

Abstract
Activating transcription factor-3 (ATF3) acts as a negative regulator of cytokine production during Gram-negative bacterial infection. A recent study reported that ATF3 provides protection from Streptococcus pneumoniae infection by activating cytokines. However, the mechanism by which S. pneumoniae induces ATF3 after infection is still unknown. In this study, we show that ATF3 was upregulated via Toll-like receptor (TLR) pathways in response to S. pneumoniae infection in vitro. Induction was mediated by TLR4 and TLR2, which are in the TLR family. The expression of ATF3 was induced by pneumolysin (PLY), a potent pneumococcal virulence factor, via the TLR4 pathway. Furthermore, ATF3 induction is mediated by p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK). Thus, this study reveals a potential role of PLY in modulating ATF3 expression, which is required for the regulation of immune responses against pneumococcal infection in macrophages.
Keywords: activating transcription factor-3 (ATF3), pneumococcal infection, pneumolysin (PLY), S. pneumoniae, Toll-like receptors (TLR)
INTRODUCTION
Activating transcription factor-3 (ATF3), a stress-inducible eukaryotic gene, is a member of the cAMP response element-binding protein (ATF/CREB) family of basic-leucine zipper (bZip) transcription factors that binds to the consensus cAMP response element (CRE) sequences of its target genes (Hai et al., 1999). ATF3 is a key regulatory factor in inflammatory responses (Gilchrist et al., 2006; 2008; 2010; Khuu et al., 2007). For example, ATF3 was highly expressed in response to lipopolysaccharide (LPS) stimulation (Gilchrist et al., 2006). LPS-induced expression of the cytokine genes is also inhibited by ATF3, indicating that ATF3 acts as a negative regulator of LPS-induced inflammation (Lai et al., 2013). In a mouse model, ATF3 was found to be a novel regulator of neutrophil migration (Boespflug et al., 2014). In this model, ATF3 repressed LPS-driven CXCL1 production, controlling the recruitment of the neutrophil to the site of inflammation. However, in humans, ATF3 is a positive regulator of interferon-γ (IFN-γ) expression, thus promoting Th1 differentiation (Filén et al., 2010). In Gram-negative bacterial infections, ATF3 is upregulated by Neisseria gonorrheae via mitogen-activated protein kinases (MAPK) signaling and suppresses the IL-6 expression during N. gonorrhoeae infection (Calton et al., 2013). In contrast, the upregulation of ATF3 confers resistance to S. pneumoniae (Gram-positive bacteria) infection via activating the production of cytokines (Nguyen et al., 2014a). However, the mechanism(s) by which S. pneumoniae stimulates ATF3 expression is still unknown.
Streptococcus pneumoniae is a major common cause of death in the USA, which causes severe, pneumococcal infections like sepsis, pneumonia, and meningitis with high rates of morbidity and mortality (File, 2004). There are over 90 identified serotypes of S. pneumoniae, and each serotype is characterized by a specific capsular polysaccharide that has extracellular neutrophil traps (Wartha et al., 2007) as well as the capacity to evade phagocytosis and complement binding (Hyams et al., 2010), thus completely escaping immunological responses. There are several pneumococcal virulence factors such as autolysin (LytA), pneumococcal surface protein A (PspA), and PspC (Koppe et al., 2012); however, pneumolysin (PLY) is one of the most critical factors in the induction of inflammation and innate immunity. During infection, S. pneumoniae releases PLY into the host environment, after which binding to Toll-like receptor 4 (TLR4) stimulates the production of inflammatory cytokines (Malley et al., 2003).
TLRs play a crucial role in the recognition of bacterial infections in mammals. These receptors are highly expressed in immune cells such as macrophages and T cells (Kabelitz, 2007). In pneumococcal infections, the recognition of virulence factors by TLRs stimulates the production of cytokines such as interleukine (IL)-1β, tumor necrosis factor (TNF)-α, and IL-17 in macrophages. This is a very important and primary innate immune response to pneumococcal infections. Among the TLR family, TLR4 and TLR2 are required for the elimination of pneumococcal infections. S. pneumoniae-derived lipoteichoic acid (LTA) binds to TLR2 and stimulates immune response to the infection (Schröder et al., 2003). Similarly, TLR4 recognizes pneumolysin (PLY) and activates protective signals against S. pneumoniae; therefore, TLR4-deficient mice were more susceptible to pneumococcal infections compared with wild-type mice (Malley et al., 2003).
A recent study reported that ATF3 provides protection against pneumococcal infection by positively regulating cytokines (Nguyen et al., 2014a). However, the mechanism(s) by which ATF3 is induced by S. pneumoniae infection remains unknown. In the present study, we showed that TLR4 and TLR2 are required for ATF3 induction. PLY, a pneumococcal virulence factor induces ATF3 expression during infection when bound to TLR4. Moreover, TLR2/4 regulate ATF3 induction via the JNK/p38 pathway. After induction, ATF3 interacts with c-Jun in the nucleus to regulate the cytokines. The findings of this study will contribute to a better understanding of the host defense mechanism against S. pneumoniae infection.
MATERIALS AND METHODS
Bacterial strains, cell cultures, and reagents
Encapsulated type 2 strain D39 S. pneumoniae (NCTC7466) was cultured in Todd-Hewitt broth as previously described (Benton et al., 1995; Choi et al., 1999). For the infection experiment, pneumococci were cultured until OD550 = 0.3 and centrifuged at 4,000 × g at 4°C for 10 min. The pellet was resuspended in 1 ml of DMEM media to a concentration of 108 CFU/ml and used to infect cells at MOI = 50. RAW 264.7 murine macrophage-like cells were obtained from the American Type Culture Collection. The cells were maintained at 37°C in a humidified incubator at 95% air-5% CO2 and grown in Dulbecco’s modified Eagle’s medium (DMEM; GE Healthcare, UK) supplemented with 10% fetal bovine serum (Bioscience) along with 1X penicillin/streptomycin (PAA Laboratories GmbH, Germany) (Tu et al., 2007). Lipopolysaccharide (serotype 026:B6, Escherichia coli) was purchased from Sigma (USA).
Overexpression and purification of PLY
The ply gene was amplified by PCR using D39 pneumococcal chromosomal DNA as a template with PLY primers (F: 5′-GGGCCCGGATCCGATGGCAAATAAAGCAGTAAATGAC-3′; R: 5′-GGCCCGCTCGAGCTAGTCATTTTCTACCTTATCCTC-3′) that incorporated BamHI and XhoI restriction enzyme sites. The PCR products were digested with the BamHI and XhoI enzymes and cloned into BamHI- and XhoI-digested pET32b(+). After transformation, E. coli BL21-containing pET32b-PLY was grown in LB medium supplemented with isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM. The histidine-tagged PLY protein was purified using an Ni-NTA column and then dialyzed against 50 mM Tris buffer (pH 7.5) for further experiments.
Transfection
Control siRNA-A (siCO), TLR4 siRNA (siTLR4), and TLR2 siRNA (siTLR2) were purchased from Santa Cruz Biotechnologies (USA). P38 siRNA (sip38) and JNK siRNA (siJNK) were purchased from Cell Signaling Technology (USA). The RAW 264.7 cells were transfected with either siCO, siTLR4 (50 nM), siTLR2 (50 nM), sip38 (100 nM), or siJNK (100 nM) using siRNA transfection reagents (TransIT-TKO, Mirus, USA). After 24 h of incubation, the transfected cells were used for the desired experiments.
Treatment with inhibitors
p38 MAPK inhibitor (SB203580) and JNK inhibitor (SP600125) were purchased from Calbiochem (USA). The RAW 264.7 cells (5 × 105) were grown overnight and then incubated with either p38 or MAPK inhibitor (10 μM) or JNK inhibitor (10 μM) for 3 h. The treated cells were then infected with the pneumococcus cells and used for further experiments.
Western blotting
Antibodies against ATF3, TLR4, TNF-α, c-Jun, and β-actin were purchased from Santa Cruz Biotechnologies, and the antibodies for p-JNK, JNK, p38, p-p38, ERK, and p-ERK were purchased from Cell Signaling Technology (USA). The concentrations of the protein samples were estimated using the DC™ protein assay kit (Bio-rad, USA). Protein samples (25–30 μg each) were separated on a 7–15% (wt/vol) polyacrylamide gel (SDS-PAGE) and transferred onto polyvinyli-denedifluoride (PVDF) membranes (Millipore, USA) for Western blotting, as previously described (Nguyen et al., 2014b).
Cytokine ELISA assay
The RAW 264.7 cells were infected with pneumococci for 1, 2, and 4 h. After the indicated time, the supernatants were collected and centrifuged at 15000 × g at 4°C for 10 min. The pellets (containing bacteria and dead cells) were discarded, and the supernatants were used for TNF-α enzyme-linked immunosorbent assays (ELISA) (BD, USA) per the manufacturer’s instructions.
Confocal microscopy
The RAW 264.7 cells (5 × 105) were grown overnight until the plates were 80% confluent and subsequently infected with pneumococcus cells for 1, 2, or 4 h. After the indicated time, 4% (vol/vol) formaldehyde was added to fix the cells. The cells were permeabilized with 1% (vol/vol) Triton X-100/PBS for 5 min and then blocked with 1% Bovine serum albumin/Phosphate buffered saline (BSA/PBS) overnight at 4°C. After blocking, the cells were incubated with primary antibody (anti-ATF3 and anti-cJun) for 1 h. Finally, the secondary antibodies labeled with fluorescein isothiocyanate (FITC) or DAPI (4′,6-diamidino-2-phenylindole) (to stain the nucleus) was added and incubated for 1 h. The samples were visualized using a confocal microscope (Carl Zeiss LSM 510 Meta DuoScan, Carl Ziess Micro Imaging GmbH, Germany).
Immunoprecipitation
The RAW 264.7 cell lysates (200 μg) were agitated with either anti-ATF3 or anti-cJun antibodies at 4°C for 4 h. Subsequently, protein A-anchored agarose (USA) was added and incubated at 4°C overnight. The mixture was centrifuged at 1000 × g to collect the protein A agarose/protein complex. The desired complex was then washed three times. Ten microliters of 5× SDS-PAGE sample buffer was added to the protein A agarose/protein complex and then boiled at 100°C for 5 min to remove beads from immunoprecipitated proteins. Finally, the supernatant was subjected to a Western blot analysis with both anti-ATF3 and anti-cJun primary antibodies.
Statistical analysis
One-way ANOVA (Holm-Sidak method) was used to analyze the statistical differences between groups. All of the results are representative of three independent experiments. Statistically significant differences were considered as P < 0.05 (* P < 0.05, ** P < 0.01, and *** P < 0.001).
RESULTS
ATF3 upregulation by S. pneumoniae infection is TLR2/4-dependent
We examined whether TLR4 is involved in the upregulation of ATF3 during pneumococcal infections. Using TLR4 siRNA (siTLR4), TLR4 expression was impaired, and the expression of ATF3 and TNF-α was subsequently checked during the infection. The results show that ATF3 expression was significantly decreased in the siTLR4-transfected cells (Fig. 1A), indicating that ATF3 expression is TLR4-dependent. It was also observed that the TNF-α levels declined in siTLR4-transfected cells (Figs. 1A and 1C).
Fig. 1.
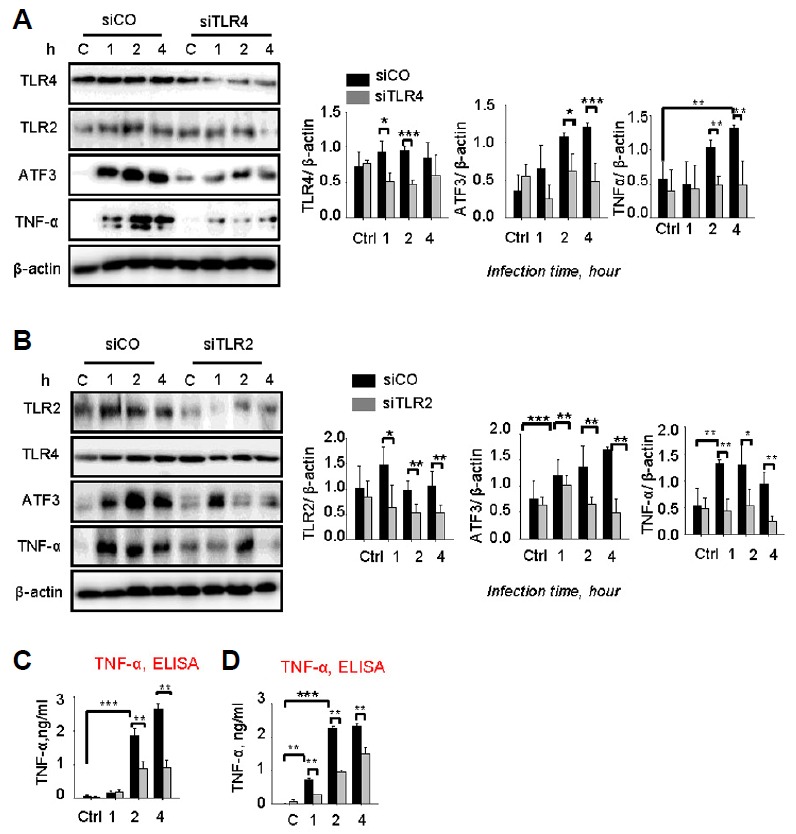
Streptococcus pneumoniae induced ATF3 via the TLR2/4 pathway. RAW 264.7 cells were transfected with siTLR4 (A, C) or siTLR2 (B, D) for 24 h and then infected with S. pneumoniae for 0, 1, 2, or 4 h. Subsequently, the cell lysates were analyzed by Western blotting. The cell supernatants from (A) and (B) were used to measure TNF-α release by TNF-α ELISA. The band densities were measured by ImageJ. The data are representative of three independent experiments and were analyzed by one-way ANOVA. Data show mean ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001.
Moreover, TLR2 also plays an important role in generating immunological responses against pneumococci (Basset et al., 2012). TLR2 expression was knocked down using TLR2 siRNA (siTLR2). We showed that siTLR2 impaired ATF3 expression during the infection (Fig. 1C). Similarly, TNF-α was also suppressed by siTLR2 (Figs. 1B and 1D), indicating that ATF3 upregulation is TLR2/4 signal-dependent.
TLR2/4 are required for phosphorylation of JNK and p38 during pneumococcal infection
In pneumococcal infection, MAPKs play an important role in generating innate immune responses and production of mediators in macrophages (Kang et al., 2009). The results obtained clearly show that phosphorylation of p38 and ERK was increased by pneumococcal infection 10 min post-infection (Figs. 2A and 2B), whereas the phosphorylation of JNK was significantly induced 30 min post-infection (Figs. 2A and 2B). Interestingly, siTLR4 treatment slightly reduced the induction of p-JNK at 30 min and 60 min post-infection, whereas treatment with siTLR2 significantly abolished the levels of p-JNK expression (Figs. 2A and 2B). Moreover, the phosphorylation of p38 markedly decreased in both siTLR4- and siTLR2-transfected cells. However, the phosphorylation of ERK was not TLR4- or TLR2-dependent (Figs. 2A and 2B). Taken together, these results indicate that S. pneumoniae-induced phosphorylation of JNK and p38 is TLR2/4-dependent.
Fig. 2.
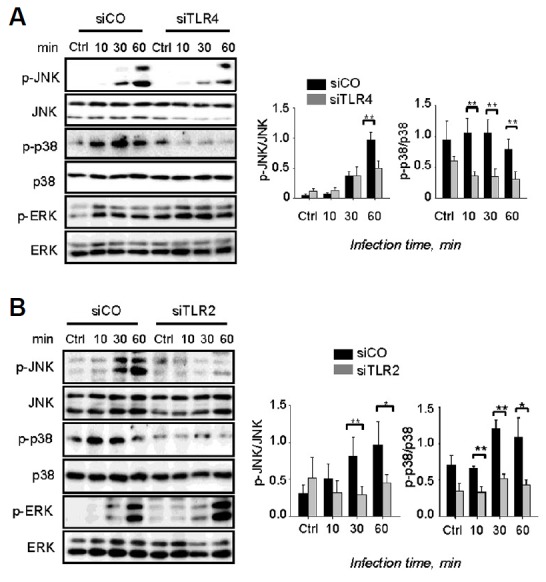
The TLR2/4 pathway is required for activation of JNK/p38. RAW 264.7 cells were transfected with siTLR4 (A) or siTLR2 (B) for 24 h and then infected with S. pneumoniae for 0, 1, 2, or 4 h. Subsequently, the cell lysates were analyzed by Western blotting. The band densities were measured by ImageJ. The data are representative of three independent experiments and were analyzed by one-way ANOVA. Data show mean ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001.
S. pneumoniae upregulated ATF3 via the JNK/p38 pathway
ATF3 was regulated by TLR4 in response to LPS induction (Gilchrist et al., 2004) and mediated by JNK and p38 kinases (Lu et al., 2007). Thus, we hypothesized that TLR4 and TLR2 might control ATF3 expression via the JNK and p38 pathways during pneumococcal infection. The specific inhibitors of p38 and JNK were used to impair their activity. The expression levels of ATF3 in the inhibitor-treated cells were then compared with the control. It was repeatedly found that S. pneumoniae infection significantly induces ATF3 expression; however, treatment of p38 and JNK inhibitors abolished this induction (Figs. 3A and 3B). sip38 and siJNK also significantly reduced ATF3 induction, whereas siCO did not, demonstrating that ATF3 expression was mediated by JNK and p38 kinase (Figs. 3C and 3D).
Fig. 3.
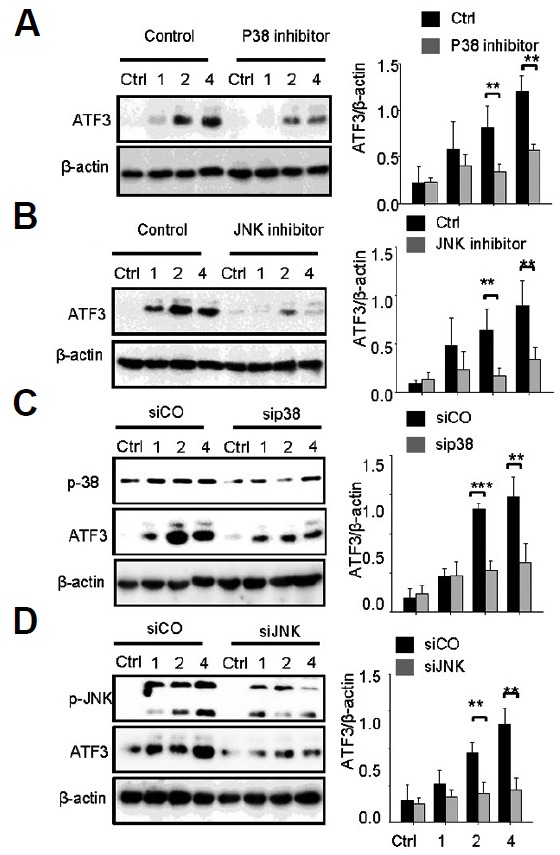
ATF3 induction is JNK/p38-dependent. RAW 264.7 cells were incubated with p38 inhibitor (A) or JNK inhibitor (B) for 1 h or transfected with sip38 (C) or siJNK (D) for 48 h, and then infected with S. pneumoniae for 0, 1, 2, or 4 h. Subsequently, the cell lysates were analyzed by Western blotting. The band densities were measured by ImageJ. The data are representative of three independent experiments and analyzed by one-way ANOVA. Data show mean ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001.
PLY induced ATF3 expression via TLR4
A previous study reported that ATF3 expression was induced by PLY in a dose- and time-dependent manner during S. pneumoniae infection (Nguyen et al., 2014a). Some studies showed that PLY triggers and promotes secretion of cytokines such as TNF-α IL-6, IL-1β via the TLR4 pathway (McNeela et al., 2010). Thus, whether ATF3 was induced by PLY via the TLR4 pathway was examined. Using the siTLR4 model, the results show that there was a significant induction of ATF3 expression by PLY after 1 h of incubation (Fig. 4A). However, this induction was abrogated by siTLR4 treatment, indicating that PLY induces ATF3 expression via the TLR4 pathway (Fig. 4A). Consistently, the TNF-α levels were also higher in PLY-incubated cells than non-treated cells. Levels of TNF-α were higher in siCo-transfected cells compared with siTLR4-transfected cells (Figs. 4A and 4C), indicating that PLY induces TNF-α via the ATF3/TLR4 pathway. Moreover, siTLR2 did not abolish PLY-induced ATF3 and TNF-α expression (Supplementary Fig. S1).
Fig. 4.
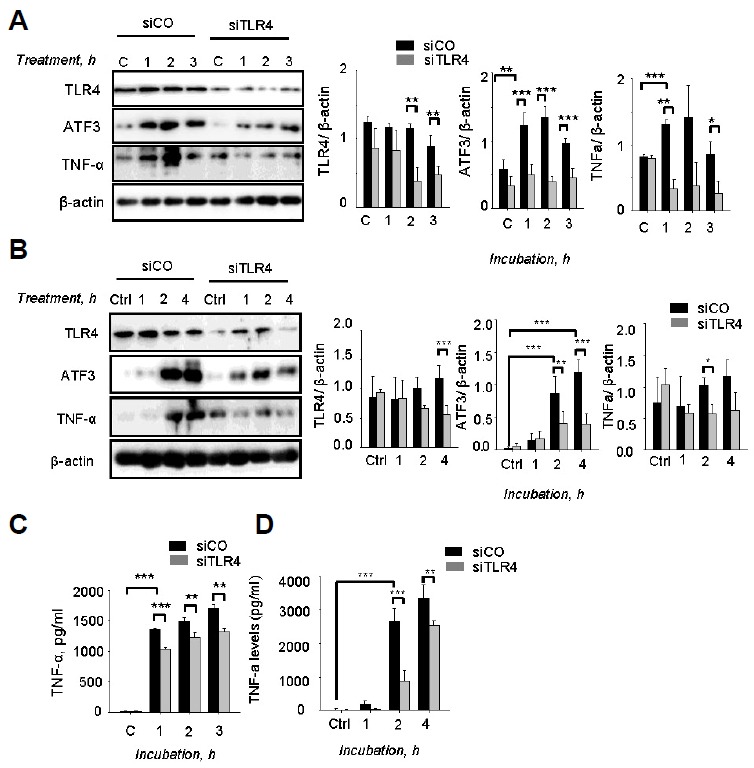
Pneumolysin induced ATF3 via TLR4. RAW 264.7 cells were transfected with siTLR4 for 24 h and then incubated with 500 ng/ml of pneumolysin (A) 50 ng/ml of LPS (B) for 0, 1, 2, or 3 h. Subsequently, the cell lysates were separated by Western blotting. (C, D) The cell supernatants from (A, B) were used to measure TNF-α release by TNF-α ELISA assay. The band densities were measured by ImageJ. The data are representative of three independent experiments and analyzed by one-way ANOVA. Data show mean ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001.
Previously, a PLY-deficient mutant strain was used to determine the role of PLY for ATF3 induction (Nguyen et al., 2014a). The results showed that ATF3 is induced by the wild-type infection at 1, 2, and 4 h postinfection, but not by the PLY mutant infection (Nguyen et al., 2014a). Moreover, we also demonstrated that purified PLY stimulated ATF3 expression dose- and time-dependently (Nguyen et al., 2014a). We also used heat-inactivated PLY and non-PLY protein such as VncR and ClpL as controls to confirm that our PLY protein was not contaminated with LPS (Nguyen et al., 2014a). Only purified PLY induced ATF3, while the other proteins, such as purified VncR and ClpL, and heat-inactivated PLY, did not; this indicates that our purified PLY was not contaminated with LPS, and that PLY is an inducer of ATF3 during pneumococcal infection (Nguyen et al., 2014a).
Furthermore, to confirm that our PLY protein was not contaminated with LPS, we used anti-PLY serum to block the PLY effect. After the PLY protein was contaminated with LPS, ATF3 induction should not be impaired by anti-PLY serum. In fact, anti-PLY serum significantly decreased the ATF3 levels (Supplementary Fig. S2), and 10 μl of anti-PLY serum inhibited nearly 90% of the ATF3 induction compared with the untreated control, demonstrating that our purified PLY was not contaminated with LPS.
Since LPS is a well-known ligand for TLR4, we also used LPS as a positive control for this experiment. Consistently, LPS also stimulates ATF3 expression via TLR4 (Figs. 4B and 4D). Taken together, our results indicated that PLY induced expression of ATF3 via TLR4 in pneumococcal infections.
S. pneumoniae stimulated interaction between ATF3 and c-Jun
As previously mentioned, ATF3 contains a leucine zipper domain which is responsible for interaction with other bZiP-containing proteins. ATF3 may act as an activator or repressor since the function of ATF3 depends on its interacting partner protein (Thompson et al., 2009). When ATF3 interacts with c-Jun, it acts as a positive regulator of cytokines (IL-8 and IL-1α) (Aung et al., 2013). During pneumococcal infection, PLY-induced ATF3 increases the production of cytokines, thus providing protection against pneumococcal infections (Nguyen et al., 2014a). In this study, we examined whether ATF3 interacts with c-Jun during pneumococcal infection. The results show that ATF3 and c-Jun were induced by S. pneumoniae. Moreover, ATF3 initiates binding with c-Jun at 1 h post-infection and strongly binds with c-Jun at 3 h post-infection (Fig. 5A). To confirm this observation, immunoprecipitation was performed. When ATF3 antibody was used for immunoprecipitation, it was found that ATF3 interacted with c-Jun (Fig. 5B). Similarly, c-Jun antibody was also used to reconfirm that ATF3 interacts with c-Jun during pneumococcal infection (Fig. 5C). Taken together, these data indicate that S. pneumoniae stimulates ATF3 and c-Jun expression; ATF3 interacts with c-Jun to regulate the expression of cytokines.
Fig. 5.

ATF3 interacts with c-Jun during pneumococcal infection. (A) The RAW 264.7 cells were infected with S. pneumoniae 0, 1, 2, or 3 h, and the cells were stained with anti-ATF3-FITC, anti-cJun-Rhodamine antibodies, and Hoechst dye and visualized using a confocal microscope. The arrows indicate cells with co-localization of ATF3 and cJun. (B, C) The RAW 264.7 cells were infected with S. pneumoniae (MOI = 100) for 0, 1, 2, or 3 h. The cell lysates were immunoprecipitated with anti-ATF3 (B) either anti-cJun (C) antibodies. Cell lysate was used as an input. As a control, normal serum was used instead of either anti-ATF3 or anti-cJun antibodies. The data are representative of two independent experiments.
DISCUSSION
ATF3 plays an important role in the inflammatory response to endotoxic shock. Several studies showed that ATF3 acts as a negative regulator of cytokine expression (Gilchrist et al., 2006). ROS-induced stimulation of ATF3 inhibited cytokine production, which makes the host more susceptible to secondary infections; however, it should be noted that this inhibition protects mice from endotoxic shock (Hoetzenecker et al., 2012). In murine cytomegalovirus (MCMV) infection, ATF3 knock-out mice showed a higher level of IFN-γ, lower hepatic viral load and less liver histopathology than those in wild-type mice (Rosenberger et al., 2008). Therefore, it protected the mice from viral infection, indicating that ATF3 negatively regulated the innate immune response to MCMV infection. In N. gonorrhoeae infection, ATF3 was upregulated by outer membrane proteins via the MAPK pathway, which inhibited IL-6 production in epithelial cells (Calton et al., 2013). In contrast, ATF3 induction by S. pneumoniae stimulated the expression of TNF-α, IL-1β, and IFN-γ cytokines in response to the infection (Nguyen et al., 2014a). However, the mechanism of ATF3 induction by S. pneumoniae infection was poorly understood. In the present study, we reported that ATF3 was induced by the TLR2/4 pathway. Moreover, p-JNK and p-p38 also played a role in the upregulation of ATF3. To our knowledge, this study demonstrates a role of ATF3 in regulating the response of macrophages to pneumococcal infection.
PLY is necessary for ATF3 expression during pneumococcal infection. Gram-negative bacteria-derived endotoxins such as LPS can induce ATF3 via activating the TLR pathway (Gilchrist et al., 2006). Several in vitro studies demonstrated that PLY is recognized by TLR4 (Malley et al., 2003). Our data also indicate that TLR4 was required for ATF3 induction by PLY. In addition, our results show that TLR2 plays a role in ATF3 induction. However, pneumococcal LTA is also known as an activator of TLR2 signaling (Dessing et al., 2008). Thus, further studies are required to define greater role of LTA in TLR2-mediated ATF3 induction.
The JNK/p38 pathway plays an important role in S. pneumoniae infection (Kang et al., 2009). JNK was induced by pneumococcal infection and stimulated IL-8 in response to the infection in epithelial cells (Schmeck et al., 2006). Additionally, p38 kinase was also induced in pneumococci-infected epithelial cells and regulated Cox-2 mediated-inflammation (N’Guessan et al., 2006). We demonstrated that S. pneumoniae induces ATF3 via JNK and p38 signaling pathways. However, ERK signaling is not required for ATF3 induction.
It has previously been reported that c-Jun, a component of AP1 transcription factor, is phosphorylated by JNK in response to nitric oxide induced-apoptosis in neuroblastoma cells (Li et al., 2004) or UV radiation (Kallunki et al., 1996). Studies also showed that pneumococcal infection induces JNK-mediated phosphorylation of c-Jun (Kenzel et al., 2006). In turn, phosphorylated c-Jun stimulates cytokine IL-8, indicating a regulatory role of c-Jun in stimulating the production of cytokine during infection. Previously, ATF3 has been reported to stimulate cytokine production during pneumococcal infection. As mentioned earlier, ATF3 acts as an activator of cytokines in hetero-dimerization with c-Jun. The data presented in this study indicate that S. pneumoniae induces an interaction between ATF3 and c-Jun, thus subsequently stimulating the production of cytokines to combat against pneumococcal infection in macrophages.
Based on the results of this study and previous findings (Nguyen et al., 2014a), we proposed a model for the role of ATF3 during pneumococcal infection in macrophages (Fig. 6). During S. pneumoniae infection, bacteria release PLY into the host environment (Supplementary Fig. S3). Then, PLY interacts with TLR4 to activate MAPK. MAPK signals are required for ATF3 induction. In the nucleus, ATF3 then interacts with the c-Jun and stimulates the production of cytokines in response to pneumococcal infection.
Fig. 6.
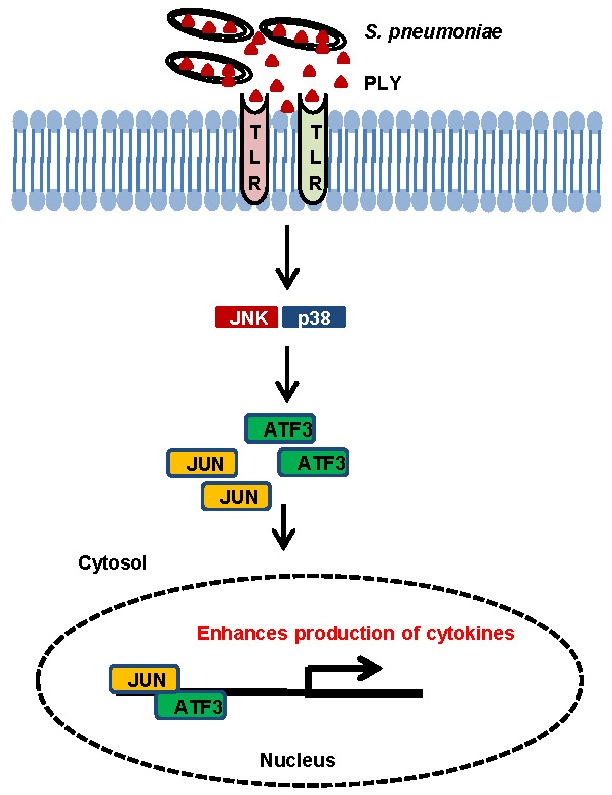
A model for ATF3 action during S. pneumoniae infection. During pneumococcal infection, PLY seems to interact with TLR4 and activates ATF3 via the TLR4/MAPK signaling pathway, which stimulates formation of an ATF3 complex with c-Jun, and this complex binds to promoters of cytokines (TNF-α, IL-1β, IFN-γ), resulting in enhanced cytokine production.
Acknowledgments
This work was supported by the Korea Health Industry Development Institute (KHIDI A08483609-02-0000-100).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Aung H.H., Lame M.W., Gohil K., An C.-I., Wilson D.W., Rutledge J.C. Induction of ATF3 gene network by triglyceride-rich lipoprotein lipolysis products increases vascular apoptosis and inflammation. Arterioscler. Thromb. Vasc. Biol. 2013;33:2088–2096. doi: 10.1161/ATVBAHA.113.301375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basset A., Zhang F., Benes C., Sayeed S., Herd M., Thompson C., Golenbock D.T., Camilli A., Malley R. Toll-like receptor (TLR) 2 mediates inflammatory responses to oligomerized RrgA pneumococcal pilus type 1 protein. J. Biol. Chem. 2012;288:2665–2675. doi: 10.1074/jbc.M112.398875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton K.A., Everson M.P., Briles D.E. A pneumolysin-negative mutant of Streptococcus pneumoniae causes chronic bacteremia rather than acute sepsis in mice. Infect. Immun. 1995;63:448–455. doi: 10.1128/iai.63.2.448-455.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boespflug N.D., Kumar S., McAlees J.W., Phelan J.D., Grimes H.L., Hoebe K., Hai T., Filippi M.-D., Karp C.L. ATF3 is a novel regulator of mouse neutrophil migration. Blood. 2014;123:2084–2093. doi: 10.1182/blood-2013-06-510909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calton C.M., Wade L.K., So M. Upregulation of ATF3 inhibits expression of the pro-inflammatory cytokine IL-6 during Neisseria gonorrhoeae infection. Cell. Microbiol. 2013;15:1837–1850. doi: 10.1111/cmi.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I.H., Shim J.H., Kim S.W., Kim S.N., Pyo S.N., Rhee D.K. Limited stress response in Streptococcus pneumoniae. Microbiol. Immunol. 1999;43:807–812. doi: 10.1111/j.1348-0421.1999.tb02474.x. [DOI] [PubMed] [Google Scholar]
- Dessing M.C., Schouten M., Draing C., Levi M., von Aulock S., van der Poll T. Role played by Toll-like receptors 2 and 4 in lipoteichoic acid-induced lung inflammation and coagulation. J. Infect. Dis. 2008;197:245–252. doi: 10.1086/524873. [DOI] [PubMed] [Google Scholar]
- File T.M., Jr. Streptococcus pneumoniae and community-acquired pneumonia: a cause for concern. Am. J. Med. 2004;117:39–50. doi: 10.1016/j.amjmed.2004.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filén S., Ylikoski E., Tripathi S., West A., Björkman M., Nyström J., Ahlfors H., Coffey E., Rao K.V.S., Rasool O., et al. Activating transcription factor 3 is a positive regulator of human IFNG gene expression. J. Immunol. 2010;184:4990–4999. doi: 10.4049/jimmunol.0903106. [DOI] [PubMed] [Google Scholar]
- Gilchrist M., Thorsson V., Li B., Rust A.G., Korb M., Kennedy K., Hai T., Bolouri H., Aderem A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- Gilchrist M., Henderson W.R., Clark A.E., Simmons R.M., Ye X., Smith K.D., Aderem A. Activating transcription factor 3 is a negative regulator of allergic pulmonary inflammation. J. Exp. Med. 2008;205:2349–2357. doi: 10.1084/jem.20072254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist M., Henderson W.R., Jr, Morotti A., Johnson C.D., Nachman A., Schmitz F., Smith K.D., Aderem A. A key role for ATF3 in regulating mast cell survival and mediator release. Blood. 2010;115:4734–4741. doi: 10.1182/blood-2009-03-213512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T., Wolfgang C., Marsee D., Allen A., Sivaprasad U. ATF3 and stress responses. Gene Expr. 1999;7:321–335. [PMC free article] [PubMed] [Google Scholar]
- Hoetzenecker W., Echtenacher B., Guenova E., Hoetzenecker K., Woelbing F., Bruck J., Teske A., Valtcheva N., Fuchs K., Kneilling M., et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat. Med. 2012;18:128–134. doi: 10.1038/nm.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyams C., Camberlein E., Cohen J.M., Bax K., Brown J.S. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect. Immun. 2010;78:704–715. doi: 10.1128/IAI.00881-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr. Opin. Immunol. 2007;19:39–45. doi: 10.1016/j.coi.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Kallunki T., Deng T., Hibi M., Karin M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell. 1996;87:929–939. doi: 10.1016/s0092-8674(00)81999-6. [DOI] [PubMed] [Google Scholar]
- Kang E.H., Gebru E., Kim M.H., Cheng H., Park S.-C. EstA protein, a novel virulence factor of Streptococcus pneumoniae, induces nitric oxide and pro-inflammatory cytokine production in RAW 264.7 macrophages through NF-κB/MAPK. Microb. Pathog. 2009;47:196–201. doi: 10.1016/j.micpath.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Kenzel S., Mancuso G., Malley R., Teti G., Golenbock D.T., Henneke P. c-Jun kinase is a critical signaling molecule in a neonatal model of group B streptococcal sepsis. J. Immunol. 2006;176:3181–3188. doi: 10.4049/jimmunol.176.5.3181. [DOI] [PubMed] [Google Scholar]
- Khuu C.H., Barrozo R.M., Hai T., Weinstein S.L. Activating transcription factor 3 (ATF3) represses the expression of CCL4 in murine macrophages. Mol. Immunol.; 2007. pp. 1598–1605. [DOI] [PubMed] [Google Scholar]
- Koppe U., Suttorp N., Opitz B. Recognition of Streptococcus pneumoniae by the innate immune system. Cell Microbiol. 2012;14:460–466. doi: 10.1111/j.1462-5822.2011.01746.x. [DOI] [PubMed] [Google Scholar]
- Lai P.-F., Cheng C.-F., Lin H., Tseng T.-L., Chen H.-H., Chen S.-H. ATF3 protects against LPS-induced inflammation in mice via inhibiting HMGB1 expression. Evid. Based Complement. Alternat. Med. 2013;2013:716481. doi: 10.1155/2013/716481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Feng Z., Porter A.G. JNK-dependent phosphor-ylation of c-Jun on serine 63 mediates nitric oxide-induced apoptosis of neuroblastoma cells. J. Biol. Chem. 2004;279:4058–4065. doi: 10.1074/jbc.M310415200. [DOI] [PubMed] [Google Scholar]
- Lu D., Chen J., Hai T. The regulation of ATF3 gene expression by mitogen-activated protein kinases. Biochem. J. 2007;401:559–567. doi: 10.1042/BJ20061081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malley R., Henneke P., Morse S.C., Cieslewicz M.J., Lipsitch M., Thompson C.M., Kurt-Jones E., Paton J.C., Wessels M.R., Golenbock D.T. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. USA. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeela E.A., Burke Á., Neill D.R., Baxter C., Fernandes V.E., Ferreira D., Smeaton S., El-Rachkidy R., McLoughlin R.M., Mori A., et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010;6:e1001191. doi: 10.1371/journal.ppat.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N’Guessan P.D., Hippenstiel S., Etouem M.O., Zahlten J., Beermann W., Lindner D., Opitz B., Witzenrath M., Rosseau S., Suttorp N., et al. Streptococcus pneumoniae induced p38 MAPK- and NF-κB-dependent COX-2 expression in human lung epithelium. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;290:L1131–L1138. doi: 10.1152/ajplung.00383.2005. [DOI] [PubMed] [Google Scholar]
- Nguyen C.T., Kim E.H., Luong T.T., Pyo S., Rhee D.-K. ATF3 confers resistance to pneumococcal infection through positive regulation of cytokine production. J. Infect. Dis. 2014a;210:1745–1754. doi: 10.1093/infdis/jiu352. [DOI] [PubMed] [Google Scholar]
- Nguyen C.T., Le N.-T., Tran T.D.-H., Kim E.-H., Park S.-S., Luong T.T., Chung K.-T., Pyo S., Rhee D.-K. S. pneumoniae ClpL modulates adherence to A549 human lung cells through Rap1/Rac1 activation. Infect. Immun. 2014b;82:3802–3810. doi: 10.1128/IAI.02012-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger C.M., Clark A.E., Treuting P.M., Johnson C.D., Aderem A. ATF3 regulates MCMV infection in mice by modulating IFN-gamma expression in natural killer cells. Proc. Natl. Acad. Sci. USA. 2008;105:2544–2549. doi: 10.1073/pnas.0712182105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeck B., Moog K., Zahlten J., van Laak V., N’Guessan P., Opitz B., Rosseau S., Suttorp N., Hippenstiel S. Streptococcus pneumoniae induced c-Jun-N-terminal kinaseand AP-1 -dependent IL-8 release by lung epithelial BEAS-2B cells. Respir. Res. 2006;7:98. doi: 10.1186/1465-9921-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder N.W.J., Morath S., Alexander C., Hamann L., Hartung T., Zähringer U., Göbel U.B., Weber J.R., Schumann R.R. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J. Biol. Chem. 2003;278:15587–15594. doi: 10.1074/jbc.M212829200. [DOI] [PubMed] [Google Scholar]
- Thompson M., Xu D., Williams B. ATF3 transcription factor and its emerging roles in immunity and cancer. J. Mol. Med. 2009;87:1053–1060. doi: 10.1007/s00109-009-0520-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu L.N., Jeong H.-Y., Kwon H.-Y., Ogunniyi A.D., Paton J.C., Pyo S.-N., Rhee D.-K. Modulation of adherence, invasion, and tumor necrosis factor alpha secretion during the early stages of infection by Streptococcus pneumoniae ClpL. Infect. Immun. 2007;75:2996–3005. doi: 10.1128/IAI.01716-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartha F., Beiter K., Albiger B., Fernebro J., Zychlinsky A., Normark S., Henriques-Normark B. Capsule and dalanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell. Microbiol. 2007;9:1162–1171. doi: 10.1111/j.1462-5822.2006.00857.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.