
Figure 1.
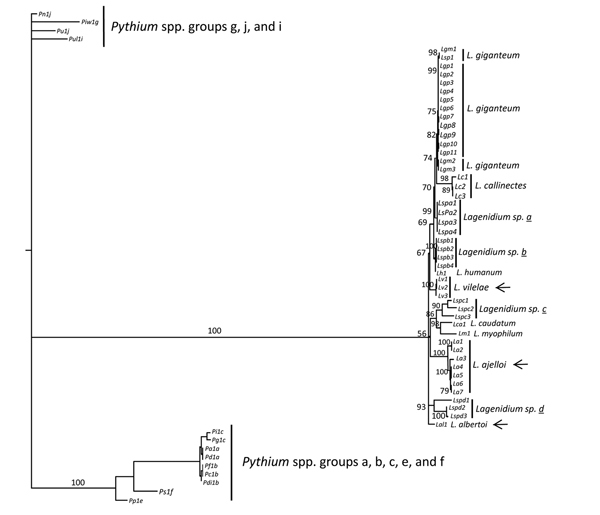
Bayesian phylogenetic analysis of concatenated 4 partial coding gene sequences (cell division cycle 42, cytochrome oxidase II, heat shock protein 90, and tubulin) and the complete internal transcribed spacers 1, 2, and 5.8S of Lagenidium DNA sequences. Thirteen Pythium species DNA sequences were included as the outgroup (groups a–c, e–g, j, and I [16 ]; Table). Support on key branches is the Bayesian probability for that branch followed by the percentage of 1,000 bootstrap resampled datasets containing the branch in neighbor-joining analyses of maximum-likelihood distances followed by the percentage of 1,000 bootstrap resampled datasets containing the branch in parsimony analyses using heuristic searches. In this analysis, the DNA sequences of L. giganteum mosquito control (Lg 1–3) and a Lagenidium sp. recovered from a nematode in Taiwan (Ls1, Lsp1 = HQ395647) clustered with L. giganteum from mammals (Lg 1–10). The pathogen of crab L. callinectes (Lc) was the sister group to the cluster. Three Lagenidium mammalian pathogenic novel species (L. ajelloi = La, L. albertoi = Lal, and L. vilelae = Lv) were placed in 3 distinctive strongly supported clades (arrows). The accession numbers, the abbreviations used to identify each species, and the Lagenidium and Pythium spp. DNA sequences are shown in the Table. ATCC, American Type Culture Collection; CBS, Centraalbureau voor Schimmelcultures.