

Abstract
Anticholinergics, blocking the muscarinic M3 receptor, are effective bronchodilators for patients with chronic obstructive pulmonary disease. Recent evidence from M3 receptor-deficient mice (M3R−/−) indicates that M3 receptors also regulate neutrophilic inflammation in response to cigarette smoke (CS). M3 receptors are present on almost all cell types, and in this study we investigated the relative contribution of M3 receptors on structural cells vs. inflammatory cells to CS-induced inflammation using bone marrow chimeric mice. Bone marrow chimeras (C56Bl/6 mice) were generated, and engraftment was confirmed after 10 wk. Thereafter, irradiated and nonirradiated control animals were exposed to CS or fresh air for four consecutive days. CS induced a significant increase in neutrophil numbers in nonirradiated and irradiated control animals (4- to 35-fold). Interestingly, wild-type animals receiving M3R−/− bone marrow showed a similar increase in neutrophil number (15-fold). In contrast, no increase in the number of neutrophils was observed in M3R−/− animals receiving wild-type bone marrow. The increase in keratinocyte-derived chemokine (KC) levels was similar in all smoke-exposed groups (2.5- to 5.0-fold). Microarray analysis revealed that fibrinogen-α and CD177, both involved in neutrophil migration, were downregulated in CS-exposed M3R−/− animals receiving wild-type bone marrow compared with CS-exposed wild-type animals, which was confirmed by RT-qPCR (1.6–2.5 fold). These findings indicate that the M3 receptor on structural cells plays a proinflammatory role in CS-induced neutrophilic inflammation, whereas the M3 receptor on inflammatory cells does not. This effect is probably not mediated via KC release, but may involve altered adhesion and transmigration of neutrophils via fibrinogen-α and CD177.
Keywords: nonneuronal acetylcholine, anticholinergics, neutrophil adhesion
acetylcholine is the primary parasympathetic neurotransmitter in the airways. It induces bronchoconstriction and mucus secretion via M3 receptors (16). Cholinergic tone is increased in patients with chronic obstructive pulmonary diseases (COPD), and this is the major reversible component of airflow obstruction in COPD (1, 12). Therefore, anticholinergics, which block M3 receptors, are effective bronchodilators for patients with COPD (1).
In addition to inducing bronchoconstriction and mucus secretion, acetylcholine has been shown to affect airway inflammation (16), which is a hallmark feature of COPD (2). In animal models of COPD, pretreatment with anticholinergics inhibits cigarette smoke-induced and lipopolysaccharide (LPS)-induced neutrophilic inflammation (19, 27). More recently, we demonstrated that these proinflammatory effects of acetylcholine are mediated via M3 receptors. Total knockout of the M3 receptor or inhibition of the M3 receptor by a pharmacological approach using the muscarinic antagonist 1,1-dimethyl-4-diphenylacetoxypiperidinium iodide (4-DAMP), which is selective for M3 receptors over M2 receptors, prevented cigarette smoke-induced neutrophilic inflammation in an early smoke-induced inflammation model in mice (15). Together, these data suggest a proinflammatory role for acetylcholine via M3 receptors.
M3 receptors are expressed abundantly throughout the airways, and almost all cell types express M3 receptors. This includes structural cells, such as airway smooth muscle, epithelial, and endothelial cells, but also inflammatory cells, such as macrophages and neutrophils (11). From in vitro studies it is known that muscarinic receptor stimulation of both structural and inflammatory cells might contribute to the proinflammatory effects of acetylcholine. For instance, stimulation of airway smooth muscle cells and epithelial cells with methacholine or acetylcholine induces the release of IL-8 (10, 20). Furthermore, acetylcholine activates human monocytes and alveolar macrophages to promote neutrophil migration, which is also observed when using macrophages from COPD patients (3, 25). It is not known whether the proinflammatory effects of acetylcholine observed in vivo are primarily mediated via M3 receptors on structural cells or via M3 receptors on inflammatory cells.
Therefore, in this study, we investigated the relative contribution of the M3 receptor on structural cells vs. inflammatory cells to cigarette smoke-induced inflammation. To distinguish between structural cells and inflammatory cells, bone marrow chimeric mice were generated. Here, we demonstrate that the proinflammatory effects of acetylcholine on neutrophilic inflammation are primarily mediated via M3 receptors on structural cells, which may involve altered adhesion and transmigration of neutrophils.
MATERIALS AND METHODS
Animals.
Homozygous, inbred, specific pathogen-free breeding colonies of muscarinic M3 receptor-deficient (M3R−/−) mice and C57Bl/6NTac wild-type mice with the same genetic background (CD45.2) were obtained from Taconic (Cambridge City, IN). The M3R−/− mice were on a 129 Sv/J background and had been backcrossed for at least 10 generations onto the C57Bl/6NTac background (8). CD45.1 congenic C57Bl/6 mice were obtained from Jackson Laboratories (Bar Harbor, ME). All animals were maintained in our own facility. Animals were housed conventionally under a 12:12-h light-dark cycle and received food and water ad libitum. All experiments were performed in accordance with the national guidelines and approved by the University of Groningen Committee for Animal Experimentation (No.: 5463G).
Generation of bone marrow chimeras.
Bone marrow cells were collected from femurs and tibia of M3R−/− mice and wild-type CD45.2 or CD45.1 mice and retro-orbitally injected into irradiated male mice (9 Gy) within 24 h after irradiation. Per mouse, 8 × 106 bone marrow cells were transplanted. Nonirradiated wild-type animals were used as additional controls; see Table 1 for an overview of the groups. After 10 wk, engraftment was analyzed by flow cytometry of peripheral blood. Seventy microliters of heparinized peripheral blood were collected 10 wk after bone marrow transplantation and at the end of the study when the mice were killed. Red blood cells were lysed, and the remaining cells were stained with antibodies directed against the following markers: CD45.2-PE (CD45.2), CD45.1-Pacific Blue (CD45.1), CD3-APC (T cells), B220-FITC (B cells), GR-1-PE/Cy7, and Mac1-PE/Cy7 (monocytes and granulocytes, respectively) (Biolegend, San Diego, CA). The stained cells were analyzed with a LSR-II flow cytometer (BD Biosciences, San Diego, CA). Engraftment was calculated as a percentage of CD45.1 vs. CD45.2 cells. Engraftment was high in all animals (>90% donor-derived cells in all lineages), and mice were included in the study 3 mo after bone marrow reconstitution (Fig. 1).
Table 1.
Overview of the experimental groups included in this study
| Background | Recipient | Irradiation | Donor | Groups |
|---|---|---|---|---|
| CD45.1 | CD45.1 | − | − | Nonirradiated control |
| CD45.1 | + | CD45.2 | Irradiated control | |
| CD45.1 | + | M3R−/− | M3R inflammatory cells | |
| CD45.2 | CD45.2 | − | − | Nonirradiated control |
| CD45.2 | + | CD45.1 | Irradiated control | |
| M3R−/− | + | CD45.1 | M3R structural cells |
M3R−/−, M3 receptor-deficient; −, without; +, with; M3R, M3 receptor.
Fig. 1.
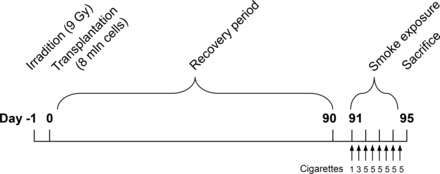
Experimental protocol. Chimeric animals were generated by sublethal irradiation (9 Gy) of recipient male C57Bl/6 mice (n = 5–12) and subsequent transplantation of 8 × 106 donor bone marrow cells via retroorbital injection within 24 h after irradiation. To allow for complete engraftment, animals were included in the protocol 3 mo after bone marrow reconstitution. Animals were exposed to cigarette smoke two times daily on 4 consecutive days by whole body exposure. Sixteen hours after the last smoke exposure, a bronchoalveolar lavage was performed, and lungs were harvested for lung tissue homogenates.
Cigarette smoke exposure.
Mice (n = 5–12/group) were exposed to cigarette smoke from Kentucky 3R4F research cigarettes (Tobacco Research Institute, University of Kentucky, Lexington, KY) on four consecutive days by whole body exposure in an early smoke-induced inflammation model, as described previously (15). Mice were exposed to mainstream smoke of one cigarette in the morning and three cigarettes in the afternoon on the first day. On day 2 to 4, mice were exposed to five cigarettes in the morning and five in the afternoon (Fig. 1). Each cigarette was smoked without a filter in 5 min at a rate of 5 l/h in a ratio with 60 l/h air using a peristaltic pump (45 rpm, Watson Marlow 323 E/D, Rotterdam, The Netherlands). Control animals were handled in the same way but instead exposed to fresh air only. Sixteen hours after the last CS exposure, animals were killed by intraperitoneal pentobarbital injection (400 mg/kg, hospital pharmacy, University Medical Center Groningen), after which the lungs were immediately lavaged, resected, and snap-frozen in liquid nitrogen. Bronchoalveolar lavage (BAL) fluid was stored at −20°C, and lung tissue was stored at −80°C.
Analysis of BAL cells and cytokines.
A BAL was performed as described previously (15). Briefly, lungs were lavaged five times with 1 ml PBS. The first fraction was used for measurement of cytokines by ELISA. Levels of keratinocyte-derived chemokine (KC), IL-6, and monocyte chemotactic protein-1 (MCP-1) were determined by a multiplex assay (R&D Systems, Minneapolis, MN). Total cell numbers were determined, and cytospins were prepared and stained with May-Grünwald and Giemsa (both Sigma, St. Louis, MO) after which a differential cell count was performed by counting at least 400 cells in duplicate in a blinded fashion.
Analysis of gene expression in lung tissue.
Total RNA was extracted from lung tissue (right superior lobe) using the RNeasy mini kit (Qiagen, Venlo, The Netherlands), as described previously (15). Equal amounts of total mRNA were then reverse transcribed, and cDNA was subjected to real-time qPCR (Westburg, Leusden, The Netherlands) or to microarray analysis using an Illumina chip and GeneSpring software. The specific forward and reverse primers used for real-time qPCR are listed in Table 2.
Table 2.
Primers used for qRT-PCR analysis
| Gene | Primer Sequence | NCBI Accession No. |
|---|---|---|
| KC | Forward: GCTGGGATTCACCTCAAGAA | NM_008176.3 |
| Reverse: AGGTGCCATCAGAGCAGTCT | ||
| IL-6 | Forward: CCGGAGAGGAGACTTCACAG | NM_031168.1 |
| Reverse: TCCACGATTTCCCAGAGAAC | ||
| MIP-2 | Forward: AAGTTTGCCTTGACCCTGAA | NM_009140.2 |
| Reverse: AGGCACATCAGGTACGATCC | ||
| LIX | Forward: GAAAGCTAAGCGGAATGCAC | NM_009141.3 |
| Reverse: GGGACAATGGTTTCCCTTTT | ||
| Fibrinogen α | Forward: AACTTCAGGTGCCCAAAATG | NM_001111048.1 |
| Reverse: AACTGAGCCCCTCAGAGTGA | ||
| CD177 | Forward: GGGAGGTGGACTGACTACCA | NM_026862.3 |
| Reverse: GGGTTCAGCAGAGAGGACAG | ||
| 18S | Forward: AAACGGCTACCACATCCAAG | NR_003278 |
| Reverse: CCTCCAATGGATCCTCGTTA |
KC, keratinocyte-derived chemokine, the mouse ortholog of IL-8, also known as CXCL1 in mice; MIP-2, macrophage inflammatory protein-2, also known as CXCL2; LIX, lipopolysaccharide-induced CXC chemokine, also known as CXCL5.
Statistical analysis.
Data are presented as means ± SE. Statistical differences between means were calculated using one- or two-way ANOVA, followed by Newman Keuls multiple-comparison tests, or by a Student's t-test with two-tailed distribution where appropriate. Differences were considered significant at P < 0.05.
RESULTS
Analysis of engraftment.
To confirm that the bone marrow transplantation was successful, engraftment was analyzed after 10 wk and at the day of death of the mice. As depicted in Fig. 2, after 10 wk engraftment was high, and this was not different between the experimental groups (data not shown). Therefore, all animals were included in the study 3 mo after bone marrow reconstitution. Engraftment was even further enhanced at the day of death. Engraftment was as follows: total cells 97.1% (ranging from 94.2 to 99.0%), monocytes 99.4% (97.5 to 100%), granulocytes 99.8% (97.8 to 100%), B cells 99.9% (99.3 to 100%), and T cells 90.6% (81.7 to 97.4%) (Fig. 2).
Fig. 2.
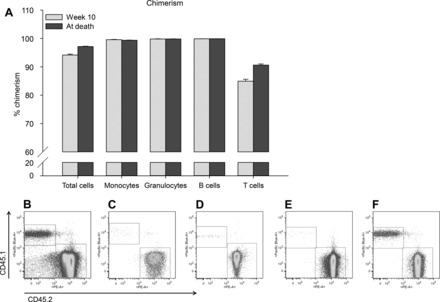
Engraftment of bone marrow cells. Engraftment was analyzed in peripheral blood of mice 10 wk after the bone marrow transplantation and at the day of death. Average engraftment levels (A) and representative FACS images for total single cells (B), monocytes (C), granulocytes (D), B cells (E), and T cells (F) at the day of death are shown; n = 68 animals.
Contribution of the M3 receptor on inflammatory cells.
To study the contribution of the M3 receptor on inflammatory cells to early cigarette smoke-induced airway inflammation, inflammatory cells in the lavage fluid of CD45.1 animals were determined after exposure to cigarette smoke. The following groups were compared: CD45.1 nonirradiated control animals, irradiated CD45.1 control animals receiving wild-type (CD45.2) bone marrow cells, and irradiated CD45.1 animals receiving M3R−/− (CD45.2) bone marrow cells (Table 1). Cigarette smoke exposure had no significant effect on macrophage and lymphocyte numbers in all groups (Fig. 3). Cigarette smoke induced a significant increase in neutrophil numbers in nonirradiated (28-fold; Fig. 3A) and irradiated control animals receiving wild-type bone marrow cells (18-fold; Fig. 3B). Interestingly, wild-type animals receiving M3R−/− bone marrow cells showed a similar increase in neutrophil number (15-fold; Fig. 3C), suggesting that the M3 receptor on inflammatory cells is not involved in cigarette smoke-induced neutrophilic inflammation.
Fig. 3.
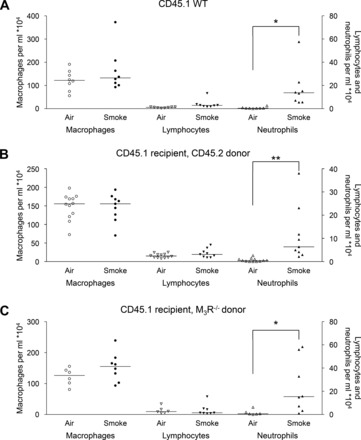
Inflammatory cells in the lavage fluid of CD45.1 animals. Mice were treated as described in Fig. 1. Sixteen hours after the last smoke exposure a bronchoalveolar lavage was performed, and macrophages, lymphocytes, and neutrophils were determined in the lavage fluid of nonirradiated wild-type (WT) animals (A), wild-type animals receiving wild-type bone marrow cells (B), and wild-type animals receiving M3 receptor-deficient (M3R−/−) bone marrow cells (C). *P < 0.05 and **P < 0.01; n = 6–12 animals.
Contribution of the M3 receptor on structural cells.
To study the contribution of the M3 receptor on structural cells to early cigarette smoke-induced airway inflammation, inflammatory cells in the lavage fluid of CD45.2 animals were determined after exposure to cigarette smoke (Fig. 4). The following groups were compared: CD45.2 nonirradiated control animals, irradiated CD45.2 control animals receiving CD45.1 bone marrow cells, and irradiated M3R−/− (CD45.2) animals receiving CD45.1 bone marrow cells (Table 1). Cigarette smoke induced a small increase in macrophage numbers in the lavage fluid in all groups (1.3- to 1.4-fold). No increase in lymphocyte numbers was observed. Cigarette smoke induced a significant increase in neutrophil numbers in nonirradiated (8-fold; Fig. 4A) and irradiated control animals receiving wild-type bone marrow cells (4-fold; Fig. 4B). In contrast, no increase in the number of neutrophils was observed in M3R−/− animals receiving wild-type bone marrow (Fig. 4C), suggesting that the M3 receptor on structural cells is involved in cigarette smoke-induced neutrophilic inflammation.
Fig. 4.
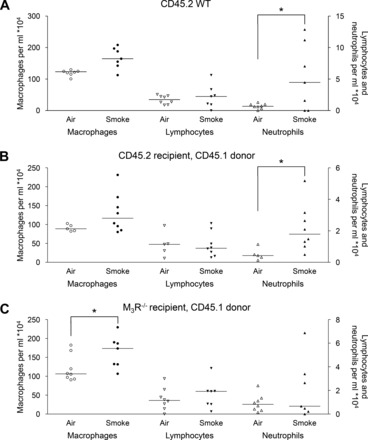
Inflammatory cells in the lavage fluid of CD45.2 animals. Mice were treated as described in Fig. 1. Sixteen hours after the last smoke exposure a bronchoalveolar lavage was performed, and macrophages, lymphocytes, and neutrophils were determined in the lavage fluid of nonirradiated wild-type animals (A), wild-type animals receiving wild-type bone marrow cells (B), and M3R−/− animals receiving wild-type bone marrow cells (C). *P < 0.05; n = 5–8 animals.
Cigarette smoke-induced cytokine release.
Subsequently, gene expression of KC (CXCL1), the mouse ortholog of IL-8 (CXCL8), was determined in lung homogenates. Cigarette smoke induced an increase in KC. Remarkably, this increase was similar in all bone marrow-transplanted groups (2.8- to 5.0-fold), as depicted in Fig. 5. Similar findings were observed at the protein level in the lavage fluid. Of note, other neutrophil chemoattractants, including macrophage inflammatory protein-2 (MIP-2, CXCL2) and LPS-induced CXC chemokine (LIX, CXCL5), were found increased in response to cigarette smoke in the CD45.1 groups only, with no interaction of genotype, whereas no increase in IL-6 or MCP-1 expression in response to cigarette smoke was observed in all strains (data not shown).
Fig. 5.
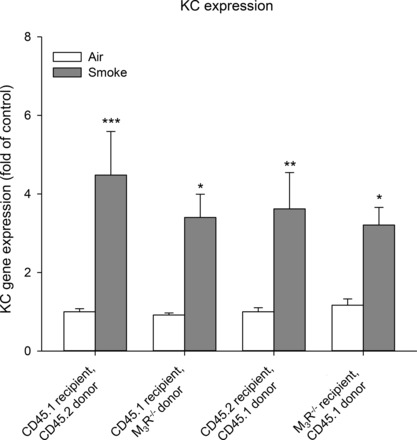
Keratinocyte-derived chemokine (KC) expression after cigarette smoke exposure. Mice were treated as described in Fig. 1. Sixteen hours after the last smoke exposure lungs were collected. Gene expression of KC in lung homogenates was determined. Results are expressed as means ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001; n = 5–12 animals.
Neutrophil adhesion.
These observations suggest that M3 receptors on structural cells do not mediate their effect via altered KC, MIP-2, or LIX production. Therefore, we considered the possibility that neutrophil adhesion might be altered after knockout of the M3 receptor on structural cells. We analyzed the expression of genes known to play a role in this process. In the airways, P-selectin, E-selectin, and L-selectin are involved in rolling of neutrophils, whereas intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, β2-integrin, and integrin-α4β1 [very late antigen-4 (VLA4)] play a role in adhesion and transmigration of neutrophils (17). However, we were not able to demonstrate regulation of these markers at the gene expression level in whole lung homogenates. Thus, no increase was observed in wild-type animals receiving wild-type bone marrow cells, nor were there any differences compared with M3R−/− animals receiving wild-type bone marrow cells (data not shown). To be able to explain the observed effects, we proceeded with a microarray gene expression analysis on whole lung homogenates of wild-type animals receiving wild-type bone marrow cells and of M3R−/− animals receiving wild-type bone marrow cells. We selected genes that were upregulated or downregulated by >1.5-fold in cigarette smoke-exposed M3R−/− animals receiving wild-type bone marrow cells compared with cigarette smoke-exposed wild-type animals receiving wild-type bone marrow cells (Table 3). Three genes were upregulated, and 12 were downregulated. Genes that were shown to have a link with neutrophilic inflammation according to the literature were analyzed by real-time qPCR. Downregulation of fibrinogen-α and CD177 was confirmed, which was lower in M3R−/− animals compared with wild-type animals, and not influenced by smoke exposure (Fig. 6). Interestingly, both genes are known to play a role in neutrophil adhesion and transmigration.
Table 3.
Genes that were upregulated or downregulated by >1.5-fold in cigarette smoke-exposed M3R−/− animals receiving wild-type bone marrow cells compared with cigarette smoke-exposed wild-type animals receiving wild-type bone marrow cells
| Gene | Definition | Regulation |
|---|---|---|
| AKR1C19 | Aldo-keto reductase family 1, member C19 | +2.0 |
| TNXB* | Tenascin XB | +2.0 |
| ERAF* | Erythroid associated factor | +1.7 |
| FGG* | Fibrinogen-γ chain | −2.1 |
| FGA* | Fibrinogen-α chain | −1.9 |
| RETNLA* | Resistin like-α | −1.9 |
| CHI3L3 | Chitinase 3-like 3 | −1.8 |
| LTF* | Lactotransferrin | −1.7 |
| CLCA3* | Chloride channel calcium-activated 3 | −1.6 |
| LOC100048554 | Similar to monocyte chemoattractant protein-2 | −1.6 |
| OTTMUSG00000000971 | Unknown | −1.6 |
| CD177* | CD177 antigen | −1.5 |
| EAR5 | Eosinophil-associated ribonuclease 5 | −1.5 |
| MFSD2 | Major facilitator superfamily domain-containing 2 | −1.5 |
| SLC26A4* | Pendrin; solute carrier family 26, member 4 | −1.5 |
Genes analyzed by real-time qPCR.
Fig. 6.
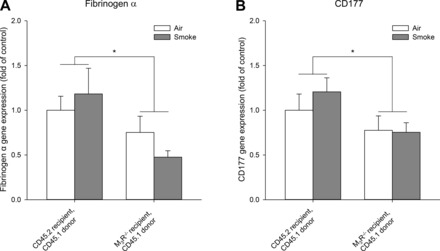
Fibrinogen-α and CD177 expression after cigarette smoke exposure. Mice were treated as described in Fig. 1. Sixteen hours after the last smoke exposure lungs were collected. Microarray analysis on lung homogenates revealed that fibrinogen-α and CD177 were downregulated after smoke exposure in M3R−/− animals receiving wild-type bone marrow cells compared with wild-type animals. Therefore, gene expression of fibrinogen-α (A) and CD177 (B) was determined by real-time qPCR. Results are expressed as means ± SE. *P < 0.05; n = 5–8 animals.
DISCUSSION
In this study, we demonstrated that the M3 receptor on structural cells plays a proinflammatory role in cigarette smoke-induced inflammation. In M3R−/− mice receiving wild-type bone marrow cells, neutrophilic inflammation in response to cigarette smoke exposure is prevented. In contrast, cigarette smoke-induced neutrophilic inflammation is still observed after knockout of the M3 receptor on inflammatory cells, suggesting that the proinflammatory effects of acetylcholine are primarily mediated via M3 receptors on structural cells. This was not regulated via inhibition of cytokine release, which was comparable in all groups, but might involve altered adhesion and transmigration of neutrophils via fibrinogen-α and CD177.
Neutrophils play a central role in COPD, and previous studies have shown that acetylcholine regulates neutrophilic inflammation via M3 receptors. Inhibition of muscarinic receptors by anticholinergics, including tiotropium, glycopyrrolate, and aclidinium, prevented neutrophilic inflammation in response to cigarette smoke exposure or LPS exposure in guinea pigs and mice (7, 19, 23, 27). Because long-acting anticholinergics are kinetically selective for M3 receptors, these findings suggest the involvement of this specific muscarinic receptor subtype. A recent study from our lab using M3R−/− mice showed that the proinflammatory effects of acetylcholine are indeed mediated via the M3 receptor. We demonstrated that cigarette smoke-induced neutrophilic inflammation was prevented in M3R−/− mice, whereas opposite effects were observed in M1R−/− and M2R−/− mice (15). In the present study we have extended these findings by demonstrating that the proinflammatory effects of acetylcholine are primarily mediated via M3 receptors on structural cells in an early smoke-induced inflammation model in mice. The involvement of the M3 receptor on structural cells in the inflammatory response is also supported by evidence from in vitro studies. Methacholine and cigarette smoke-induced IL-8 release from airway smooth muscle cells can be inhibited by the M3 antagonists 4-DAMP and DAU-5884 (10). Moreover, acetylcholine-induced IL-8 release from bronchial epithelial cells can be inhibited by 4-DAMP (20). Strikingly, acetylcholine-induced effects on inflammatory cells were also shown to be mediated via M3 receptors. 4-DAMP inhibited acetylcholine-induced neutrophil chemotactic activity from macrophages (22), as well as neutrophil chemotaxis from LPS-activated macrophages from COPD patients (25). These in vitro studies suggest a direct effect of acetylcholine on both structural cells and inflammatory cells. Apparently, the observed effects of acetylcholine on inflammatory cells in vitro are not relevant to the in vivo situation, as we demonstrate that cigarette smoke-induced inflammation is still observed after knockout of the M3 receptor on inflammatory cells.
Although there was no increase in neutrophil numbers in M3R−/− animals receiving wild-type bone marrow cells after exposure to cigarette smoke compared with wild-type animals, the increase in the neutrophil chemoattractant KC was similar to the increase observed in wild-type animals. This suggests that neutrophil recruitment is altered after knockout of the M3 receptor. Neutrophil recruitment is a complex cellular process that occurs through a cascade of multiple steps, including tethering, rolling, adhesion, crawling, and subsequent transmigration of neutrophils (17). An initial screen on genes known to be involved in the process of neutrophil recruitment, including selectins, ICAM-1, VCAM-1, β2-integrin, and VLA4, could not explain the observed increase in KC in M3R−/− animals receiving wild-type bone marrow cells, without an increase in neutrophil numbers. However, with the use of a microarray analysis, two genes that were recently linked to the process of neutrophil recruitment, fibrinogen-α and CD177 (5, 21), were shown to be downregulated in M3R−/− animals compared with wild-type animals.
Fibrinogen-α, besides its role in blood hemostasis, regulates the adhesive behavior of neutrophils. Fibrinogen can bind to β2-integrins and thereby alter the recruitment of neutrophils (5, 9). Neutrophil recruitment in response to platelet-activating factor is altered in fibrinogen-α knockout mice. Specifically, the number of rolling neutrophils and the number of adhering neutrophils was decreased in fibrinogen-α knockout mice compared with wild-type mice (5). Therefore, a reduction in fibrinogen-α levels in M3R−/− mice might affect neutrophil recruitment in response to cigarette smoke exposure by reducing the rolling and adhesion of neutrophils.
CD177 is a neutrophil-specific antigen expressed by a subpopulation of neutrophils. CD177 can bind to platelet endothelial cell adhesion molecule (PECAM)-1 and thereby alter the recruitment of neutrophils (21, 24). PECAM-1 is known to be involved in neutrophil transmigration (17). In vitro, transmigration of neutrophils through endothelial cells in response to IL-8 or N-formyl-methionyl-leucyl-phenylalanine was inhibited after inhibition of the CD177-PECAM-1 interaction (21). Interestingly, the same study showed that CD177-positive neutrophils transmigrated more efficiently than CD177-negative neutrophils, indicating the potential relevance of CD177 for transmigration. These findings were confirmed by Kuckleburg et al., who demonstrated that inhibition of CD177 or inhibition of the interaction between CD177 and PECAM-1 inhibits neutrophil transmigration under both static and flow conditions (18). Therefore, a reduction in CD177 levels in M3R−/− mice might affect neutrophil recruitment in response to cigarette smoke exposure by reducing the transmigration of neutrophils.
The gene expression of neither fibrinogen-α nor CD177 was regulated by cigarette smoke, indicating that this is a constitutive difference between the wild-type and M3R−/− mice. Expression levels of both genes were lower in the M3R−/− strain compared with wild-type mice, which further supports the observation that neutrophillic inflammation is altered in M3R−/− mice receiving wild-type bone marrow and not in wild-type animals receiving M3R−/− bone marrow. Interestingly, both genes were recently identified as potential inflammatory biomarkers for COPD. Fibrinogen was identified as a biomarker in the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints study (6), and an elevated level of fibrinogen was independently and significantly associated with mortality in patients with COPD (4). CD177 levels were increased in the airways of Cynomolgus monkeys after ozone challenge (14), indicating the potential relevance of both genes for COPD pathophysiology. It is beyond the scope of the current manuscript to investigate the functional roles of CD177 and fibrinogen-α in detail; however, our results and the above-mentioned clinical data certainly provide strong rationale for future studies in this area.
The fact that KC levels were increased in M3R−/− mice receiving wild-type bone marrow cells in our study also has other implications. Previously, we have shown that increased KC release in response to cigarette smoke exposure is prevented after total knockout of the M3 receptor (15). KC can be released from airway structural cells, including airway smooth muscle cells and epithelial cells, and from inflammatory cells, including macrophages. The finding that KC release is prevented after total knockout of the M3 receptor, but not after knockout of the M3 receptor on structural cells only, suggests a role for inflammatory cells in this response. The suggestion that acetylcholine induces the release of KC from inflammatory cells is supported by in vitro studies showing enhanced neutrophil recruitment after stimulation of macrophages with acetylcholine (22, 25). However, knockout of the M3 receptor only on inflammatory cells does not prevent neutrophilic inflammation, whereas knockout of the M3 receptor on structural cells does. Therefore, the contribution of the M3 receptor on inflammatory cells to the proinflammatory effect of acetylcholine is only small, and this is mainly dependent on M3 receptors on structural cells.
Acetylcholine is not only released as a neurotransmitter but can also be released from nonneuronal cells. Almost all cell types in the airways have been shown to express synthesizing enzymes for acetylcholine, suggesting acetylcholine production (26). It has been proposed that this nonneuronal acetylcholine might contribute to inflammation, although direct evidence is still limited (13, 16). Results from our study indicate that nonneuronal acetylcholine acting on inflammatory cells does not significantly contribute to the proinflammatory effects of acetylcholine. There might be a role for nonneuronal acetylcholine released from airway structural cells, including epithelial cells; however, future studies are needed to investigate this in more detail.
In conclusion, we demonstrate that acetylcholine regulates inflammation via M3 receptors on structural cells. This emphasizes the important role of airway structural cells in neutrophilic inflammation and the pathophysiology of COPD, and the contribution of acetylcholine to this response. Whether anticholinergic therapy also affects inflammation in patients with COPD still needs to be elucidated, but our results indicate that this might be an additional beneficial effect of M3 selective anticholinergics.
GRANTS
We thank the Netherlands Lung Foundation for financial support (Grant No.: 3.2.08.014).
DISCLOSURES
P. S. Hiemstra, H. Meurs, H. A. Kerstjens, and R. Gosens have received research grants from Boehringer Ingelheim.
AUTHOR CONTRIBUTIONS
L.E.M.K., R.P.v.O., M.N.H., M.v.d.B., P.S.H., H.M., H.A.M.K., and R.G. conception and design of research; L.E.M.K., R.P.v.O., A.D.-A., and I.S.T.B. performed experiments; L.E.M.K., R.P.v.O., and M.v.d.B. analyzed data; L.E.M.K., M.N.H., P.S.H., H.M., H.A.M.K., and R.G. interpreted results of experiments; L.E.M.K. prepared figures; L.E.M.K. drafted manuscript; L.E.M.K., R.P.v.O., M.N.H., M.v.d.B., P.S.H., J.W., H.M., H.A.M.K., and R.G. edited and revised manuscript; L.E.M.K., R.P.v.O., A.D.-A., I.S.T.B., M.N.H., M.v.d.B., P.S.H., J.W., H.M., H.A.M.K., and R.G. approved final version of manuscript.
REFERENCES
- 1.Barnes PJ. The role of anticholinergics in chronic obstructive pulmonary disease. Am J Med 117, Suppl 12A: 24S–32S, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med 343: 269–280, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Buhling F, Lieder N, Kuhlmann UC, Waldburg N, Welte T. Tiotropium suppresses acetylcholine-induced release of chemotactic mediators in vitro. Respir Med 101: 2386–2394, 2007. [DOI] [PubMed] [Google Scholar]
- 4.Celli BR, Locantore N, Yates J, Tal-Singer R, Miller BE, Bakke P, Calverley P, Coxson H, Crim C, Edwards LD, Lomas DA, Duvoix A, MacNee W, Rennard S, Silverman E, Vestbo J, Wouters E, Agusti A, Investigators ECLIPSE Inflammatory biomarkers improve clinical prediction of mortality in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 185: 1065–1072, 2012. [DOI] [PubMed] [Google Scholar]
- 5.de Almeida VV, Silva-Herdade A, Calado A, Rosario HS, Saldanha C. Fibrinogen modulates leukocyte recruitment in vivo during the acute inflammatory response. Clin Hemorheol Microcirc In press. [DOI] [PubMed] [Google Scholar]
- 6.Dickens JA, Miller BE, Edwards LD, Silverman EK, Lomas DA, Tal-Singer R, Evaluation of COPD Longitudinally to Identify Surrogate Endpoints (ECLIPSE) study investigators. COPD association and repeatability of blood biomarkers in the ECLIPSE cohort (Abstract). Respir Res 12: 146, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dominguez-Fandos D, Ferrer E, Puig-Pey R, Carreno C, Prats N, Aparici M, Musri MM, Gavalda A, Peinado VI, Miralpeix M, Barbera JA. Effects of aclidinium bromide in a cigarette smoke-exposed Guinea pig model of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 50: 337–346, 2014. [DOI] [PubMed] [Google Scholar]
- 8.Fisahn A, Yamada M, Duttaroy A, Gan JW, Deng CX, McBain CJ, Wess J. Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron 33: 615–624, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest 113: 1596–1606, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gosens R, Rieks D, Meurs H, Ninaber DK, Rabe KF, Nanninga J, Kolahian S, Halayko AJ, Hiemstra PS, Zuyderduyn S. Muscarinic M3 receptor stimulation increases cigarette smoke-induced IL-8 secretion by human airway smooth muscle cells. Eur Respir J 34: 1436–1443, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD (Abstract) 7: 73, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gross NJ, Skorodin MS. Role of the parasympathetic system in airway obstruction due to emphysema. N Engl J Med 311: 421–425, 1984. [DOI] [PubMed] [Google Scholar]
- 13.Gwilt CR, Donnelly LE, Rogers DF. The non-neuronal cholinergic system in the airways: an unappreciated regulatory role in pulmonary inflammation? Pharmacol Ther 115: 208–222, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Hicks A, Kourteva G, Hilton H, Li H, Lin TA, Liao W, Li Y, Wei X, March T, Benson J, Renzetti LM. Cellular and molecular characterization of ozone-induced pulmonary inflammation in the Cynomolgus monkey. Inflammation 33: 144–156, 2010. [DOI] [PubMed] [Google Scholar]
- 15.Kistemaker LE, Bos IS, Hylkema MN, Nawijn MC, Hiemstra PS, Wess J, Meurs H, Kerstjens HA, Gosens R. Muscarinic receptor subtype-specific effects on cigarette smoke-induced inflammation in mice. Eur Respir J 42: 1677–1688, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Kistemaker LE, Oenema TA, Meurs H, Gosens R. Regulation of airway inflammation and remodeling by muscarinic receptors: perspectives on anticholinergic therapy in asthma and COPD. Life Sci 91: 1126–1133, 2012. [DOI] [PubMed] [Google Scholar]
- 17.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13: 159–175, 2013. [DOI] [PubMed] [Google Scholar]
- 18.Kuckleburg CJ, Tilkens SB, Santoso S, Newman PJ. Proteinase 3 contributes to transendothelial migration of NB1-positive neutrophils. J Immunol 188: 2419–2426, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pera T, Zuidhof A, Valadas J, Smit M, Schoemaker RG, Gosens R, Maarsingh H, Zaagsma J, Meurs H. Tiotropium inhibits pulmonary inflammation and remodelling in a guinea pig model of COPD. Eur Respir J 38: 789–796, 2011. [DOI] [PubMed] [Google Scholar]
- 20.Profita M, Bonanno A, Siena L, Ferraro M, Montalbano AM, Pompeo F, Riccobono L, Pieper MP, Gjomarkaj M. Acetylcholine mediates the release of IL-8 in human bronchial epithelial cells by a NFkB/ERK-dependent mechanism. Eur J Pharmacol 582: 145–153, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Sachs UJ, Andrei-Selmer CL, Maniar A, Weiss T, Paddock C, Orlova VV, Choi EY, Newman PJ, Preissner KT, Chavakis T, Santoso S. The neutrophil-specific antigen CD177 is a counter-receptor for platelet endothelial cell adhesion molecule-1 (CD31). J Biol Chem 282: 23603–23612, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Sato E, Koyama S, Okubo Y, Kubo K, Sekiguchi M. Acetylcholine stimulates alveolar macrophages to release inflammatory cell chemotactic activity. Am J Physiol Lung Cell Mol Physiol 274: L970–L979, 1998. [DOI] [PubMed] [Google Scholar]
- 23.Shen LL, Liu YN, Shen HJ, Wen C, Jia YL, Dong XW, Jin F, Chen XP, Sun Y, Xie QM. Inhalation of glycopyrronium inhibits cigarette smoke-induced acute lung inflammation in a murine model of COPD. Int Immunopharmacol 18: 358–364, 2014. [DOI] [PubMed] [Google Scholar]
- 24.Stroncek DF. Neutrophil-specific antigen HNA-2a, NB1 glycoprotein, and CD177. Curr Opin Hematol 14: 688–693, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Vacca G, Randerath WJ, Gillissen A. Inhibition of granulocyte migration by tiotropium bromide (Abstract). Respir Res 12: 24, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol 154: 1558–1571, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wollin L, Pieper MP. Tiotropium bromide exerts anti-inflammatory activity in a cigarette smoke mouse model of COPD. Pulm Pharmacol Ther 23: 345–354, 2010. [DOI] [PubMed] [Google Scholar]