibrils. The work identified methylated curcumin 232 (Figure 23) as an oligomerization inhibitor (IC50 0.15 μM) with 6–7 fold improvement over curcumin (IC50 1.0 μM). MAC 233 (IC50 0.8 μM) likewise proved nearly equivalent to curcumin by assay. In addition, a preliminary SAR provides some clues for future workers: (1) A least one enone group in the spacer between aryl rings is necessary to observe anti-Aβ aggregation; (2) Unsaturation in the linker is also required for inhibition; (3) Both methoxy and hydroxyl substitutions at the m- and p-positions elicit de-aggregation, a positive combination being a curcumin reversal (m-OH, p-OMe, 234 IC50 0.6 μM). It is interesting that the MAC variation 233 seems to offer little in the way of potency as it does for cancer and inflammation. A more elaborate SAR is likely to uncover improved and perhaps highly potent MAC candidates at this early stage of discovery.
Figure 23.

Potential oligomerization inhibitors against the Aβ peptide and fibrils (232–234).
Oxidative stress is believed to be a major factor in the progression of AD by causing neurological degeneration and death [163]. An effective antioxidant against AD-related oxidative stress is an attractive strategy. An early study examined the antioxidant capacities of curcumin and (2E,6E)-2,6-Bis(3,5-dimethoxybenzylidene)cyclohexanone 235 (MCH, Figure 24) [164]. The compound is relatively stable by comparison to curcumin and shows enhanced anti-oxidative performance in vitro. Rat pheochromocytoma PC12 cells, a model for neurological cells under Aβ oxidative stress, were found to be protected from H2O2 injury by maintaining endogenous antioxidant enzyme activity, scavenging for reactive oxygen species and shielding cells from H2O2-induced cytotoxicity and apoptosis. In vivo experiments for MCH were not conducted, but the in vitro results suggest a promising avenue for further exploration.
Figure 24.

A potential Aβ peptide blocker (235) and an in vitro chloroquine-sensitive (CQ-S) and chloroquine-resistant (CQ-R) P. falciparum MAC antimalarial (236).
8. Malaria
Malaria is a serious infectious disease characterized by fevers, chills and flu-like illness. It is transmitted by Anopheles mosquitos infected with five known species of Plasmodium parasites, which include P. falciparum, P. vivax, P. malariae, P. ovale and P. knowlesi. Three of the five species have been reported to exhibit drug-resistant behavior, while Plasmodium parasites are gaining resistance against an increasing number of different anti-malarial drugs. With the growing issue of drug-resistance, it is of paramount interest to search for alternative anti-malarial drugs that are effective and inexpensive.
Several studies indicate that curcumin and its analogs may well serve as effective anti-malarial agents. According to Reddy and co-workers, curcumin delivers significant anti-malarial activity both in vitro and in vivo [165]. Curcumin inhibits chloroquine-resistant Plasmodium falciparum in culture in a dose-dependent manner (IC50 5 μM). In the in vivo experiments, curcumin was orally administered to P. berghei-infected mice in a dose known to be safe for humans. The drug treatment resulted in an 80%–90% reduction in blood parasitemia, and curcumin treated mice experienced an overall survival rate of 29% compared to 0% for the non-treated control. In another study by Mishra and colleagues, in vitro experiments were performed to examine the effectiveness of curcumin and its analogs against red blood cell cultures containing either chloroquine-sensitive (CQ-S) or chloroquine-resistant (CQ-R) P. falciparum [166]. The agent was found to significantly inhibit the growth of both CQ-S (IC50 7.9 μM) and CQ-R (IC50 29.6 μM) in red blood cell cultures. Several structural modifications of curcumin were also tested against both CQ-S and CQ-R, and many exhibited enhanced inhibitory activity. A pyrazole analog 236 (Figure 24) demonstrated the most effective inhibitory performance among the compounds tested (CQ-S (IC50 0.48 μM) and CQ-R (IC50 3.9 μM)). The authors speculated that the improved anti-malarial activity of 226 arises from increased cellular uptake and retention as well as greater inhibitory potency against the target itself.
A more recent study evaluated the antimalarial activity of MACs against the two strains of P. falciparum, and found that the compounds exhibit significant anti-malarial behavior in an in vitro setting [167]. Twenty different MACs were tested against P. falciparum cultures and six of them were found to provide excellent inhibition of both CQ-R and CQ-S (237, 238, IC50 < 1 μM, Figure 25). Considering the increased bioavailability and stability of MACs as well as their enhanced anti-malarial abilities, this class of molecules offers an intriguing direction for further molecular design opportunities.
Figure 25.

Potent in vitro CQ-R and CQ-S inhibitors of the P. falciparum parasite (237–238).
9. Summary
A host of MACs has shown potent anti-tumorigenic activity when tested by in vitro and in vivo models spanning a wide range of cancers and inflammatory conditions. Although a number of structure-activity relationship (SAR) studies reveal useful correlations within a limited structural set of analogs [168], a truly broad analysis has yet to be achieved. Likewise, few of the reported structure activity studies make an attempt to deal with the subtle impact of 3D geometry as implied by Figure 4. To be sure, the size and electronegativity of substituents on the terminal aromatic rings (Figure 10) and the rigidity of the spacers between these rings may influence the in vitro biological actions of MAC’s. In addition, four other features have surfaced as advantageous elements for overall improved activity: (1) A doubly conjugated carbonyl is generally more potent than a single enone; (2) Double or triple methoxy substitution on the terminal aromatic rings, often accompanied by an aromatic OH group, frequently provides increased activity; (3) Terminal pyridine rings furnish both superior potency and solubility; (4) A piperidone ring at the center of a MAC not only imparts solubility, but also creates the opportunity to append extensions through an amide-like linker with additional beneficial properties. Assuming protein binding sites can tolerate such structural modification, specific and selective targeting might be an option. One example illustrated by in vivo testing involved the coupling of EF24 (6) to coagulation factor VIIa followed by protein-protein interaction with tissue factor leading to endocytosis and targeted delivery of the drug to the cytoplasm of MDA-MB-231 breast cancer cells. To date, this has not yet blossomed into a predictive and fruitful pathway for investigation, most likely because there are no X-ray structures of the corresponding ligand-protein complexes at present. It goes without saying that the same situation exists for the parent template with no single macromolecule-curcumin solid-state structure having been solved. In spite of the paucity of well-determined MAC-protein structural models, a growing number of investigations highlight that MACs are superior to curcumin in vitro by a 10–20 fold potency gain across a wide range of cancer cell lines, inflammation models and against numerous signaling proteins and transcription factors. Likewise, murine-based in vivo studies demonstrate clearly that MACs with appropriate pharmacokinetic and ADME properties (Absorption, Distribution, Metabolism and Excretion) can be conceived, prepared and tested for potential relief of human cancer, inflammatory diseases and other conditions. Similarly, by virtue of their improved physical properties, MACs have successfully demonstrated good PK profiles in mice and promoted tumor regression in cancer xenografts as well as orthotopic models in vivo; almost always more efficacious by comparison to curcumin. Though the literature is not yet deep, chemical and biological efforts to create MACs with the potential to relieve TB, Alzheimer’s, malaria and other maladies is underway. Based on the available data, it would appear that many of the underlying biochemical mechanisms controlling the latter conditions are common. One can be hopeful and optimistic that new developmental drugs might be capable of cross-treating any number of them.
Acknowledgments
We are grateful to the NIH (R01CA165306) for support to make this work possible.
Abbreviation
- MACs
monocarbonyl analogs of curcumin
- Aβ
amyloid β
- AD
Alzheimer’s Disease
- AKT
protein kinase B
- ALR
aldose reductase
- AP-1
the activator protein 1
- ATP
adenosine triphosphate
- COX-2
cyclooxygenase
- DNMT
DNA methyltransferase
- ERK
extracellular-signal regulated kinase
- HIF
hypoxia inducible factor
- HSP
heat shock protein
- HUVEC
human umbilical vein endothelial cells
- IL
interleukin
- i.p.
intraperitoneal
- i.v.
intravenous
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MID
minimum inhibitory concentration
- MTD
maximum tolerance dose
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- LOX
lipoxygenase
- LPS
lipopolysaccharide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NCI
National Cancer Institute
- NF-κB
nuclear factor κB
- NO
nitric oxide
- NSAID
nonsteroidal anti-inflammatory drug
- ODC
ornithine decarboxylase
- p38
stress-activated protein kinase
- PGE
prostaglandine E
- p.o.
per os (by mouth)
- RAW264.7
mouse macrophage-like cell line
- ROS
reactive oxygen species
- SAR
structure-activity relationship
- STAT
signal transducer and activator of transcription
- TB
tuberculosis
- TF
tissue factor
- TNF
tumor necrosis factor
- TPA
12-O-tetradecanoyl-13-acetate
- VEGF
vascular endothelial growth factor
- VHL
von Hippel-Lindau
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- 1.Li S., Yuan W., Deng G., Wang P., Yang P., Aggarwal B.B. Chemical Composition and Product Quality Control of Turmeric (Curcuma longa L.) Pharm. Crops. 2011;2:28–54. doi: 10.2174/2210290601102010028. [DOI] [Google Scholar]
- 2.ClinicalTrials.gov. [(accessed on 27 October 2014)]; Available online: https://clinicaltrials.gov/
- 3.Mishra S.B., Mukerjee A., Singh S. Turmeric: A time tested folk medicine with Ayurvedic perspective. J. Pharm. Biomed. Res. 2011;1:67–70. [Google Scholar]
- 4.Anand P., Kunnumakkara A.B., Newman R.A., Aggarwal B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007;4:807–818. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- 5.Shen L., Ji H.-F. The pharmacology of curcumin: Is it the degradation products? Trends Mol. Med. 2012;18:138–144. doi: 10.1016/j.molmed.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Gordon O.N., Schneider C. Vanillin and ferulic acid: not the major degradation products of curcumin. Trends Mol. Med. 2012;18:361–363. doi: 10.1016/j.molmed.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen L., Ji H.-F. Low stability remedies the low bioavailability of curcumin. Trends Mol. Med. 2012;18:363–364. doi: 10.1016/j.molmed.2012.05.002. [DOI] [Google Scholar]
- 8.Liang G., Shao L., Wang Y., Zhao C., Chu Y., Xiao J., Zhao Y., Li X., Yang S. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg. Med. Chem. 2009;17:2623–2631. doi: 10.1016/j.bmc.2008.10.044. [DOI] [PubMed] [Google Scholar]
- 9.Fossey S.L., Bear M.D., Lin J., Li C., Schwartz E.B., Li P.-K., Fuchs J.R., Fenger J., Kisseberth W.C., London C.A. The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer. 2011;11:e112. doi: 10.1186/1471-2407-11-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao C., Liu Z., Liang G. Promising curcumin-based drug design: Mono-carbonyl analogues of curcumin (MACs) Curr. Pharm. Des. 2013;19:2114–2135. [PubMed] [Google Scholar]
- 11.Yu W., Xiao H., Lin J., Li C. Discovery of novel STAT3 small molecule inhibitors via in silico site-directed fragment-based drug design. J. Med. Chem. 2013;56:4402–4412. doi: 10.1021/jm400080c. [DOI] [PubMed] [Google Scholar]
- 12.Zhu S., Moore T.W., Lin X., Morii N., Mancini A., Howard R.B., Culver D., Arrendale R.F., Reddy P., Evers T.J., et al. Synthetic curcumin analog EF31 inhibits the growth of head and neck squamous cell carcinoma xenografts. Integr. Biol. (Camb.) 2012;4:633–640. doi: 10.1039/c2ib20007d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu S., Moore T.W., Morii N., Howard R.B., Culver D., Arrendale R.F., Reddy P., Evers T.J., Zhang H., Sica G., et al. Synthetic curcumin analog UBS109 inhibits the growth of head and neck squamous cell carcinoma xenografts. Curr. Cancer Drug Targets. 2014;14:380–393. doi: 10.2174/1568009614666140312163524. [DOI] [PubMed] [Google Scholar]
- 14.Nagaraju G.P., Zhu S., Wen J., Farris A., Adsay V., Diaz R., Snyder J.P., Shoji M., El-Rayes B. Novel Synthetic Curcumin Analogs EF31 and UBS109 are Potent DNA Hypomethyating Agents in Pancreatic Cancer. Cancer Lett. 2013;341:195–203. doi: 10.1016/j.canlet.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Shibata H., Yamakoshi H., Sato A., Ohori H., Kakudo Y., Kudo C., Takahashi Y., Watanabe M., Takano H., Ishioka C., et al. Newly synthesized curcumin analog has improved potential to prevent colorectal carcinogenesis in vivo. Cancer Sci. 2009;100:956–960. doi: 10.1111/j.1349-7006.2009.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kudo C., Yamakoshi H., Sato A., Nanjo H., Ohori H., Ishioka C., Iwabuchi Y., Shibata H. Synthesis of 86 species of 1,5-diaryl-3-oxo-1,4-pentadienes analogs of curcumin can yield a good lead in vivo. BMC Pharmacol. 2011;11 doi: 10.1186/1471-2210-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato A., Kudo C., Yamakoshi H., Uehara Y., Ohori H., Ishioka C., Iwabuchi Y., Shibata H. Curcumin analog GO-Y030 is a novel inhibitor of IKKb that suppresses NF-κB signaling and induces apoptosis. Cancer Sci. 2011;102:1045–1051. doi: 10.1111/j.1349-7006.2011.01886.x. [DOI] [PubMed] [Google Scholar]
- 18.Reid J.M., Buhrow S.A., Gilbert J.A., Jia L., Shoiji M., Snyder J.P., Ames M.M. Mouse pharmacokinetics and metabolism of the curcumin analog, 4-Piperidione,3,5-bis[(2-fluorophenyl)methylene]-acetate(3E,5E) (EF-24; NSC 716993) Cancer Chemother. Pharmacol. 2014;73:1137–1146. doi: 10.1007/s00280-014-2447-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore T.W., Zhu S., Shoji M., Snyder J.P. Liver S9 fraction-derived metabolites of curcumin analog UBS109. ACS Med. Chem. Lett. 2014;5:288–292. doi: 10.1021/ml4002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ireson C., Orr S., Jones D.J.L., Verschoyle R., Lim C.-L., Luo J.-L., Howells L., Plummer S., Jukes R., Williams M., et al. Characterization of metabolites of the chemopreventive agent curcumin in human and rat hepatocytes and in the rat in vivo, and evaluation of their ability to inhibit phorbol ester-induced prostaglandin E2 production. Cancer Res. 2001;61:1058–1064. [PubMed] [Google Scholar]
- 21.Hoehle S.I., Pfeiffer E., Sólyom A., Metzler M. Metabolism of curcuminoids in tissue slices and subcellular fractions from rat liver. J. Agric. Food Chem. 2006;54:756–764. doi: 10.1021/jf058146a. [DOI] [PubMed] [Google Scholar]
- 22.Bukhari S.N.A., Jantan I.B., Jasamai M., Ahmad W. Synthesis and Biological Evaluation of Curcumin Analogues. J. Med. Sci. 2013;13:501–513. doi: 10.3923/jms.2013.501.513. [DOI] [Google Scholar]
- 23.Adams B.K., Ferstl E.M., Davis M.C., Herold M., Kurtkaya S., Camalier R.F., Hollingshead M.G., Kaur G., Sausville E.A., Rickles F.R., et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg. Med. Chem. 2004;12:3871–3883. doi: 10.1016/j.bmc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Tan K.L., Koh S.B., Ee P.L.R., Khan M., Go M.L. Curcumin analogs with potent and selective anti-proliferative activity on acute promyelocytic leukemia: Involvement of accumulated misfolded nuclear receptor co-repressor (N-CoR) protein as a basis of selective activity. ChemMedChem. 2012;7:1567–1579. doi: 10.1002/cmdc.201200293. [DOI] [PubMed] [Google Scholar]
- 25.Khairia M., Youssef K.M., El-Sherbeny M.A. Synthesis and antitumor activity of some curcumin analogs. Arch. Pharm. Chem. 2005;338:181–189. doi: 10.1002/ardp.200400939. [DOI] [PubMed] [Google Scholar]
- 26.Brown A., Shi Q., Moore T.W., Younghyoun Y., Prussia A., Maddox C., Liotta D.D., Shim H., Snyder J.P. Monocarbonyl Curcumin Analogs: Heterocyclic Pleiotropic Kinase Inhibitors that Mediate Anti-Cancer Properties. J. Med. Chem. 2013;56:3456–3466. doi: 10.1021/jm4002692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bairwa K., Grover J., Kania M., Jachak S.M. Recent developments in chemistry and biology of curcumin analogues. RSC Adv. 2014;4:13946–13978. doi: 10.1039/c4ra00227j. [DOI] [Google Scholar]
- 28.Zhang Y., Zhao C., He W., Wang Z., Fang Q., Xiao B., Liu Z., Liang G., Yang S. Discovery and evaluation of asymmetrical monocarbonyl analogs of curcumin as anti-inflammatory agents. Drug Des. Dev. Ther. 2014;8:373–382. doi: 10.2147/DDDT.S58168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Padhye S., Chavan D., Paandey S., Deshpande J., Swamy K.V., Sarkar F.H. Perspectives on Chemopreventive and Therapeutic Potential of Curcumin Analogs in Medicinal Chemistry. Mini Rev. Med. Chem. 2010;10:372–387. doi: 10.2174/138955710791330891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agrawal D.K., Mishra P.K. Curcumin and Its Analogues: Potential Anticancer Agents. Med. Res. Rev. 2010;30:818–860. doi: 10.1002/med.20188. [DOI] [PubMed] [Google Scholar]
- 31.Vyas A., Dandawate P., Padhye S., Ahmad A., Sarkar F. Perspectives on new synthetic curcumin analogs and their potential anticancer properties. Curr. Pharm. Des. 2013;19:2047–2069. [PMC free article] [PubMed] [Google Scholar]
- 32.Cridge B.K., Larsen L., Rosengren R.J. Curcumin and its derivatives in breast cancer: Current developments and potential for the treatment of drug-resistant cancers. Oncol. Discov. 2013;1:1–9. doi: 10.7243/2052-6199-1-6. [DOI] [Google Scholar]
- 33.Shao W.Y., Cao Y.N., Yu Z.W., Pan W.J., Qiu X., Bu X.Z., An L.K., Huang Z.S., Guand L.Q., Chan A.S.C. Facile preparation of new unsymmetrical curcumin derivatives by solid-phase synthesis strategy. Tetrahedron Lett. 2006;47:4085–4089. doi: 10.1016/j.tetlet.2006.03.145. [DOI] [Google Scholar]
- 34.Huang J.-D., Tang Q.-Q., Chen X.-Y., Ye Y., Wang Y. (1E,4E)-1,5-Bis(2,6-difluorophenyl)penta-1,4-dien-3-one. Acta Crystallogr. Sect. E Struct. Rep. Online. 2011;67:o758. doi: 10.1107/S1600536811007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaftory M., Tanaka K., Toda F. Reactions in the solid state. 2. The crystal structures of the inclusion complexes of 1,1,6,6-tetraphenylhexa-2,4-diyne-1,6-diol with benzylideneacetophenone and 2,5-diphenylhydroquinone with dibenzylideneacetone. J. Org. Chem. 1985;50:2154–2158. doi: 10.1021/jo00212a031. [DOI] [Google Scholar]
- 36.Biradha K., Nangia A., Desiraju G.R., Carrell C.J., Carrell H.J. C-HLO Hydrogen bonded multi-point recognition in molecular assemblies of dibenzylidene ketones and 1,3,5-trinitrobenzenes. J. Mater. Chem. 1997;7:1111–1122. doi: 10.1039/a607106f. [DOI] [Google Scholar]
- 37.Biradha K., Sharma C.V.K., Paneerselvam K., Shimoni L., Carrrell H.L., Zacharias D.E., Desiraju G.R. Solid State Supramolecular Assembly via C-H--O Hydrogen Bonds: Crystal Structures of the Complexes of 1,3,5-Trinitrobenzene with Dibenzylideneacetone and 2,5-Dibenzylidene-cyclopentanone. Chem. Commun. 1993:1473–1475. [Google Scholar]
- 38.Turowska-Tyrk I. Monitoring initial structural changes in a crystal during photo-induced disapperance of its diffracting propertites. Chem. Phys. 2003;288:241–247. doi: 10.1016/S0301-0104(03)00029-6. [DOI] [Google Scholar]
- 39.Shan Y., Zhou H., Huang S.D. 1,5-bis(4-methoxyphenyl)penta-1,4-dien-3-one. Z. Kristallogr. New Cryst. Struct. 1999;214:381–382. [Google Scholar]
- 40.Butcher R.J., Yathirajan H.S., Sarojini B.K., Narayana B., Raj K.K.V. (1E,4E)-1-(3-Nitrophenyl)-5-phenylpenta-1,4-dien-3-one. Acta Crystallogr. Sect. E Struct. Rep. Online. 2006;62:o1973. doi: 10.1107/S1600536806014024. [DOI] [Google Scholar]
- 41.Samshuddin S., Butcher R.J., Akkurt M., Narayana B., Sarojini B.K., Yathirajan H.S. (1E,4E)-1-(3-Nitrophenyl)-5-phenylpenta-1,4-dien-3-one. Acta Crystallogr. Sect. E Struct. Rep. Online. 2012;68:o74. doi: 10.1107/S1600536811052548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao C.-G., Chen X.-Y., Hu J., Zhang L., Zhao Y.-J., Yang S.-L., Liang G. Synthesis and Crystal Structure of Monocarbonyl Analogs of Curcumin. Chin. J. Struct. Chem. 2012;31:491–498. doi: 10.1002/cjoc.201100252. [DOI] [Google Scholar]
- 43.Zhao C., Yang J., Liang D., Liang D., Tang Q., Zhang Y., Liang G., Li X. Synthesis, Crystal Structure and Anti-tumor Properties of Fluorine-Containing Curcumin Analogues. Chin. J. Org. Chem. 2010;30:289–294. [Google Scholar]
- 44.Jia Z., Quail J.W., Arora V.K., Dimmock J.R. Structures of 3,5-bis(benzylidene)-4-piperidone hydrochloride (I) and its N-methyl analog (II) Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1988;44:2114–2117. doi: 10.1107/S0108270188008728. [DOI] [Google Scholar]
- 45.Lagisetty P., Powell D.R., Awasthi V. Synthesis and structural determination of 3,5-bis(2-fluorobenzylidene)-4-piperidone analogs of curcumin. J. Mol. Struct. 2009;936:23–28. doi: 10.1016/j.molstruc.2009.07.016. [DOI] [Google Scholar]
- 46.Leonova E.S., Makarov M.V., Rybalkina E.Y., Nayani S.L., Tongwa P., Fonari A., Timofeeva T.V., Odinets I.L. Structure-cytotoxicity relationship in a series of N-phosphorus substituted E,E-3,5-bis(3-pyridinylmethylene)- and E,E-3,5-bis(4-pyridinylmethylene)piperid-4-ones. Eur. J. Med. Chem. 2010;45:5926–5934. doi: 10.1016/j.ejmech.2010.09.058. [DOI] [PubMed] [Google Scholar]
- 47.Vatsadze S.Z., Kovalkina M.A., Svirdenkova N.V., Zyk N.V., Chuarakov A.V., Kuz’mina L.G., Howard J.A.K. Novel dienone-based ligands for the synthesis of coordination polymers. Cryst. Eng. Commun. 2004;6:112–115. doi: 10.1039/b402418d. [DOI] [Google Scholar]
- 48.Abaee M.S., Mojtahedi M.M., Zahedi M.M., Mesbah A.W., Ghandchi N.M., Massa W. Synthesis of Bis (arylmethylidene) thiopyranones and Crystal Structure of the Phenyl Derivative. Phosphorus Sulfur Silicon Relat. Elem. 2007;182:2891–2895. doi: 10.1080/10426500701542759. [DOI] [Google Scholar]
- 49.Abaee M.S., Mojtahedi M.M., Sharifi R., Zahedi M.M., Mesbah A.W., Massa W. (3E,5E)-3,5-Bis(4-hydroxybenzylidene)oxan-4-one. J. Chem. Res. 2008:388–389. [Google Scholar]
- 50.Du Z.-Y., Huang H.-R., Lu Y.-J., Zhang K., Fang Y.-X. (3E,5E)-3,5-Bis(4-hydroxy-3,5-dimethoxybenzylidene)oxan-4-one monohydrate. Acta Crystallogr. Sect. E Struct. Rep. Online. 2010;66:o3334. doi: 10.1107/S1600536810041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karthikeyan N.S., Sathiyanarayanan K.I., Aravindan P.G., Giridharan P. Synthesis, crystal structure, and anticancer properties of cyclic monocarbonyl analogs of curcumin. Med. Chem. Res. 2011;20:81–87. doi: 10.1007/s00044-009-9284-7. [DOI] [Google Scholar]
- 52.Jia Z., Quail J.W., Arora V.K., Dimmock J.R. Structure of 3,5-bis(benzylidene)-1-methyl-4-piperidone methobromide hemiethanol solvate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1989;45:1117–1118. doi: 10.1107/S0108270189000569. [DOI] [Google Scholar]
- 53.This result was achieved by searching the Cambridge Structural Database (CSD) for compound 15 (R = H).
- 54.Theocharis C.R., Jones W., Thomas J.M., Motevalli M., Hursthouse M.B. The solid-state photodimerisation of 2,5-dibenzylidenecyclopentanone (DBCP); a topochemical reaction that yields an amorphous product. J. Chem. Soc. Perkin Trans 2. 1984:71–76. [Google Scholar]
- 55.Arshad I., Ashraf S., Abbas A., Hameed S., Lo K.M., Naseer M.M. Conformational isomerism in a conformational polymorph of 2,5-dibenzylidenecyclopentanone: Crystallographic and quantum chemical structures. Eur. Chem. Bull. 2014;3:587–592. [Google Scholar]
- 56.Kawamata J., Inoue K., Inabe T. Molecular and crystal structures of novel second-order nonlinear optical crystals: a,a'-Dibenzylidenecycloalkanones. Bull. Chem. Soc. Jpn. 1998;71:2777. doi: 10.1246/bcsj.71.2777. [DOI] [Google Scholar]
- 57.Ganesh T., Guza R.C., Bane S., Ravindra R., Shanker N., Lakdawala A.S., Snyder J.P., Kingston D.G.I. The bioactive Taxol conformation on β-tubulin: Experimental evidence from highly active constrained analogs. Proc. Nat. Acad. Sci. USA. 2004;101:10006–10011. doi: 10.1073/pnas.0403459101. The favored structures: Tubulin-bound Taxol conformations in CDCl3 (4%) and D2O/DMSO-d6 (2%) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thepchatri P., Eliseo T., Cicero D.O., Myles D., Snyder J.P. Relationship among ligand conformations in solution, in the solid state, and at the Hsp90 binding site: Geldanamycin and radicicol. J. Am. Chem. Soc. 2007;129:3127–3134. doi: 10.1021/ja064863p. The favored structures: Hsp90- bound conformers of geldanamycin (4%) and radicicol (21%) [DOI] [PubMed] [Google Scholar]
- 59.Cox B.D., Prosser A.R., Katzman B.M., Alcaraz A.A., Liotta D.C., Wilson L.J., Snyder J.P. Anti-HIV Small Molecule Binding in the Peptide Sub-Pocket of the CXCR4:CVX15 Crystal Structure. ChemBioChem. 2014;15:1614–1620. doi: 10.1002/cbic.201402056. The favored structure: Proposed CXCR4-bound AMD11070 (14%) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kasinski A.L., Du Y., Thomas S.L., Zhao J., Sun S.-Y., Khuri F.R., Wang C.-Y., Shoji M., Sun A., Snyder J.P., et al. Inhibition of IkB kinase nuclear factor-κB signaling pathway by 3,5-bis(2-flurobenzylidene) piperidin-4-one (EF24), a novel monoketone analog of curcumin. Mol. Pharmacol. 2008;74:654–661. doi: 10.1124/mol.108.046201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olivera A., Moore T.W., Hu F., Brown A.P., Sun A., Liotta D.C., Snyder J.P., Yoon Y., Shim H., Marcus A.I., et al. Inhibition of the NF-κB signaling pathway by the curcumin analog, 3,5-Bis-(2-pyridinylmethylidene)-4-piperidone (EF31): Anti-inflammatoryand anti-cancer properties. Int. Immunol. Pharmacol. 2012;12:368–377. doi: 10.1016/j.intimp.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas S.L., Zhao J., Li Z., Lou B., Du Y., Purcell J., Snyder J.P., Khuri F.R., Liotta D.C., Fu H. Activation of the p38 pathway by a novel monoketone curcumin analog, EF24, suggests a potential combination strategy. Biochem. Pharmacol. 2010;80:1309–1316. doi: 10.1016/j.bcp.2010.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adams B.K., Cai J., Armstrong J. EF24, a novel synthetic curcumin analog induces apoptosis in cancer cells via a redox dependent mechanism. Anticancer Drugs. 2005;16:263–275. doi: 10.1097/00001813-200503000-00005. [DOI] [PubMed] [Google Scholar]
- 64.Vilekar P., Awasthi S., Natarajan A., Anant S., Awasthi V. EF24 suppresses maturation and inflammatory response in dendritic cells. Int. Immunol. 2012;24:455–464. doi: 10.1093/intimm/dxr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang G., Li X., Chen L., Yang S., Wu X., Studer E., Gurley E., Hylemon P.B., Ye F., Li Y., et al. Synthesis and anti-inflammatory activities of mono-carbonyl analogues of curcumin. Bioorg. Med. Chem. Lett. 2008;18:1525–1529. doi: 10.1016/j.bmcl.2007.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhao C., Yang J., Wang Y., Liang D., Yang X., Li X., Wu J., Wu X., Yang S., Li X., et al. Synthesis of mono-carbonyl analogues of curcumin and their effects on inhibition of cytokine release in LPS-stimulated RAW 264.7 macrophages. Bioorg. Med. Chem. 2010;18:2388–2393. doi: 10.1016/j.bmc.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 67.Wang Y., Yu C., Pan Y., Li J., Zhang Y., Yang S., Zhang H., Li X., Liang G. A novel compound C12 inhibits inflammatory cytokine production and protects from inflammatory injury in vivo. PLoS One. 2011;6:e24377. doi: 10.1371/journal.pone.0024377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y., Yu C., Pan Y., Yang X., Huang Y., Feng Z., Li X., Yang S., Liang G. A novel synthetic mono-carbonyl analogue of curcumin, A13, exhibits anti-inflammatory effects in vivo by inhibition of inflammatory mediators. Inflammation. 2011;35:594–604. doi: 10.1007/s10753-011-9350-4. [DOI] [PubMed] [Google Scholar]
- 69.Pan Y., Wang Y., Cai L., Cai Y., Hu J., Yu C., Li J., Feng Z., Yang S., Li X., et al. Inhibition of high glucose-induced inflammatory response and macrophage infiltration by a novel curcumin derivative prevents renal injury in diabetic rats. Br. J. Pharmacol. 2012;166:1169–1182. doi: 10.1111/j.1476-5381.2012.01854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pan Y., Zhu G., Wang Y., Cai L., Cai Y., Hu J., Li Y., Yan Y., Wang Z., Li X., et al. Attenuation of high-glucose-induced inflammatory response by a novel curcumin derivative B06 contributes to its protection from diabetic pathogenic changes in rat kidney and heart. J. Nutr. Biochem. 2013;24:146–155. doi: 10.1016/j.jnutbio.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 71.Wu J., Li J., Cai Y., Pan Y., Ye F., Zhang Y., Zhao Y., Yang S., Li X., Liang G. Evaluation and Discovery of novel synthetic chalcone derivatives as anti-inflammatory agents. J. Med. Chem. 2011;54:8110–8123. doi: 10.1021/jm200946h. [DOI] [PubMed] [Google Scholar]
- 72.Weber W.M., Hunsaker L.A., Roybal C.N., Bobrovnikova-Marjon E.V., Abcouwer S.F., Royer R.E., Deck L.M., Vander Jagt D.L. Activation of NF-κB is inhibited by curcumin and related enones. Bioorg. Med. Chem. 2006;14:2450–2461. doi: 10.1016/j.bmc.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 73.Yadav B., Taurin S., Rosengren R.J., Schumacher M., Diederich M., Somers-Edgar T.J., Larsen L. Synthesis and cytotoxic potential of heterocyclic cyclohexanone analogues of curcumin. Bioorg. Med. Chem. 2010;18:6701–6707. doi: 10.1016/j.bmc.2010.07.063. [DOI] [PubMed] [Google Scholar]
- 74.Cao B., Wang Y., Ding K., Neamati N., Long Y.Q. Synthesis of the pyridinyl analogues of dibenzylideneacetone (pyr-dba) via an improved Claisen-Schmidt condensation, displaying diverse biological activities as curcumin analogues. Org. Biomol. Chem. 2012;10:1239–1245. doi: 10.1039/c1ob06773g. [DOI] [PubMed] [Google Scholar]
- 75.Katsori A.M., Chatzopoulou M., Dimas K., Kontogiorgis C., Patsilinakos A., Trangas T., Hadjipavlou-Litina D. Curcumin analogues as possible anti-proliferative & anti-inflammatory agents. Eur. J. Med. Chem. 2011;46:2722–2735. doi: 10.1016/j.ejmech.2011.03.060. [DOI] [PubMed] [Google Scholar]
- 76.Johansson M.H. Reversible michael additions: Covalent inhibitors and prodrugs. Mini Rev. Med. Chem. 2012;12:1330–1344. doi: 10.2174/13895575112091330. [DOI] [PubMed] [Google Scholar]
- 77.Serafimova I.M., Pufall M.A., Krishnan S., Duda K., Cohen M.S., Maglathlin R.L., McFarland J.M., Miller R.M., Frödin M., Taunton J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012;8:471–476. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schlesier K., Harwat M., Bohm V., Bitsch R. Assessment of anti-oxidant activity by using different in vitro methods. Free Radic. Res. 2002;36:177–187. doi: 10.1080/10715760290006411. [DOI] [PubMed] [Google Scholar]
- 79.Liu Z., Tang L., Zou P., Zhang Y., Wang Z., Fang Q., Jianag L., Chen G., Xu Z., Zhang H., et al. Synthesis and biological evaluation of allylated and prenylated mono-carbonyl analogs of curcumin as anti-inflammatory agents. Eur. J. Med. Chem. 2014;74:671–682. doi: 10.1016/j.ejmech.2013.10.061. [DOI] [PubMed] [Google Scholar]
- 80.Nurrochmad A., Supardjan A.M., Sardjiman Inhibition of cyclooxygenase by cyclovalone and its three analogue compounds. Majalah Farmasi Indonesia. 1998;9:180–185. [Google Scholar]
- 81.Gafner S., Lee S., Cuendet M., Barthelemy S., Vergnes L., Labidalle S., Mehta R.G., Boone C.W., Pezzuto J.M. Biologic evaluation of curcumin and structural derivatives in cancer chemoprevention model systems. Phytochemistry. 2004;65:2849–2859. doi: 10.1016/j.phytochem.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 82.Weber W.M., Hunsaker L.A., Abcouwer S.F., Deck L.M., Vander Jagt D.L. Anti-oxidant activities of curcumin and related enones. Bioorg. Med. Chem. 2005;13:3811–3820. doi: 10.1016/j.bmc.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 83.Weber W.M., Hunsaker L.A., Gonzales A.M., Heynekamp J.J., Orlando R.A., Deck L.M., Vander Jagt D.L. TPA-induced upregulation of activator protein-1 can be inhibited or enhanced by analogs of the natural product curcumin. Biochem. Pharmacol. 2006;72:928–940. doi: 10.1016/j.bcp.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 84.Liang G., Yang S., Zhou H., Shao L., Huang K., Xiao J., Huang Z., Li X. Synthesis, crystal structure and anti-inflammatory properties of curcumin analogues. Eur. J. Med. Chem. 2009;44:915–919. doi: 10.1016/j.ejmech.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 85.Spartan Student Edition. [(accessed on 3 December 2014)]. Available online: http://www.scientificsoftware-solutions.com/product.php?productid=17747.
- 86.Dimmock J.R., Padmanilayam M.P., Puthucode R.N., Nazarali A.J., Motaganahalli N.L., Zello G.A., Quail J.W., Oloo E.O., Kraatz H.B., Prisciak J.S., et al. A conformational and structure activity relationship study of cytotoxic 3,5-bis(arylidene)-4-piperidones and related N-acryloyl analogues. J. Med. Chem. 2001;44:586–593. doi: 10.1021/jm0002580. [DOI] [PubMed] [Google Scholar]
- 87.Dimmock J.R., Jha A., Zello G.A., Quail J.W., Oloo E.O., Nienaber K.H., Kowalczyk E.S., Allen T.M., Santos C.L., Clercq E.D., et al. Cytotoxic N-[4-(3-aryl-3-oxo-1-propenyl) phenylcarbonyl]-3, 5-bis (phenylmethylene)-4-piperidones and related compounds. Eur. J. Med. Chem. 2002;37:961–972. doi: 10.1016/S0223-5234(02)01414-9. [DOI] [PubMed] [Google Scholar]
- 88.Thomas S.L., Zhong D., Zhou W., Malik S., Liotta D., Snyder J.P., Hamel E., Giannakakou P. EF24, a novel curcumin analog, disrupts the microtubule cytoskeleton and inhibits HIF-1. Cell Cycle. 2008;7:2409–2417. doi: 10.4161/cc.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ohori H., Yamakoshi H., Tomizawa M., Shibuva M., Kakudo Y., Takahashi A., Takahashi S., Kato S., Suzuki T., Ishioka C., et al. Synthesis and biological analysis of new curcumin analogues bearing an enhanced potential for the medicinal treatment of cancer. Mol. Cancer Ther. 2006;5:2563–2570. doi: 10.1158/1535-7163.MCT-06-0174. [DOI] [PubMed] [Google Scholar]
- 90.Chandru H., Sharada A.C., Bettadaiah B.K., Kumar C.S.A., Rangappa K.S., Jayashree K. In vivo growth inhibitory and anti-angiogenic effects of synthetic novel dienone cyclopropoxy curcumin analogs on mouse Ehrlich ascites tumor. Bioorg. Med. Chem. 2007;15:7696–7703. doi: 10.1016/j.bmc.2007.08.051. [DOI] [PubMed] [Google Scholar]
- 91.Robinson T.P., Ehlers T., Hubbard R.B., IV, Bai X., Arbiser J.L., Goldsmith D.J., Bowen J.P. Design, synthesis and biological evaluation of angiogenesis inhibitors: Aromatic enones and dienone analogues of curcumin. Bioorg. Med. Chem. 2003;13:115–117. doi: 10.1016/S0960-894X(02)00832-6. [DOI] [PubMed] [Google Scholar]
- 92.Robinson T.P., Hubbard R.B., IV, Ehlers T., Arbiser J.L., Goldsmith D.J., Bowen J.P. Synthesis and biological evaluation of aromatic enones related to curcumin. Bioorg. Med. Chem. 2005;13:4007–4013. doi: 10.1016/j.bmc.2005.03.054. [DOI] [PubMed] [Google Scholar]
- 93.Woo H.B., Shin W.S., Leed S., Ahna C.M. Synthesis of novel curcumin mimics with asymmetrical units and their anti-angiogenic activity. Bioorg. Med. Chem. Lett. 2005;15:3782–3786. doi: 10.1016/j.bmcl.2005.05.064. [DOI] [PubMed] [Google Scholar]
- 94.Jha A., Mukherjee C., Prasad A.K., Parmar V.S., de Clercq E., Balzarini J., Stables J.P., Manavathu E.K., Shrivastav A., Sharma R.K., et al. E,E,E-1-(4-Arylamino-4-oxo-2-butenoyl)-3,5-bis-(arylidene)-4-piperidones: A topographical study of some novel potent cytotoxins. Bioorg. Med. Chem. 2007;15:5854–1565. doi: 10.1016/j.bmc.2007.05.065. [DOI] [PubMed] [Google Scholar]
- 95.Melphalan, a Nitrogen Mustard Alkylating Agent. [(accessed on 26 October 2014)]. Available online: http://en.wikipedia.org/wiki/Melphalan.
- 96.Fuchs J.R., Pandit B., Bhasin D., Etter J.P., Regan N., Abdelhamid D., Li C., Lin J., Li P.-K. Structure-activity relationship studies of curcumin analogues. Bioorg. Med. Chem. Lett. 2009;19:2065–2069. doi: 10.1016/j.bmcl.2009.01.104. [DOI] [PubMed] [Google Scholar]
- 97.Suarez J.A.Q., Rando D.G., Santos R.P., Gonçalves C.P., Ferreira E., de Carvalho J.E., Kohn L., Maria D.A., Faião-Flores F., Michalik D., et al. New antitumoral agents I: In vitro anticancer activity and in vivo acute toxicity of synthetic 1,5-bis(4-hydroxy-3-methoxyphenyl)-1,4- pentadien-3-one and derivatives. Bioorg. Med. Chem. 2010;18:6275–6281. doi: 10.1016/j.bmc.2010.07.026. [DOI] [PubMed] [Google Scholar]
- 98.Yamakoshi H., Ohori H., Kudo C., Sato A., Kanoh N., Ishioka C., Shibata H., Iwabuchi Y. Structure-activity relationship of C5-curcuminoids and synthesis of their molecular probes thereof. Bioorg. Med. Chem. 2010;18:1083–1092. doi: 10.1016/j.bmc.2009.12.045. [DOI] [PubMed] [Google Scholar]
- 99.Xiao J., Wang Y., Peng J., Guo L., Hu J., Cao M., Zhang X., Zhang H., Wang Z., Li X., et al. A synthetic compound, 1,5-bis(2-methoxyphenyl)-penta-1,4-dien-3-one (B63), induces apoptosis and activates endoplasmic reticulum stress in non-small cell lung cancer cells. Int. J. Cancer. 2012;131:1455–1465. doi: 10.1002/ijc.27406. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y., Xiao J., Zhou H., Yang S., Wu X., Jiang C., Zhao Y., Liang D., Li X., Liang G. A novel mono-carbonyl analogue of curcumin,(1E,4E)-1,5-bis(2, 3-dimethoxyphenyl) penta-1,4- dien-3-one, induced cancer cell H460 apoptosis via activation of endoplasmic reticulum stress signaling pathway. J. Med. Chem. 2011;54:3768–3778. doi: 10.1021/jm200017g. [DOI] [PubMed] [Google Scholar]
- 101.Hutzen B., Friedman L., Sobo M., Lin L., Cen L., De A.S., Yamakoshi H., Shibata H., Iwabuchi Y., Lin J. Curcumin analogue GO-Y030 inhibits STAT3 activity and cell growth in breast and pancreatic carcinomas. Int. J. Oncol. 2009;35:867–872. doi: 10.3892/ijo_00000401. [DOI] [PubMed] [Google Scholar]
- 102.Lin L., Hutzen B., Ball S., Foust E., Sobo M., Deangelis S., Pandit B., Friedman L., Li C., Li P.K., et al. New curcumin analogues exhibit enhanced growth-suppressive activity and inhibit AKT and signal transducer and activator of transcription 3 phosphorylation in breast and prostate cancer cells. Cancer Sci. 2009;100:1719–1727. doi: 10.1111/j.1349-7006.2009.01220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thakur A., Manohar S., V’elez Gerena C.E., Zayas B., Kumar V., Malhotra S.V., Rawat D.S. Novel 3,5-bis(arylidene)-4-piperidone based monocarbonyl analogs of curcumin: Anticancer activity evaluation and mode of action study. Med. Chem. Commun. 2014;5:576. doi: 10.1039/c3md00399j. [DOI] [Google Scholar]
- 104.Sun L., Liu J., Lin S.S., Shi W.T., Zhu J., Liang G., Yuan S.T. Potent anti-angiogenic activity of B19-α monocarbonyl analogue of curcumin. Chin. J. Nat Med. 2014;12:8–14. doi: 10.1016/S1875-5364(14)60002-9. [DOI] [PubMed] [Google Scholar]
- 105.Liu Z., Sun Y., Ren L., Huang Y., Cai Y., Weng Q., Shen X., Li X., Liang G., Wang Y. Evaluation of a curcumin analog as an anti-cancer agent inducing ER stress-mediated apoptosis in non-small cell lung cancer cells. BMC Cancer. 2013;13:494–506. doi: 10.1186/1471-2407-13-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wei X., Du Z.-Y., Zheng X., Cui X.-X., Conney A.H., Zhang K. Synthesis and evaluation of curcumin-related compounds for anticancer activity. Eur. J. Med. Chem. 2012;53:235–245. doi: 10.1016/j.ejmech.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 107.Wei X., Zhou D., Wang H., Ding N., Cui X.-X., Wang H., Verano M., Zhang K., Conney A.H., Zheng X., et al. Effects of Pyridine Analogs of Curcumin on Growth, Apoptosis and NF-κB Activity in Prostate Cancer PC-3 Cells. Anticancer Res. 2013;33:1343–1350. [PubMed] [Google Scholar]
- 108.Zhou D.-Y., Zhang K., Conney A.H., Ding N., Cui X.-X., Wang H., Verano M., Zhao S.-Q., Fan Y.-X., Zheng X., et al. Synthesis and Evaluation of Curcumin-Related Compounds Containing Benzyl Piperidone for Their Effects on Human Cancer Cells. Chem. Pharm. Bull. 2013;61:1149–1155. doi: 10.1248/cpb.c13-00507. [DOI] [PubMed] [Google Scholar]
- 109.Samaan N., Zhong Q., Fernandez J., Chen G., Hussain A.; M., Zheng S., Wang G., Chen Q.-H., Pohl J., Alizadeh A.R., et al. Design, synthesis, and evaluation of novel heteroaromatic analogs of curcumin as anti-cancer agents. Eur. J. Med. Chem. 2014;75:123–131. doi: 10.1016/j.ejmech.2014.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shoji M., Sun A., Kisiel W., Lu Y.J., Shim H., McCarey B.E., Nichols N., Parker E.T., Pohl J., Alizadeh A.R., et al. Targeting Tissue Factor-Expressing Tumor Angiogenesis and Tumors with EF24 Conjugated to Factor VIIa. J. Drug Target. 2008;16:185–197. doi: 10.1080/10611860801890093. [DOI] [PubMed] [Google Scholar]
- 111.Liu H., Liang Y., Wang L., Tian L., Song R., Han T., Pan S., Liu L. In Vivo and In Vitro Suppression of Hepatocellular Carcinoma by EF24, a Curcumin Analog. PLoS One. 2012;7:e48075. doi: 10.1371/journal.pone.0048075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yang C.H., Yue J., Sims M., Pfeffer L.M. The Curcumin Analog EF24 Targets NF-κB and miRNA-21, and Has Potent Anticancer Activity In Vitro and In Vivo. PLoS One. 2013;8:e71130. doi: 10.1371/journal.pone.0071130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang D., Wang Y., Dong L., Huang Y., Yuana J., Ben W., Yang Y., Ning N., Lu M., Guan Y. Therapeutic role of EF24 targeting glucose transporter 1-mediated metabolism and metastasis in ovarian cancer cells. Cancer Sci. 2013;104:1690–1696. doi: 10.1111/cas.12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rath K.S., McCann G.A., Cohn D.E., Rivera B.K., Kuppusamy P., Selvendiran K. Safe and targeted anticancer therapy for ovarian cancer using a novel class of curcumin analogs. J. Ovarian Res. 2013;6:35. doi: 10.1186/1757-2215-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Subramaniam D., May R., Sureban S.M., Lee K.B., George R., Kuppusamy P., Ramanujam R.P., Hideg K., Dieckgraefe B.K., Houchen C.W., et al. Diphenyl difluoroketone: A curcumin derivative with potent in vivo anticancer activity. Cancer Res. 2008;68:1962–1969. doi: 10.1158/0008-5472.CAN-07-6011. [DOI] [PubMed] [Google Scholar]
- 116.The HCT-116 human colon tumor xenograft study [111] reported the animals to be treated i.p. with 200 microg/kg (μg/kg) body weight daily for 23 days. This dose is to be compared with that used in almost all other studies, namely 10–200 mg/kg. The robust effects of this diminutive dose remains a paradox.
- 117.Yamaguchi M., Moore T.W., Sun A., Snyder J.P., Shoji M. Novel curcumin analogue UBS 109 potently stimulates osteoblastogenesis and suppresses osteoclastogenesis: Involvement in Smad activation and NF-κB inhibition. Integr. Biol. (Camb.) 2012;4:905–913. doi: 10.1039/c2ib20045g. [DOI] [PubMed] [Google Scholar]
- 118.Yamaguchi M., Zhu S., Zhang S., Wu D., Moore T.M., Snyder J.P., Shoji M. Curcumin analogue UBS109 prevents bone loss in breast cancer bone metastasis mouse model: Involvement in osteoblastogenesis and osteoclastogenesis. Cell Tissue Res. 2014;357:245–252. doi: 10.1007/s00441-014-1846-4. [DOI] [PubMed] [Google Scholar]
- 119.Nagaraju G.P., Zhu S., Ko J.E., Ashritha N., Kandimalla R., Snyder J.P., Shoji M., El-Rayes B.F. Antiangiogenic effects of a novel synthetic curcumin analogs in pancreatic cancer. Cancer Lett. 2014 doi: 10.1016/j.canlet.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 120.Yadav B., Taurin S., Larsen L., Rosengren R.J. RL66 a second-generation curcumin analog has potent in vivo and in vitro anticancer activity in ER-negative breast cancer models. Int. J. Oncol. 2012;41:1723–1732. doi: 10.3892/ijo.2012.1625. [DOI] [PubMed] [Google Scholar]
- 121.Antibiotic/Antimicrobial Resistance. [(accessed on 20 September 2014)]; Available online: http://www.cdc.gov/drugresistance/
- 122.White House orders plan for antibiotic resistance. [(accessed on 21 September 2014)]; Available online: http://apnews.excite.com/article/20140918/us--obama-antibiotics-828e636215.html.
- 123.Mahady G.B., Pendland S.L., Yun G., Lu Z.Z. Turmeric (curcuma longa) and curcumin inhibit the growth of Helicobacter pylori, a group 1 carcinogen. Anticancer Res. 2002;22:4179–4181. [PubMed] [Google Scholar]
- 124.Rudrappa T., Bais H.P. Curcumin, a known phenolic from curcuma longa, attenuates the virulence of Pseudomonas aeruginosa PAO1 in whole plant and animal pathogenicity models. J. Agric. Food Chem. 2008;56:1955–1962. doi: 10.1021/jf072591j. [DOI] [PubMed] [Google Scholar]
- 125.Parvathy K.S., Negi P.S., Srinivas P. Antioxidant, antimutagenic and antibacterial activities of curcumin-β-diglucoside. Food Chem. 2009;115:265–271. doi: 10.1016/j.foodchem.2008.12.036. [DOI] [Google Scholar]
- 126.Rai D., Singh J.K., Roy N., Panda D. Curcumin inhibits FtsZ assembly: An attractive mechanism for its antibacterial activity. Biochem. J. 2008;410:147–155. doi: 10.1042/BJ20070891. [DOI] [PubMed] [Google Scholar]
- 127.FtsZ. [(accessed on 4 December 2014)]. Available online: http://en.wikipedia.org/wiki/FtsZ.
- 128.Kaur S., Modi N.H., Panda D., Roy N. Probing the binding site of curcumin in Escherichia coli and Bacillus subtilis FtsZ—A structural insight to unveil antibacterial activity of curcumin. Eur. J. Med. Chem. 2010;45:4209–4214. doi: 10.1016/j.ejmech.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 129.Beuria T.K., Singh P., Surolia A., Panda D. Promoting assembly and bundling of FtsZ as a strategy to inhibit bacterial cell division; a new approach for developing novel antibacterial drugs. Biochem. J. 2009;423:61–69. doi: 10.1042/BJ20090817. [DOI] [PubMed] [Google Scholar]
- 130.Plaza A., Keffer J.L., Bifulco G., Lloyd J.R., Bewley C.A. Chrysophaentins A–H, antibacterial bisdiarylbutene macrocycles that inhibit the bacterial cell division protein FtsZ. J. Am. Chem. Soc. 2010;132:9069–9077. doi: 10.1021/ja102100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chatterji B.P., Jindal B., Srivastava S., Panda D. Microtubules as antifungal and antiparasitic drug targets. Expert Opin. Ther. Pat. 2011;21:167–186. doi: 10.1517/13543776.2011.545349. [DOI] [PubMed] [Google Scholar]
- 132.Liang G., Yang S., Jiang L., Zhao Y., Shao L., Xiao J., Ye F., Li Y., Li X. Synthesis and anti-bacterial properties of mono-carbonyl analogues of curcumin. Chem. Pharm. Bull. 2008;56:162–167. doi: 10.1248/cpb.56.162. [DOI] [PubMed] [Google Scholar]
- 133.Selvakumar B., Venkataraman R. Synthesis and Biological Evaluation of Some Curcumin Analogs and Their Derivatives. Rasāyan J. Chem. 2010;3:260–265. [Google Scholar]
- 134.Vilekar P., Ding C., Lagisetty P., Awasthi V., Awasthi S. Antibacterial Activity of Synthetic Curcumin Derivatives: 3,5-Bis(benzylidene)-4-Piperidone (EF24) and EF24-Dimer Linked via Diethylenetriaminepentacetic Acid (EF2DTPA) Appl. Biochem. Biotechnol. 2014;172:3363–3373. doi: 10.1007/s12010-014-0741-5. [DOI] [PubMed] [Google Scholar]
- 135.WHO; [(accessed on 26 September 2014)]. Tuberculosis. Available online: http://www.who.int/mediacentre/factsheets/fs104/en/ [Google Scholar]
- 136.Centers for Disease Control and Prevention; [(accessed on 25 September 2014)]. Treatment. Available online: http://www.cdc.gov/tb/topic/treatment/default.htm. [Google Scholar]
- 137.WHO; [(accessed on 27 September 2014)]. How many TB Cases have been Successfully Treated? Available online: http://www.who.int/gho/tb/epidemic/treatment/en/ [Google Scholar]
- 138.WHO; [(accessed on 27 September 2014)]. Drug-resistant Tuberculosis Now at Record Levels. Available online: http://www.who.int/mediacentre/news/releases/2010/drug_resistant_tb_20100318/en/ [Google Scholar]
- 139.Schraufstätter E., Bernt H. Antibacterial Action of Curcumin and Related Compounds. Nature. 1949;164:456–457. doi: 10.1038/164456a0. [DOI] [PubMed] [Google Scholar]
- 140.Pasca M.R., Guglierame P., Arcesi F., Bellinzoni M., Rossi E.D., Riccardi G. Rv2686c-Rv2687c-Rv2688c, an ABC Fluoroquinolone Efflux Pump in Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2004;48:3175–3178. doi: 10.1128/AAC.48.8.3175-3178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Silva P.E.A., Bigi F., Santangelo M.D.L.P., Romano M.I., Martin C., Cataldi A., Ainsa J.A. Characterization of P55, a Multidrug Efflux Pump in Mycobacterium Bovis and Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2001;45:800–804. doi: 10.1128/AAC.45.3.800-804.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Balganesh M., Dinesh N., Sharma S., Kuruppath S., Nair A.V., Sharma U. Efflux Pumps of Mycobacterium Tuberculosis Play a Significant Role in Antituberculosis Activity of Potential Drug Candidates. Antimicrob. Agents Chemother. 2012;56:2643–2651. doi: 10.1128/AAC.06003-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dube D., Kukshal V., Srivastava S.K., Tripathi R.P., Ramachandran R. NAD+-dependent DNA Ligase (Rv3014c) From M. tuberculosis: Strategies for Inhibitor Design. Med. Chem. Res. 2008;17:189–198. doi: 10.1007/s00044-007-9052-5. [DOI] [Google Scholar]
- 144.Takeuchi T., Ishidoh T., Iijima H., Kuriyama I., Shimazaki N., Koiwai O., Kuramochi K., Kobayashi S., Sugawara F., Sakaguchi K., et al. Structural Relationship of Curcumin Derivatives Binding to the BRCT Domain of Human DNA Polymerase Lambda. Genes Cells. 2006;11:223–235. doi: 10.1111/j.1365-2443.2006.00937.x. [DOI] [PubMed] [Google Scholar]
- 145.Agrawal D.K., Saikia D., Tiwari R., Ojha S., Shanker K., Kumar J.K., Gupta A.K., Tandon S., Negi A.S., Khanuja S.P.S. Demethoxycurcumin and its semisynthetic analogues as antitubercular Agents. Planta Med. 2008;74:1828–1831. doi: 10.1055/s-0028-1088335. [DOI] [PubMed] [Google Scholar]
- 146.Marathe S.A., Dasgupta I., Gnanadhas D.P., Chakravortty D. Multifaceted Roles of Curcumin: Two Sides of a Coin! Expert Opin. Biol. Ther. 2011;11:1485–1499. doi: 10.1517/14712598.2011.623124. [DOI] [PubMed] [Google Scholar]
- 147.Dimmock J.R., Kandepu N.M., Das U., Zello G.A., Nienaber K.H. Antimycobacterial arylidenecyclohexanones and related Mannich bases. Pharmazie. 2004;59:502–505. doi: 10.1002/chin.200447216. [DOI] [PubMed] [Google Scholar]
- 148.Das S., Das U., Bandy B., Gorecki D.K.J., Dimmock J.R. 2-[4-(4-Methoxyphenylcarbonyloxy)benzylidene]-6-dimethylaminomethyl cyclohexanone hydrochloride: A Mannich base which inhibits the growth of some drug-resistant strains of Mycobacterium tuberculosis. Pharmazie. 2010;65:849–850. [PMC free article] [PubMed] [Google Scholar]
- 149.Das U., Das S., Bandy B., Sables J.P., Dimmock J.R. N-Aroyl-3,5-bis(benzylidene)-4-piperidones: A novel class of antimycobacterial agents. Bioorg. Med. Chem. 2008;16:3602–3607. doi: 10.1016/j.bmc.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Baldwin P., Reeves A.Z., Powell K.R., Napier R.J., Swimm A.I., Sun A., Giesler K., Bommarius B., Shinnick T.M., Snyder J.P., et al. Monocarbonyl analogs of curcumin inhibit growth of antibiotic sensitive and resistant strains of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2015 doi: 10.1016/j.ejmech.2015.01.020. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kim J.-J., Lee H.-M., Shin D.-M., Kim W., Yuk J.-M., Jin H.S., Lee S.-H., Cha G.-H., Kim J.-M., Lee Z.-W., et al. Host Cell Autophagy Activated by Antibiotics Is Required for Their Effective Antimycobacterial Drug Action. Cell Host Microbe. 2012;11:457–468. doi: 10.1016/j.chom.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 152.Zhou T., Bai Y., Sun A., Liotta D., Snyder J.P., Fu H., Huang B. Autophagy and apoptosis in hepatocellular carcinoma induced by EF25-(GSH)2: A novel curcumin analog. PLoS One. 2014;9:e107876. doi: 10.1371/journal.pone.0107876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.WHO; [(accessed on 27 September 2014)]. Dementia. Available online: http://www.who.int/mediacentre/factsheets/fs362/en/ [Google Scholar]
- 154.Anand P., Thomas S.G., Kunnumakkara A.B., Sundaram C., Harikumar K.B., Sung B., Tharakan S.T., Misra K., Priyadarsini I.K., Rajasekharan K.N., et al. Biological Activities of Curcumin and Its Analogues (Congeners) Made by Man and Mother Nature. Biochem. Pharmacol. 2008;76:1590–1611. doi: 10.1016/j.bcp.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 155.Yang F. Curcumin Inhibits Formation of Amyloid Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid in Vivo. J. Biol. Chem. 2004;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 156.A Pilot Study of Curcumin and Ginkgo for Treating Alzheimer’s Disease. [(accessed on 26 September 2012)]; Available online: http://clinicaltrials.gov/ct2/show/NCT00164749?term=curcumin+and+alzheimers+disease&rank=1.
- 157.Ringman J.M., Frautschy S.A., Cole G.M., Masterman D.L., Cummings J.L. A potential role of the curry spice curcumin in Alzheimer’s disease. Curr. Alzheimer Res. 2005;2:131–136. doi: 10.2174/1567205053585882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Garcia-Alloza M., Borrelli L.A., Rozkalne A., Hyman B.T., Bacskai B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007;102:1095–1104. doi: 10.1111/j.1471-4159.2007.04613.x. [DOI] [PubMed] [Google Scholar]
- 159.Landau M., Sawaya M.R., Faull K.F., Laganowsky A., Jiang L., Sievers S.A., Liu J., Barrio J.R., Eisenberg D. Towards a pharmacophore for Amyloid. PLoS Biol. 2011;9:e1001080. doi: 10.1371/journal.pbio.1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yanagisawa D., Shirai N., Amatsubo T., Taguchi H., Hirao K., Urushitani M., Morikawa S., Inubushi T., Kato M., Kato F., et al. Relationship between the tautomeric structures of curcumin derivatives and their Aβ-binding activities in the context of therapies for Alzheimer’s disease. Biomaterials. 2010;32:4179–4185. doi: 10.1016/j.biomaterials.2010.01.142. [DOI] [PubMed] [Google Scholar]
- 161.Payton F., Peter Sandusky P., Alworth W.L. NMR Study of the solution structure of curcumin. J. Nat. Prod. 2007;70:143–146. doi: 10.1021/np060263s. [DOI] [PubMed] [Google Scholar]
- 162.Orlando R.A., Gonzales A.M., Royer R.E., Deck L.M., Vander Jagt D.L. A chemical analog of curcumin as an improved inhibitor of amyloid Abeta oligomerization. PLoS One. 2012;7:e31869. doi: 10.1371/journal.pone.0031869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Markesbery W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997;23:134–147. doi: 10.1016/S0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 164.Ao G., Chu X., Ji Y., Wang J. Antioxidant properties and PC12 cell protective effects of a novel curcumin analogue (2E,6E)-2,6-bis(3,5-Dimethoxybenzylidene)cyclohexanone (MCH) Int. J. Mol. Sci. 2014;15:3970–3988. doi: 10.3390/ijms15033970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Reddy R.C., Vatsala P.G., Keshamouni V.G., Padmanaban G., Rangarajan P.N. Curcumin for malaria therapy. Biochem. Biophys. Res. Commun. 2005;326:472–474. doi: 10.1016/j.bbrc.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 166.Mishra S., Karmodiya K., Surolia N., Surolia A. Synthesis and exploration of novel curcumin analogues as anti-malarial agents. Bioorg. Med. Chem. 2008;16:2894–2902. doi: 10.1016/j.bmc.2007.12.054. [DOI] [PubMed] [Google Scholar]
- 167.Manohar S., Khan S.I., Kandi S.K., Raj K., Sun G., Yang X., Molina A.D.C., Ni N., Wang B., Rawat D.S. Synthesis, antimalarial activity and cytotoxic potential of new monocarbonyl analogues of curcumin. Bioorg. Med. Chem. Lett. 2013;23:112–116. doi: 10.1016/j.bmcl.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 168.Reference 27 (Bairwa et al. 2014) overviews a sampling of recent SAR studies.