

ABSTRACT
Microsporidia are a group of obligate intracellular parasites that are best known for their unique infection mechanism and their unparalleled levels of genomic reduction and compaction. We sequenced the genome of Ordospora colligata, a gut parasite of the microcrustacean Daphnia sp. and the closest known relative to the microsporidia characterized by the most extreme genomic reduction, the model genus Encephalitozoon. We found that the O. colligata genome is as compact as those of Encephalitozoon spp., featuring few introns and a similar complement of about 2,000 genes, altogether showing that the extreme reduction took place before the origin of Encephalitozoon spp. and their adaptation to vertebrate hosts. We also found that the O. colligata genome has acquired by horizontal transfer from its animal host a septin that is structurally analogous to septin 7, a protein that plays a major role in the endocytosis-based invasion mechanism of the fungal pathogen Candida albicans. Microsporidian invasion is most often characterized by injection through a projectile tube, but microsporidia are also known to invade cells by inducing endocytosis. Given the function of septins in other systems, we hypothesize that the acquired septin could help O. colligata induce its uptake by mimicking host receptors.
Importance The smallest known eukaryotic genomes are found in members of the Encephalitozoon genus of microsporidian parasites. Their extreme compaction, however, is not characteristic of the group, whose genomes can differ by an order of magnitude. The processes and evolutionary forces that led the Encephalitozoon genomes to shed so much of their ancestral baggage are unclear. We sequenced the genome of Ordospora colligata, a parasite of the water flea Daphnia sp. and the closest known relative of Encephalitozoon species, and show that this extreme reduction predated the split between the two lineages. We also found that O. colligata has acquired a septin gene by host-to-parasite horizontal transfer and predicted that the encoded protein folds like a septin 7, which plays a major role in endocytosis. We hypothesize that this acquisition could help O. colligata parasitize its hosts by facilitating endocytic infection, a mechanism that occurs in microsporidia but that is not yet well understood.
Importance
The smallest known eukaryotic genomes are found in members of the Encephalitozoon genus of microsporidian parasites. Their extreme compaction, however, is not characteristic of the group, whose genomes can differ by an order of magnitude. The processes and evolutionary forces that led the Encephalitozoon genomes to shed so much of their ancestral baggage are unclear. We sequenced the genome of Ordospora colligata, a parasite of the water flea Daphnia sp. and the closest known relative of Encephalitozoon species, and show that this extreme reduction predated the split between the two lineages. We also found that O. colligata has acquired a septin gene by host-to-parasite horizontal transfer and predicted that the encoded protein folds like a septin 7, which plays a major role in endocytosis. We hypothesize that this acquisition could help O. colligata parasitize its hosts by facilitating endocytic infection, a mechanism that occurs in microsporidia but that is not yet well understood.
INTRODUCTION
Microsporidia are obligate intracellular parasites that are related to fungi and characterized by a distinctive infection apparatus. Infection is mediated by resistant spores that contain a long and coiled filament that is attached to the spore apex, where the cell wall is the thinnest. When triggered, the filament rapidly bursts from the spore and everts to become a tube, following a rapid increase in internal osmotic pressure caused by aquaporin-mediated swelling (1). Microsporidia can enter a host cells using two different modes, both involving the polar tube (2, 3). In the first mode, the polar tube of an external spore ejects and acts like a molecular hypodermic needle and pierces the closest host cell in its trajectory. In the second mode, the microsporidian spore is taken up by host endocytosis but then quickly evades degradation by discharging its polar tube to escape the endocytic vacuole (3). The mechanism triggering endocytosis is unclear, and the potential use of specific receptors for that purpose has not been observed (3). In both cases, the polar tube permits the transfer of the infectious cytoplasm into that of the host cell (4).
Microsporidian genomes have also attracted some attention due to their small size, but, in fact, they range in size by more than an order of magnitude (from 2.3 to >24 Mbp) (5). The smallest known microsporidian genomes, which are the smallest in any eukaryote, are found within members of the Encephalitozoonidae, a lineage that primarily infects humans and other mammals, where they can cause various mild to severe systemic diseases. Encoding roughly 2,000 genes and ranging between 2.3 and 2.9 Mbp in size (6–9), Encephalitozoon genomes are extremely compact, with few introns, very short intergenic regions, and no transposable elements. Although these reduced genomes are the best studied of any microsporidian genomes, it is still unclear why they have been so drastically altered: reduction is sometimes linked to their obligate intracellular parasitic lifestyle (10, 11), but only vaguely, and this is not obviously consistent with the 10-fold variability in other microsporidia.
The closest known relative of members of the Encephalitozoonidae is Ordospora colligata, which infects the microcrustacean Daphnia magna (12). Infections are typically located in the anterior part of the host’s midgut, where they can reach very high intensities, with nearly every gut epithelium cell being infected (13). The consequences of infection are not as severe as those seen with some microsporidia, but the reproductive success of infected females is reduced by 20%, and they die earlier than uninfected controls (14). Infected hosts release transmission-stage parasites with their feces, and the free-floating spores are then ingested by other filter-feeding Daphnia spp. The parasite has been reported from D. magna populations in Europe and the Middle East (13, 15).
As the sister to the Encephalitozoon genome, the O. colligata genome might offer some important clues to the origin and evolution of this model for genomic reduction and compaction. Here we describe the complete sequence of the nuclear genome of O. colligata OC4 and compare it to those of its relatives from the lineage Encephalitozoonidae. We show that the structures and contents of the O. colligata Oc4 and Encephalitozoon genomes are remarkably similar, with only a few distinct chromosomal reorganizations and very limited differences in gene content, showing that the extreme reduction characterizing Encephalitozoon genomes significantly preceded the origin of the genus. The most surprising difference between the two is that the O. colligata genome has acquired a Daphnia-derived septin by horizontal gene transfer (HGT). This protein is structurally analogous to septin 7 and retains transmembrane domains, suggesting that it could be located at the spore surface. In the fungal pathogen Candida albicans, the invasion mechanism is mediated by proteins called invasins that interact with the host septin 7 to induce endocytosis of the parasite by the host endothelial microfilaments (16). The presence of a Daphnia-derived septin 7 in O. colligata could facilitate its attachment to epithelial cells of Daphnia spp. by binding directly to its N-cadherin-like surface receptors and increase the likelihood of infection either simply by maintaining proximity to the host or more directly by triggering the host’s endocytosis mechanism.
RESULTS
Genome structure of O. colligata.
The assembled O. colligata OC4 genome resulted in a total of 2,290,528 bp of unique sequence distributed in 15 contigs (627× average coverage). The 12 largest O. colligata single-copy contigs are structurally similarly to those of the 11 Encephalitozoon chromosomes, although there is evidence of some large transpositions such as the equivalents of chromosomes V and IX split and attached to other loci (Fig. 1). In contrast, the remaining three small contigs (5,456 to 16,597 bp) are repeated up to four times in the genome based on their respective coverage data and correspond to subtelomeric regions found in the Encephalitozoon species that could not be linked unambiguously by PCR.
FIG 1 .

Chromosomal reorganization between the O. colligata and Encephalitozoon cuniculi genomes. The O. colligata OC4 and E. cuniculi GBM1 contigs are identified by Arabic and uppercase Roman numerals, respectively, and color coded accordingly (outer ring). G+C percentages are plotted underneath the corresponding contig designations (middle ring). Syntenic clusters conserved between the contigs are connected by ribbons (inner ring); ribbons are colored according to the E. cuniculi contigs. All chromosomal rearrangements in O. colligata relative to E. cuniculi were validated by PCR; the four sequenced E. cuniculi GBM1, ECI, ECII, and ECIII strains share the same genome architecture (7). For simplicity, O. colligata small and repeated contigs 13, 14, and 15 are not shown. AT-rich loci not found in other microsporidia are indicated by asterisks. The figure was plotted with Circos (69).
One of the main structural differences between the O. colligata and Encephalitozoon genomes pertains to the telomeric regions. In the Encephalitozoon genomes, the rRNA operons are present in the subtelomeric regions of each chromosome, for a total of at least 22 copies (6–9). In contrast, the rRNA operons are present only four times in the O. colligata genome, indicating that its subtelomeric regions are structured differently from those in Encephalitozoon species. The sharp increase in GC richness in the Encephalitozoon subtelomeric regions compared with their chromosome cores (Fig. 1) was not observed in O. colligata; however, the cores themselves display the same arcing G+C% pattern as the Encephalitozoon species (Fig. 1) (7, 9), peaking in the central portions. At 38.2%, the overall G+C content of the O. colligata genome is lower than that of Encephalitozoon species and, perhaps not coincidentally, closest to that of the invertebrate pathogen E. romaleae (Table 1).
TABLE 1 .
General features of the O. colligata and Encephalitozoon genomes
| Feature | Value(s) |
||||
|---|---|---|---|---|---|
| O. colligata OC4 | E. cuniculi GB-M1a | E. intestinalis ATCC 50506a | E. hellem ATCC 50504a | E. romaleae SJ-2008a | |
| No. of chromosomesb | 10 | 11 | 11 | 11 | 11 |
| Estimated total genome size (Mbp) | 3.0 | 2.9 | 2.3 | 2.5 | 2.5 |
| Assembled-genome size (Mbp) | 2.3 | 2.5 | 2.2 | 2.3 | 2.2 |
| Genome coverage (%) | 77 | 86 | 96 | 92 | 88 |
| G+C content (%) | 38.2 | 47 | 41.4 | 43.4 | 40.3 |
| Gene density (gene/kbp) | 0.82 | 0.83 | 0.91 | 0.89 | 0.84 |
| Mean gene length (bp) | 1041 | 1041 | 999 | 1018 | 1061 |
| Mean intergenic length (bp)b | 176 | 166 | 100 | 106 | 130 |
| No. of SSU-LSU rRNA genesc | 4 | 22 | 22 | 22a | 22 |
| No. of 5S rRNA genes | 3 | 3 | 3 | 3 | 3 |
| No. of ncRNAsd | 10 | NA | 14 | 11 | NA |
| No. of tRNAs | 46 | 46 | 46 | 46 | 46 |
| No. of tRNA introns (sizes in bp) | 2 (11, 42) | 2 (12, 41) | 2 (12, 41) | 2 (12, 41) | 2 (12, 41) |
| No. of splic. introns (size range in bp) | 30 (23–77) | 36 (23–76) | 36 (23–76) | 36 (23–76) | 36 (23–76) |
| Predicted no. of ORFs | 1820 | 2010 | 1944 | 1928 | 1835 |
The E. cuniculi and E. romaleae values are from Pombert et al. (6). The E. intestinalis and E. hellem values are from the 2014 accession updates.
The numbers of chromosomes for O. colligata were estimated based on the chromosomal reorganizations observed with the Encephalitozoon species.
The numbers of small-subunit and large-subunit (SSU-LSU) rRNA genes were inferred based on their overall coverage relative to other genes.
NA, the ncRNAs in E. cuniculi and E. romaleae were not assessed. They are likely similar in number and content to those found in O. colligata, E. intestinalis, and E. hellem.
Evolution of gene content in O. colligata and Encephalitozoon spp.
To first confirm the relationship between O. colligata and Encephalitozoon spp. that has previously been inferred from small-subunit (SSU) rRNA (17), we reconstructed the phylogeny based on 104 proteins that are shared between all microsporidian species investigated. This analysis robustly confirmed the positioning of O. colligata at the base of the Encephalitozoon species (see Fig. S1 in the supplemental material) (17), a phylogenetic affiliation that is also supported by gene content and metabolic profiling (see below). O. colligata is not closely related to Hamiltosporidium tvaerminnensis (formerly called Octosporea bayeri), which also infects Daphnia spp. (18) and, in some parts of their species range, coinfects the same host individual as O. colligata (19). Daphnia spp. are susceptible to many microsporidian species, which are widespread across the phylogenetic tree (17, 20, 21).
Annotating all open reading frames using homology to known proteins and positional orthology with E. cuniculi resulted in a total of 1,801 discrete protein-coding genes found in the O. colligata assembly. Overall, this makes the O. colligata genome only slightly less compact than those of its Encephalitozoon relatives, with very similar coding density (0.82 genes/kb versus 0.83 genes/kb in E. cuniculi), gene content, and lack of repeats and similar intron distribution (Table 1). Most of the genes that were identified are shared with Encephalitozoon species, even in those cases where Encephalitozoon spp. differ from other microsporidia. For example, O. colligata is incapable of endogenous RNA interference and lacks the Dicer and Argonaute proteins found in the 6-Mbp+ genomes of the microsporidian species Nosema ceranae, Spraguea lophii, and Trachipleistophora hominis (22–24). Unsurprisingly, the folate-related genes that were acquired by HGT in the Encephalitozoon lineage leading to E. hellem and E. romaleae (6) are also not found in O. colligata. However, the two ricin B-lectin domain-containing paralogs that are conserved across microsporidian species (22) and duplicated in tandem in the E. cuniculi strains (from ECU08_1700 to ECU08_1730) (7, 9) are also absent from the O. colligata genome. Instead, a single open reading frame (ORF) (M896_091670) that does not display any significant homology to genes encoding other proteins is located in the corresponding locus, suggesting that the ricin b-lectin paralogs were either amplified in Encephalitozoon spp. or perhaps lost or heavily modified and reduced in O. colligata.
Up to 95% of the O. colligata proteome is shared with the Encephalitozoon species (Fig. 2). In contrast, only 74% identity is found in Nosema species (a member of the sister group corresponding to the O. colligata/Encephalitozoon clade), with percentages decreasing rapidly as one moves further away in the tree, for an averaged pairwise proportion of 50% (highest, 100%; lowest, 16%). However, these low percentages do not necessarily mean that the genes are nonhomologous, since they may simply reflect the high rate of sequence divergence occurring in microsporidia. None of the O. colligata proteins that are absent from Encephalitozoon spp. have been found in other microsporidia, and, with the exception noted below, none have identifiable functions. These unique protein-coding genes are not restricted to subtelomeric regions; many are inserted within the cores of the O. colligata chromosome between genes arrayed in otherwise syntenic fashion with other Encephalitozoon spp. However, in 14 cases that included the M896_091670 gene inserted in lieu of the b-lectin genes, positional orthologs that are, however, dissimilar in sequences are found between the O. colligata and Encephalitozoon cores, suggesting that the corresponding genes may not be unique to O. colligata but may rather be divergent beyond recognition.
FIG 2 .

Pairwise distribution and metabolic profiling of the microsporidian proteome. The phylogenetic positions in the cladogram are derived from our phylogenetic inferences (see Fig. S1 in the supplemental material). Nodes recovered in all of the bootstrap replicates are indicated by asterisks, whereas values indicate levels of support for the corresponding nodes. The early-diverging microsporidian Mitosporidium and the Cryptomycota Rozella spp. were used as outgroups in this analysis. Assembled-genome sizes are indicated in Mbp between each branch and the corresponding microsporidian taxa. In the adjacent heat map, light and dark colors indicate the percentages of proteins from the branching species that are shared with the species indicated on top. Darker colors indicate higher levels of conservation; conservation is reduced as we move further across the phylogenetic tree. For each species, the total number of predicted proteins is indicated in the center of the figure. “uORF” (unknown ORF) refers to the number of proteins for which no gene ontology (GO) could be ascribed. pFUNC refers to predicted proteins with at least one associated GO in InterProScan 5 analyses. On the right, the metabolic profiles (in percentages) derived from the detected GO are categorized according to KEGG pathways and drawn to scale. Ami, amino acid metabolism; Car, carbohydrate metabolism; Cgd, cell growth and death; Cmc, cell motility cytoskeleton; Ene, energy metabolism; Fsd, folding, sorting, and degradation; Lip, lipid metabolism; Mem, membrane transport; Mcv, metabolism of cofactors and vitamins; Mtp, metabolism of terpenoids and polyketides; Mis, miscellaneous; Nuc, nucleotide metabolism; Rep, replication and repair; Sig, signal transduction; Tsc, transcription; Tsl, translation; Tsc, transport and catabolism. N. antheraeae, Nosema antheraeae; N. apis, Nosema apis; V. corneae, Vittaforma corneae; A. locustae, Antonospora locustae; A. algerae, Anncaliia algerae; E. aedis, Edhazardia aedis; V. culicis floridensis, Vavraia culicis floridensis; N. parisii, Nematocida parisii; N. sp. 1, Nematocida sp. 1; M. daphniae, Mitosporidium daphniae; R. allomysis, Rozella allomysis.
About 58% of the O. colligata proteome can be assigned putative functions, consistent with other microsporidia, for which 29% to 61% of the proteome has identifiable functions. The relative distributions of functions across metabolic pathways for identifiable proteins in microsporidia are similar across most species (Fig. 2; see also Table S1 in the supplemental material), with an elevated proportion of proteins involved in amino acid and carbohydrate metabolism in the genomes from basal Rozella and Mitosporidium species, which is congruent with their closer positioning to other nonpathogenic fungi. Surprisingly, Nosema bombycis also displays an elevated proportion of proteins involved in carbohydrate metabolism, perhaps resulting from its recent abundant genomic duplications (25). The human pathogen Enterocytozoon bieneusi displays a large proportion of genes for amino acid biosynthesis, signal transduction, and translation-related components, the latter of which represent the highest proportion in any sequenced microsporidian, 45% more than the second highest tally in the gene-rich genomes of Mitosporidium and Cryptomycota Rozella species (Fig. 1; see also Table S1).
To investigate the levels of divergence between the O. colligata and Encephalitozoon protein-coding genes, we aligned all shared orthologs and calculated the nucleotide diversity within Encephalitozoon species (Pi) and between O. colligata and Encephalitozoon spp. [K(JC)] (see Table S2 in the supplemental material). In both absolute [K(JC)] rates and relative [K(JC)/Pi] rates, polar tube protein 2 (PTP2; M896_060250) stands out as one of the fastest-evolving genes between the two genera. This contrasts sharply with polar tube protein 1 (PTP1; M896_060260) and polar tube protein 3 (PTP3; M896_121330), which are located on the adjacent upstream locus and on a different chromosome, respectively, and which display similar nucleotide diversities within and between the two genera. The nucleotide changes observed for PTP2 are not silent; PTP2 displays very little similarity with its Encephalitozoon orthologs at the amino acid level, with 25% pairwise identity at most over the aligned regions. Its N-terminal signal peptide has been preserved, but the 8-amino-acid-long lysine-rich domain located inside the core of the Encephalitozoon PTP2 proteins (26) is only barely recognizable in O. colligata (KPKKKKSK versus VPVKEKAR, respectively). Other highly variable genes between the two genera with identifiable functions include an endochitinase (M896_021680; orthologous to ECU09_1320) involved in spore wall maintenance, a phosphoacetylglucosamine mutase (M896_010540; orthologous to ECU01_0650) involved in carbohydrate metabolism, and a ubiquitin carboxyl-terminase hydrolase (M896_060900; orthologous to ECU06_0910) involved in ubiquitin conjugation. O. colligata contains two additional copies of another ubiquitin carboxyl-terminase hydrolase orthologous to ECU03_0580. One of these, M896_031130, is syntenic with its Encephalitozoon orthologs, but the other, M896_051260, is paralogous and likely arose from intragenomic duplication. The M896_031130/ECU03_0580 orthologs are not particularly divergent, but the paralogous M896_051260 is highly derived.
Horizontal gene transfer of host-derived septin gene to O. colligata.
In addition to subtelomeres, the O. colligata genome has three other regions of unusually low G+C content, but these are not common to Encephalitozoon spp. or other microsporidia (Fig. 1 and 3). In all three cases, the overall G+C percentage is about 10% lower than the content in the surrounding regions. Two of the three segments (on contigs 5 and 8) are inserted within blocks of genes that are otherwise syntenic with the Encephalitozoon genomes, whereas the third (on contig 2) is located at one of the junctions of a major intrachromosomal transposition involving chromosome 2 (Fig. 3). We confirmed the assembly of all three regions by PCR and Sanger sequencing, excluding potential assembly errors.
FIG 3 .
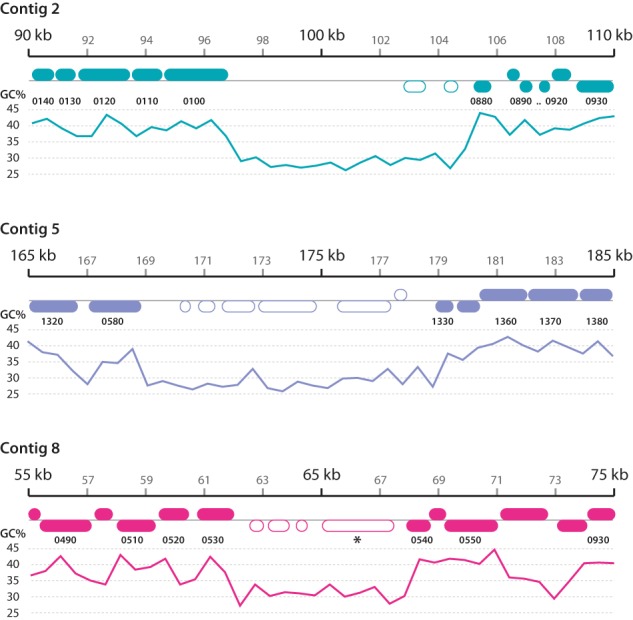
O. colligata genomic loci that are absent from other microsporidia. The regions shown under the 20-kbp scale bars are color coded according to Fig. 1 and are drawn to scale, with the corresponding G+C content plotted underneath. Numbers between the chromosomal loci and G+C plots indicate the corresponding loci in E. cuniculi (e.g., 0140, ECU02_0140; 0490, ECU08_0490). Proteins that are not found in other microsporidia are indicated by empty rounded rectangles. The M896_080490 protein homologous to the Daphnia pulex DAPPUDRAFT_204173 protein is indicated by an asterisk.
The presumed transposition breakpoint is relatively gene poor, but the other two regions are of comparable density to that of the genome as a whole and encode several potential open reading frames (Fig. 3). All but a few these ORFs are of unknown function. M896_020810 has low similarity to magnesium transporters, and M896_051300 and M896_051310 feature indistinct coiled coil motifs. In contrast, however, the O. colligata M896_080490 protein located within the low-GC region on chromosome 8 is homologous to genes encoded in the genomes of Daphnia pulex (E value of 4e-53 with DAPPUDRAFT_204173; 60% similarity over 256 aligned amino acid residues) and Daphnia magna (E value of 2e-50; 63% similarity over 287 aligned amino acid residues) (reference 27 and unpublished data). Phylogenetic inferences derived from the alignment of M896_080490 with similar sequences from the NCBI NR database (E value BLASTP cutoff value of 1E-40) cluster this protein firmly within clades of Daphnia pulex paralogs (Fig. 4). The D. pulex and D. magna genomes contain large AT-rich stretches, suggesting that the AT-rich segments may all have been acquired by O. colligata from its host, but there is no sequence similarity to support this conjecture in the other cases.
FIG 4 .
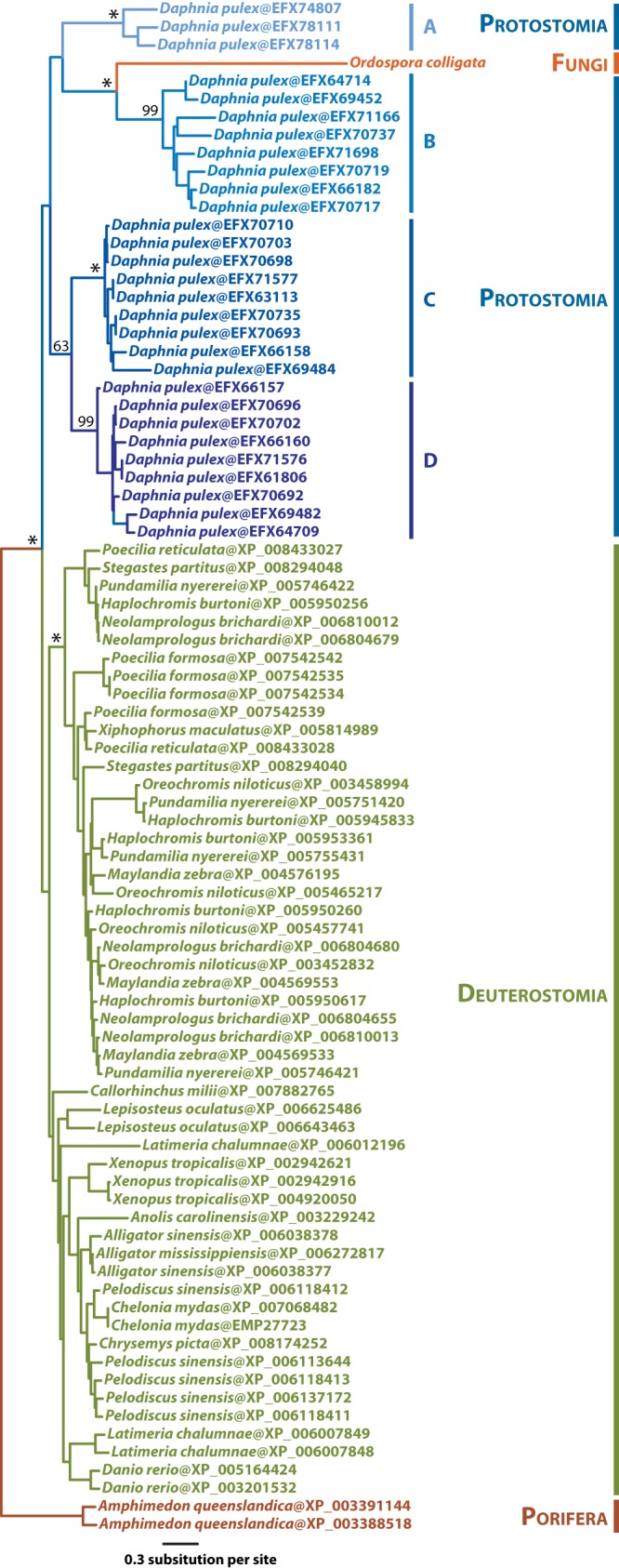
Maximum likelihood phylogenetic inferences of O. colligata’s septin 7 (M896_080490) and similar protein sequences. All protein sequences displaying similarity greater than the selected cutoff (E value cutoff 1E to 40; accession date, 20 August 2014) were retrieved from the NCBI NR database. The best ML tree (LG+Γ4) is shown here. The major nodes retrieved in all bootstrap replicates are indicated by asterisks, whereas numbers indicate the corresponding levels of bootstrap support (values lower than 60 are not shown). Daphnia paralogous clades are color coded with a bluish gradient and labeled A to D. Their specific functions (e.g., that of septin 2, 4, 6, or 7) are not clear. Branch lengths are drawn to scale.
The functional identity of M896_080490 is of particular interest. The Daphnia homologues from clades A to D (Fig. 4; see also Fig. S2) are annotated as GTP-binding cell division proteins (IPR000038), but in InterProScan searches, M896_080490 is correlated with scores for septins and GTPases encompassing 29 subfamilies across 46 eukaryotic species. M896_080490 is made up of three domains with detailed signature motifs: a GTP-binding cell division protein (IPR000038; residues 103 to 518; 6.6E-39), a P-loop-containing nucleoside triphosphate hydrolase (IPR027417; residues 177 to 397; 2.36E-18), and a general substrate transporter (IPR016196; residues 618 to 753; 3.40E-05). The first two overlapping motifs were independently confirmed by three-dimensional (3D) folding predictions (Fig. 5), whereas the third is compatible with the presence of transmembrane helices predicted by TMHMM, TopPred, and TMpred analyses. Hydrophobicity analyses further strongly suggest that the N-terminal portion of the protein, corresponding to the septin/GTPase motifs, is exposed outside the membrane (see Data S1 at https://github.com/JFP-Laboratory/mBio-2015/). Two domains, spanning residues 1 to 221 and residues 222 to 794, were predicted using the Ginzu hierarchical screening method. The first domain has weak (Ginzu confidence of 0.0735) and likely spurious structural homology with DNA cleavage/binding domains (2NRRA_101) and the hemolysin binding component of Bacillus cereus toxins (2NRJA), but the second has a much stronger match (Ginzu confidence of 0.607009) as a septin hydrolase/GTPase, similar to human septin 7 (Protein Data Bank [PDB] entry 2QAG.1.C). No signal peptide suggestive of secretion was predicted. Interestingly, two other septins (M896_011310 and M896_100270) were found to be among the fastest-evolving genes between the O. colligata and Encephalitozoon species, with K(JC)/Pi ratios of 2.70 and 2.66, respectively.
FIG 5 .

Three-dimensional model of M896_080490, a septin 7 analog acquired by O. colligata from its Daphnia host by horizontal transfer. (Upper left) The backbone of this 794-residue protein is colored green, alpha helices are colored blue, and beta sheets are colored red. (Lower left) Overlay between the O. colligata septin 7 (in blue) and human septin 7 (2QAG.1.C; in red) tridimensional models. (Upper right) SWISS-MODEL QMEAN4 scoring comparison of the contiguous 192 residues (20.75% sequence identity) of the model to its template 2QAG.1.C for a nonredundant set of proven PDB structures. The amino acid length refers to the length of the correlated sequence section, not the length of the protein. The normalized QMEAN4 score is −10.24. (Lower right) Heat map dot plot representation of the amino acid residues conserved between M896_080490 and 2QAG.1.C as implemented in FFFAS (61). The highest to lowest similarities between amino acids are shown from blue to red. Optimally aligned contiguous amino acids are connected by green lines. The contiguous 192 residues represented in the upper right panel are located between M896_080490 amino acid residues 400 and 600.
DISCUSSION
The Encephalitozoon genus has been held as a model for extreme genome reduction and compaction, and the patterns left by these processes over the evolution of the genus have been well studied (6, 8, 9, 28). Explanations for why these genomes are so compact are less clear, but the 10-fold variation in genome size in microsporidia suggests that it is not intrinsically related to intracellular parasitism. Here, we show that the extreme reduction in the Encephalitozoon genome actually predates the origin of the genus and is common to the Daphnia-infecting sister lineage represented by O. colligata. Indeed, in terms of gene density, content, and overall structure, the O. colligata genome is hardly distinguishable from those of Encephalitozoon spp. The impressive level of synteny observed between the two genera strongly argues against independent rounds of reductions, such that the genome of the last common ancestor of the O. colligata and Encephalitozoon species was almost assuredly similar in every aspect.
Of the differences in gene content we do describe, however, the septin 7 homologue stands out as being of particular functional and evolutionary interest. Obligate intracellular parasites have limited opportunities to exchange genetic material, given that their sheltered environment offers few opportunities to exchange genes with organisms other than the host. Turning a host’s proteome against itself is a strategy that has been used by both prokaryotic and eukaryotic pathogens throughout the course of evolution, although clear-cut cases of host-to-parasite HGT in eukaryotes are still rare in general (29, 30). Horizontal gene transfer has been shown in microsporidians, despite their genome-reductionist tendencies, and some of their newly acquired genes have obvious possible benefits with respect to infection or survival outside their host (31, 32). Only a single case of HGT from an animal host has been reported in microsporidia, however (6, 33), and here we show that a septin 7 in O. colligata not only is animal derived but is apparently closely related to homologues in D. pulex and D. magna genomes. The directionality of this transfer is clear; no homologues are found in other fungi, and the transfer is likely recent as well. The horizontal transfer seems to have occurred after the split between the O. colligata and Encephalitozoon spp. and to have resulted in the acquisition of additional transmembrane domains since its origin. The alternative, acquisition by their common ancestor and then loss in the members of the Encephalitozoonidae, seems unlikely given their host range histories.
The function of the O. colligata septin is unknown, but other fungal pathogens offer intriguing possibilities related to cell division and compartmentalization (34). Intuitively, cell division proteins form valuable acquisition targets for vertically transmitted parasites to modulate the reproductive cycle of hosts. Here, however, the function of the HGT-acquired septin is unlikely to be involved in cell division: unlike the microsporidian Daphnia pathogen Perezia diaphanosomae, which can infect both gut and reproductive organs (12), O. colligata is a strictly horizontally transmitted gut epithelium pathogen that has never been encountered in reproductive organs. Thus, a secreted septin would not disrupt host reproduction. There is also no evidence that this HGT-acquired septin is secreted and, considering the evolutionary distance between the parasite and its hosts, it is unlikely that this protein would play a role in O. colligata’s own cell division. One could, however, imagine a function in compartmentalization, for example, inducing endocytosis by the host. Encephalitozoon species can invade their host by endocytosis (2, 3), and in the fungal pathogen Candida albicans, the endocytic invasion is initiated by proteins called invasins that interact with the host septin 7, a major effector (35), which initiates a molecular cascade, ultimately inducing the uptake of the pathogen (16). The septin acquired by O. colligata is structurally analogous to septin 7 and features additional transmembrane motifs, suggesting that it could be localized at the proteinaceous exospore. An externally exposed septin could camouflage O. colligata and bypass the need for invasins by binding to cell surface N-cadherins of Daphnia spp., by recruiting other components of the host septin 2/6/7 complex, or by interacting directly with the microfilaments from the gut epithelium to facilitate entry into the host by endocytosis. Alternatively, a surface septin could also facilitate infection by simply helping to keep the parasite in close proximity to the host cell surface.
Unfortunately, little is known about the infection process in O. colligata (or in many other microsporidia for that matter), so more direct evidence is required to determine if O. colligata is capable of infection by self-induced endocytosis and, if so, if the HGT-acquired septin is surface localized and facilitates this process. There is no guarantee that the gene has remained functional since being acquired by HGT, and while the septin core appears to have been conserved in O. colligata (see Fig. S2 in the supplemental material), the long branch it displays in phylogenetic analyses (Fig. 4) suggests that it is evolving at a fast pace. In any case, because Encephalitozoon species are capable of infecting their host by endocytosis (3) and yet lack the septin 7 found in O. colligata, we infer that this gene is not essential to the underlying mechanism. However, considering the number of diverse microsporidian species that infect Daphnia spp., the presence of a functional septin 7 in O. colligata could confer to it a competitive advantage over its parasitic relatives. The distribution of this gene in related taxa is also of interest for efforts to help determine how recent the HGT was: it may have originated relatively early and been lost in Encephalitozoon spp. or may be present within only a limited number of genotypes.
MATERIALS AND METHODS
Tissue culture and DNA purification.
O. colligata isolate OC4 was cultivated in Daphnia magna clone ELK1-1 (England-LadyKirk-pond 1) (36) and used for further laboratory procedures. Approximately 1,000 female hosts infected with O. colligata OC4 were homogenized in 10 mM Tris-HCl (pH 7) and then filtered sequentially through a 40-µm-pore-size nylon mesh and an 8-µm-pore-size cellulose nitrate membrane adapted to a syringe. The filtrate was centrifuged at 4,000 rpm for 10 min and resuspended in 10 mM Tris-HCl (pH 7). The solution, containing spores and other tissue debris, was centrifuged in 60% Percoll (Sigma Aldrich) at 14,000 rpm for 5 min, and the pellet was washed 3 times with 10 mM Tris-HCl (pH 7) to obtain a clear spore solution. Spores were incubated with lysozyme (Sigma Aldrich) (2.5 mg/ml) at 37°C for 1 h to lysate contaminant bacteria. An additional step of contaminant cell lysis was performed by adding a lysis buffer (1% SDS; 2% Triton X-100; 1 mg/ml proteinase K; 10 mM Tris-HCl; 1 mM EDTA; 100 mM NaCl; pH 7.0) to the mixture and incubating it at 56°C for 1 h. The lysate was centrifuged at 14,000 rpm to recover the spores, which were treated with DNase I (Sigma Aldrich) at 37°C overnight to eliminate contaminating DNA. The enzyme was inactivated with EGTA (50 mM) at 95°C for 30 min, and the spores recovered by centrifugation were frozen and thawed several times. The O. colligata purified spores were used for DNA isolation using a DNeasy tissue-extracting kit (Qiagen). Before sequencing, genomic DNA was assessed for quality by Qubit fluorometric quantification (Life Technologies).
Sequencing.
The O. colligata purified DNA Illumina 100-bp paired-end (PE) libraries (212-bp inserts, 43-bp average standard deviation, 40,096,994 PE reads, 8,019,398,800 bp total) were prepared using TruSeq SBS V5 chemistry and sequenced by Fasteris (Geneva, Switzerland) on an Illumina HiSeq 2000 instrument. Reads were processed and the adapters removed with CASAVA pipeline version 1.8.1 (mean quality score, 33.90; 85.37% of all bases ≥ Q30). Read quality was further assessed with FASTQC (37).
Genome assembly.
Paired-end reads were filtered by calculation of quality scores with Sickle 1.210 (Bioinformatics Core, University of California, Davis [https://github.com/najoshi/sickle]) and the filtered reads iteratively assembled de novo with Ray 2.0-rc8 using odd k-mer values (from 19 to 31) on 32 Infiniband QDR-connected Intel Xeon nodes (256 Nehalem X5560 processing cores at 2.8 GHz). Microsporidian contigs were filtered from host and other low-level contaminants in the resulting datasets with BLAST homology searches (38) using Encephalitozoon genomes (BLASTN) and proteins (tBLASTn) as queries. Microsporidian contigs from each k-mer assembly were concatenated and merged with Consed (39) and then visually inspected for potential discrepancies between the various k-mer assemblies. Merged contigs were extended using our in silico chromosome walking approach (7) with the addSolexaReads.pl script from the Consed package. Ambiguous regions in the assemblies were amplified by PCR using flanking primers and then validated by Sanger sequencing. Final assemblies were verified by read mapping with Bowtie 1.0 (40) and visual inspection with Tablet 1.13.07.31 (41).
Genome annotation.
Transfer and ribosomal RNAs were positioned on the O. colligata contigs with tRNAscan-SE 1.3.1 (42) and RNAmmer 1.2 (43), respectively. Open reading frames were first positioned on the sequences with Artemis 16.0.0 (44) built-in tools. Start methionines were then refined using multiple-sequence alignments of orthologs with MAFFT 7.058 b (45) and the presence of CCC/GGG-like transcription signals as described by Peyretaillade et al. (46). Noncoding RNAs (ncRNAs) were positioned using a combination of BLASTN homology searches, syntenic information from Encephalitozoon genomes, and RFAM (47) searches as implemented in Artemis. Putative protein-coding gene functions were ascribed by homology searches against UniProt (48), Pfam (49) searches, and InterProScan 5 (50) analyses. Microsatellites were searched for with WebSat (51) using default parameters.
Protein analyses and 3D structure predictions.
The presence of signal peptides in primary amino acid sequences was searched for with SignalP 4.1 (52). Transmembrane domain searches and hydrophobicity analyses were performed with TMHMM 2.0 (53), TopPred 2 (54), and TMpred (55) as implemented on the CBS (http://www.cbs.dtu.dk/services/), Institut Pasteur (http://mobyle.pasteur.fr/), and Expasy (http://www.expasy.org) web portals, respectively. The M896_080490 primary amino acid sequence was converted from FASTA to PDB format and first modeled three-dimensionally with ROSETTA3 version 2014wk05 (56). This resulted in an unfolded chain of little value but containing several directional changes indicating a degree of secondary structure. The sequence was then run through Robetta (57), and two domains were determined via Ginzu analysis. These domains were queried against the NCBI nonredundant database (NR) using PSI-BLAST (58) to determine potential sequence homology to previously filed protein structures. Due to its higher confidence score, the second domain was analyzed with Web-based InterPro software (59). The highest-correlating gene ontologies (GOs) were analyzed with respect to previously predicted functions and correlated with the results of the Robetta analysis. The model was created with the most highly correlating base model via SWISS-MODEL (60) (Fig. 3) and correlated with its parent template by dot plot analysis as implemented in FFAS03 (61).
Phylogenetic analyses.
For phylogenomic inferences, the microsporidian protein sequences were retrieved from the MicrosporidiaDB (62), SilkPathDB (http://silkpathdb.swu.edu.cn/silkpathdb/), and GenBank databases. Orthologous sequences were identified by BLASTP searches at an E value cutoff of 1E-20 using the O. colligata proteins as queries. Orthologs were aligned with MAFFT L-INS-I (45), and the ambiguous positions in the resulting alignments were filtered out with BMGE (63) using the default parameters. Maximum likelihood (ML) inference analyses were performed with PHYML 3.0 (64) using the LG model of amino acid substitutions with four gamma categories. A total of 100 bootstrap replicate experiments were performed. Bootstrap replicates were generated with Seqboot and node percentages calculated with Consense from the PHYLIP 3.695 package (65). For horizontal gene transfer inference determinations, the M896_080490 orthologous and paralogous sequences were retrieved from the NCBI nonredundant (NR) database (accession date, 20 August 2014) using BLASTP searches with an E value cutoff of 1E-40. The retrieved sequences were aligned with MUSCLE (66), and the resulting alignment was filtered with BMGE using the default parameters. ML inference and bootstrap replicate experiments were performed as described above.
Pairwise distribution and metabolic profiling.
For each species, local protein databases were generated using MAKEBLASTDB from the NCBI BLAST+ 2/2/28 package. The presence or absence of genes in comparisons between species was determined by evaluating pairwise BLASTP hits (E value cutoff, 1e-10) for all possible combinations. Metabolic profiles were inferred from InterProScan 5 (50) analyses performed on each protein data set; for each protein, the gene ontologies retrieved were filtered to remove duplicates and concatenated into higher hierarchies derived from the KEGG orthology pathways using custom Perl scripts.
Nucleotide diversity.
The O. colligata protein-coding genes and their Encephalitozoon orthologs were aligned by codon comparisons performed with MACSE (67). For each alignment, the nucleotide diversity (Pi) between Encephalitozoon species and their divergence from O. colligata [K(JC)] were inferred using the polymorphism and divergence in functional regions tool implemented in DnaSP 5/10/01 (68) with O. colligata as the outgroup.
Accession numbers.
The O. colligata data were released in the NCBI database under BioProject PRJNA210314, BioSample SAMN02867507, and accession number JOKQ00000000.
SUPPLEMENTAL MATERIAL
Phylogenetic position of O. colligata. The best ML tree (LG+Γ4) shown is derived from 104 proteins (36,973 amino acid [aa] positions) that are shared between the 32 taxonomic units. The levels of bootstrap support are indicated above the corresponding nodes, with asterisks highlighting nodes that were recovered in all of the performed replicate experiments. The early-diverging microsporidian Mitosporidium and the Cryptomycota Rozella spp. were used as outgroups. Download
Multiple sequence alignment of O. colligata septin 7 and its Daphnia homologues from clades A and B (Fig. 4). Only the conserved regions are shown for brevity. Conserved residues are color coded according to their amino acid properties. The underlying conservation, quality, and consensus plots were generated with Jalview (http://www.jalview.org/). Download
{kind=link}
Functional distribution of the microsporidian proteome across KEGG metabolic pathways.
Nucleotide divergences between the O. colligata protein-coding genes and their Encephalitozoon homologues.
ACKNOWLEDGEMENT
This work was supported by a grant from the Canadian Institutes of Health Research to P.J.K. (MOP-42517; http://www.cihr-irsc.gc.ca/).
Footnotes
Citation Pombert J, Haag KL, Beidas S, Ebert D, Keeling PJ. 2015. The Ordospora colligata genome: evolution of extreme reduction in microsporidia and host-to-parasite horizontal gene transfer. mBio 6(1):e02400-14. doi:10.1128/mBio.02400-14.
REFERENCES
- 1.Keeling PJ, Fast NM. 2002. Microsporidia: biology and evolution of highly reduced intracellular parasites. Annu Rev Microbiol 56:93–116. doi: 10.1146/annurev.micro.56.012302.160854. [DOI] [PubMed] [Google Scholar]
- 2.Franzen C. 2004. Microsporidia: how can they invade other cells? Trends Parasitol 20:275–279. doi: 10.1016/j.pt.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Franzen C. 2005. How do microsporidia invade cells? Folia Parasitol (Praha) 52:36–40. [DOI] [PubMed] [Google Scholar]
- 4.Xu Y, Weiss LM. 2005. The microsporidian polar tube: a highly specialised invasion organelle. Int J Parasitol 35:941–953. doi: 10.1016/j.ijpara.2005.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corradi N, Selman M. 2013. Latest progress in microsporidian genome research. J Eukaryot Microbiol 60:309–312. doi: 10.1111/jeu.12030. [DOI] [PubMed] [Google Scholar]
- 6.Pombert J-F, Selman M, Burki F, Bardell FT, Farinelli L, Solter LF, Whitman DW, Weiss LM, Corradi N, Keeling PJ. 2012. Gain and loss of multiple functionally related, horizontally transferred genes in the reduced genomes of two microsporidian parasites. Proc Natl Acad Sci U S A 109:12638–12643. doi: 10.1073/pnas.1205020109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pombert J-F, Xu J, Smith DR, Heiman D, Young S, Cuomo CA, Weiss LM, Keeling PJ. 2013. Complete genome sequences from three genetically distinct strains reveal high intraspecies genetic diversity in the microsporidian Encephalitozoon cuniculi. Eukaryot Cell 12:503–511. doi: 10.1128/EC.00312-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corradi N, Pombert J-F, Farinelli L, Didier ES, Keeling PJ. 2010. The complete sequence of the smallest known nuclear genome from the microsporidian Encephalitozoon intestinalis. Nat Commun 1:77. doi: 10.1038/ncomms1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katinka MD, Duprat S, Cornillot E, Méténier G, Thomarat F, Prensier G, Barbe V, Peyretaillade E, Brottier P, Wincker P, Delbac F, El Alaoui H, Peyret P, Saurin W, Gouy M, Weissenbach J, Vivarès CP. 2001. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 414:450–453. doi: 10.1038/35106579. [DOI] [PubMed] [Google Scholar]
- 10.Corradi N, Slamovits CH. 2011. The intriguing nature of microsporidian genomes. Brief Funct Genomics 10:115–124. doi: 10.1093/bfgp/elq032. [DOI] [PubMed] [Google Scholar]
- 11.Peyretaillade E, El Alaoui H, Diogon M, Polonais V, Parisot N, Biron DG, Peyret P, Delbac F. 2011. Extreme reduction and compaction of microsporidian genomes. Res Microbiol 162:598–606. doi: 10.1016/j.resmic.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Larsson JIR, Ebert D, Vávra J.. 1997. Ultrastructural study and description of Ordospora colligata gen. et sp. nov. (microspora, Ordosporidae fam. nov.), a new microsporidian parasite of Daphnia magna (Crustacea, Cladocera). Eur J Protistol 33:432–443. doi: 10.1016/S0932-4739(97)80055-7. [DOI] [Google Scholar]
- 13.Ebert D. 2005. Ecology, epidemiology, and evolution of parasitism in Daphnia. NCBI. http://www.ncbi.nlm.nih.gov/books/NBK2036/.
- 14.Ebert D, Lipsitch M, Mangin KL. 2000. The effect of parasites on host population density and extinction: experimental epidemiology with Daphnia and six microparasites. Am Nat 156:459–477. doi: 10.1086/303404. [DOI] [PubMed] [Google Scholar]
- 15.Goren L, Ben-Ami F. 2013. Ecological correlates between cladocerans and their endoparasites from permanent and rain pools: patterns in community composition and diversity. Hydrobiologia 701:13–23. doi: 10.1007/s10750-012-1243-5. [DOI] [Google Scholar]
- 16.Phan QT, Eng DK, Mostowy S, Park H, Cossart P, Filler SG. 2013. Role of endothelial cell septin 7 in the endocytosis of Candida albicans. mBio 4:-13. doi: 10.1128/mBio.00542-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vossbrinck CR, Debrunner-Vossbrinck BA. 2005. Molecular phylogeny of the microsporidia: ecological, ultrastructural and taxonomic considerations. Folia Parasitol (Praha) 52:131–142. doi: 10.14411/fp.2005.017. [DOI] [PubMed] [Google Scholar]
- 18.Haag KL, Larsson JI, Refardt D, Ebert D. 2011. Cytological and molecular description of Hamiltosporidium tvaerminnensis gen. et sp. nov., a microsporidian parasite of Daphnia magna, and establishment of Hamiltosporidium magnivora comb. Parasitology 138:447–462. doi: 10.1017/S0031182010001393. [DOI] [PubMed] [Google Scholar]
- 19.Ebert D, Hottinger JW, Pajunen VI. 2001. Temporal and spatial dynamics of parasite richness in a Daphnia metapopulation. Ecology 82:3417–3434. doi: 10.1890/0012-9658(2001)082[3417:TASDOP]2.0.CO;2. [DOI] [Google Scholar]
- 20.Weigl S, Körner H, Petrusek A, Seda J, Wolinska J. 2012. Natural distribution and co-infection patterns of microsporidia parasites in the Daphnia longispina complex. Parasitology 139:870–880. doi: 10.1017/S0031182012000303. [DOI] [PubMed] [Google Scholar]
- 21.Haag KL, James TY, Pombert J-F, Larsson R, Schaer TM, Refardt D, Ebert D. 2014. Evolution of a morphological novelty occurred before genome compaction in a lineage of extreme parasites. Proc Natl Acad Sci U S A 111:15480–15485. doi: 10.1073/pnas.1410442111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell SE, Williams TA, Yousuf A, Soanes DM, Paszkiewicz KH, Williams BA. 2013. The genome of Spraguea lophii and the basis of host-microsporidian interactions. PLoS Genet 9:e1003676. doi: 10.1371/journal.pgen.1003676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cornman RS, Chen YP, Schatz MC, Street C, Zhao Y, Desany B, Egholm M, Hutchison S, Pettis JS, Lipkin WI, Evans JD. 2009. Genomic analyses of the microsporidian Nosema ceranae, an emergent pathogen of honey bees. PLoS Pathog. 5:e1000466. doi: 10.1371/journal.ppat.1000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinz E, Williams TA, Nakjang S, Noël CJ, Swan DC, Goldberg AV, Harris SR, Weinmaier T, Markert S, Becher D, Bernhardt J, Dagan T, Hacker C, Lucocq JM, Schweder T, Rattei T, Hall N, Hirt RP, Embley TM. 2012. The genome of the obligate intracellular parasite Trachipleistophora hominis: new insights into microsporidian genome dynamics and reductive evolution. PLoS Pathog. 8:e1002979. doi: 10.1371/journal.ppat.1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan G, Xu J, Li T, Xia Q, Liu S, Zhang G, Li S, Li C, Liu H, Yang L, Liu T, Zhang X, Wu Z, Fan W, Dang X, Xiang H, Tao M, Li Y, Hu J, Li Z, Lin L, Luo J, Geng L, Wang L, Long M, Wan Y, He N, Zhang Z, Lu C, Keeling PJ, Wang J, Xiang Z, Zhou Z. 2013. Comparative genomics of parasitic silkworm Microsporidia reveal an association between genome expansion and host adaptation. BMC Genomics 14:186. doi: 10.1186/1471-2164-14-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouzahzah B, Nagajyothi F, Ghosh K, Takvorian PM, Cali A, Tanowitz HB, Weiss LM. 2010. Interactions of Encephalitozoon cuniculi polar tube proteins. Infect Immun 78:2745–2753. doi: 10.1128/IAI.01205-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, Bauer DJ, Cáceres CE, Carmel L, Casola C, Choi J-H, Detter JC, Dong Q, Dusheyko S, Eads BD, Fröhlich T, Geiler-Samerotte KA, Gerlach D, Hatcher P, Jogdeo S, Krijgsveld J, Kriventseva EV, Kültz D, Laforsch C, Lindquist E, Lopez J, Manak JR, Muller J, Pangilinan J, Patwardhan RP, Pitluck S, Pritham EJ, Rechtsteiner A, Rho M, Rogozin IB, Sakarya O, Salamov A, Schaack S, Shapiro H, Shiga Y, Skalitzky C, Smith Z, Souvorov A, Sung W, Tang Z, Tsuchiya D, et al.. 2011. The ecoresponsive genome of Daphnia pulex. Science 331:555–561. doi: 10.1126/science.1197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selman M, Sak B, Kváč M, Farinelli L, Weiss LM, Corradi N. 2013. Extremely reduced levels of heterozygosity in the vertebrate pathogen Encephalitozoon cuniculi. Eukaryot Cell 12:496–502. doi: 10.1128/EC.00307-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao H, Xu C, Lu H-L, Chen X, St Leger RJ, Fang W. 2014. Host-to-pathogen gene transfer facilitated infection of insects by a pathogenic fungus. PLoS Pathog. 10:e1004009. doi: 10.1371/journal.ppat.1004009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson MT, Seifert HS. 2011. Opportunity and means: horizontal gene transfer from the human host to a bacterial pathogen. mBio 2:e00005–11. doi: 10.1128/mBio.00005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fast NM, Law JS, Williams BA, Keeling PJ. 2003. Bacterial catalase in the microsporidian Nosema locustae: implications for microsporidian metabolism and genome evolution. Eukaryot Cell 2:1069–1075. doi: 10.1128/EC.2.5.1069-1075.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsaousis AD, Kunji ER, Goldberg AV, Lucocq JM, Hirt RP, Embley TM. 2008. A novel route for ATP acquisition by the remnant mitochondria of Encephalitozoon cuniculi. Nature 453:553–556. doi: 10.1038/nature06903. [DOI] [PubMed] [Google Scholar]
- 33.Selman M, Pombert J-F, Solter L, Farinelli L, Weiss LM, Keeling P, Corradi N. 2011. Acquisition of an animal gene by microsporidian intracellular parasites. Curr Biol 21:R576–R577. doi: 10.1016/j.cub.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bridges AA, Gladfelter AS. 2014. Fungal pathogens are platforms for discovering novel and conserved septin properties. Curr Opin Microbiol 20:42–48. doi: 10.1016/j.mib.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakahira M, Macedo JN, Seraphim TV, Cavalcante N, Souza TA, Damalio JC, Reyes LF, Assmann EM, Alborghetti MR, Garratt RC, Araujo AP, Zanchin NI, Barbosa JA, Kobarg J. 2010. A draft of the human septin interactome. PLoS One 5:e13799. doi: 10.1371/journal.pone.0013799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Refardt D, Ebert D. 2007. Inference of parasite local adaptation using two different fitness components. J Evol Biol 20:921–929. doi: 10.1111/j.1420-9101.2007.01307.x. [DOI] [PubMed] [Google Scholar]
- 37.Andrews S. 2012. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 38.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 39.Gordon D, Abajian C, Green P. 1998. Consed: a graphical tool for sequence finishing. Genome Res 8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 40.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using tablet for visual exploration of second-generation sequencing data. Brief Bioinform 14:193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 42.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lagesen K, Hallin P, Rødland EA, Staerfeldt H-H, Rognes T, Ussery DW. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 45.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peyretaillade E, Gonçalves O, Terrat S, Dugat-Bony E, Wincker P, Cornman RS, Evans JD, Delbac F, Peyret P. 2009. Identification of transcriptional signals in Encephalitozoon cuniculi widespread among microsporidia phylum: support for accurate structural genome annotation. BMC Genomics 10:607. doi: 10.1186/1471-2164-10-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. 2013. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41:D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.The UniProt Consortium 2014. Activities at the universal protein resource (UniProt). Nucleic Acids Res 42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. 2014. Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong S-Y, Lopez R, Hunter S. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martins WS, Lucas DC, Neves KF, Bertioli DJ. 2009. WebSat—a web software for microsatellite marker development. Bioinformation 3:282–283. doi: 10.6026/97320630003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 53.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 54.Von Heijne G. 1992. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol 225:487–494. doi: 10.1016/0022-2836(92)90934-C. [DOI] [PubMed] [Google Scholar]
- 55.Hofmann K, Stoffel W. 1993. TMbase—a database of membrane-spanning protein segments. Biol Chem Hoppe Seyler 374:166. doi: 10.1515/bchm3.1993.374.1-6.143. [DOI] [Google Scholar]
- 56.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YE, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popović Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. 2011. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim DE, Chivian D, Baker D. 2004. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32:W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and psi-blast: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJA, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong S-Y. 2012. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A. 2011. FFAS server: novel features and applications. Nucleic Acids Res 39:W38–W44. doi: 10.1093/nar/gkr441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aurrecoechea C, Brestelli J, Brunk BP, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer ET, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Srinivasamoorthy G, Stoeckert CJ, Thibodeau R, Treatman C, Wang H. 2010. EuPathDB: a portal to eukaryotic pathogen databases. Nucleic Acids Res 38:D415–D419. doi: 10.1093/nar/gkp941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Criscuolo A, Gribaldo S. 2010. BMGE (block mapping and gathering with entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol 10:210. doi: 10.1186/1471-2148-10-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 65.Felsenstein J. 2014. PHYLIP (phylogeny inference package), version 3.695.Department of Genome Sciences, University of Washington, Seattle, WA. [Google Scholar]
- 66.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ranwez V, Harispe S, Delsuc F, Douzery EJ. 2011. MACSE: multiple alignment of coding sequences accounting for frameshifts and stop codons. PLoS One 6:e22594. doi: 10.1371/journal.pone.0022594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Librado P, Rozas J. 2009. DnaSP V5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 69.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic position of O. colligata. The best ML tree (LG+Γ4) shown is derived from 104 proteins (36,973 amino acid [aa] positions) that are shared between the 32 taxonomic units. The levels of bootstrap support are indicated above the corresponding nodes, with asterisks highlighting nodes that were recovered in all of the performed replicate experiments. The early-diverging microsporidian Mitosporidium and the Cryptomycota Rozella spp. were used as outgroups. Download
Multiple sequence alignment of O. colligata septin 7 and its Daphnia homologues from clades A and B (Fig. 4). Only the conserved regions are shown for brevity. Conserved residues are color coded according to their amino acid properties. The underlying conservation, quality, and consensus plots were generated with Jalview (http://www.jalview.org/). Download
Functional distribution of the microsporidian proteome across KEGG metabolic pathways.
Nucleotide divergences between the O. colligata protein-coding genes and their Encephalitozoon homologues.