

Abstract
The enzyme phosphite dehydrogenase (PTDH) catalyzes the NAD+-dependent conversion of phosphite to phosphate and represents the first biological catalyst that has been characterized to carry out the enzymatic oxidation of phosphorus. Despite over a decade’s worth of investigation into both the mechanism of its unusual reaction, as well as its utility in cofactor regeneration, there has been a lack of any structural data on PTDH. Here we present the co-crystal structure of an engineered thermostable variant of PTDH bound to NAD+ (1.7 Å resolution), as well as four other co-crystal structures of thermostable PTDH and its variants with different ligands (all between 1.85 – 2.3 Å resolution). These structures provide a molecular framework for understanding prior mutational analysis, and point to additional residues, located in the active site, that may contribute to the enzymatic activity of this highly unusual catalyst.
INTRODUCTION
In spite of the fact that a number of bacteria have been shown to carry out oxidation or reduction of phosphorus compounds (1), enzyme mediated redox chemistry of phosphorus remains poorly characterized. Enzymatic catalysis of phosphorus redox chemistry has been reported in the NAD+-dependent phosphite oxidoreductase from Pseudomonas fluorescens 195 (2, 3), and in the hypophosphite oxidase from Bacillus caldolyticus (4). More recently, selection of strains capable of surviving on hypophosphite or phosphite as the sole phosphorus source led to the identification of Pseudomonas stutzeri WM88 as an efficient oxidizer of reduced phosphorus compounds (5). Genetic characterization of this strain demonstrated that the oxidation of hypophosphite to phosphate occurs in two distinct steps, and the gene clusters responsible for these steps have been identified (6). The htx locus encodes a 2-oxoglutarate-dependent dioxygenase that catalyzes the oxidation of hypophosphite to phosphite (7). The ptx locus encodes a gene product PtxD (PTDH; phosphite dehydrogenase) that catalyzes the NAD+-dependent oxidation of phosphite (Scheme 1) (8). Homogeneous preparations of recombinant PDTH were shown to catalyze the oxidation of phosphite to phosphate, coupled with the reduction of NAD+ to NADH, the first characterization of enzymatic oxidation of phosphorus.
Scheme 1.
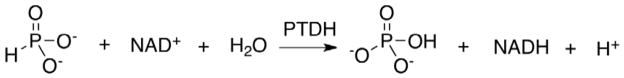
Overall scheme of reaction catalyzed by PTDH.
Sequence comparisons place PTDH within the family of NAD+-dependent D-2-hydroxyacid dehydrogenases (9) and multiple sequence alignments reveal that PTDH shares 26–33% identity with other members of this family. Based on the existing structures of D-2-hydroxyacid dehydrogenases (10–14) and by analogy to the reaction mechanism catalyzed by these enzymes, three conserved catalytic residues have been identified as essential for catalysis by PTDH (8). A conserved histidine residue (His-292 in PTDH) is proposed to act as a general base in activating the nucleophilic water molecule for attack onto phosphite, Glu-266 is believed to both orient and modulate the pKa of the active site histidine, and Arg-237 is proposed to bind and orient the substrate phosphite prior to nucleophilic attack (15). Steady state analyses of site-specific variants at each of these residues demonstrate that mutation at His-292 and non-conservative mutations at Arg-237 severely compromises catalysis. Replacement of Glu-266 by Gln results in an unexpected improvement in kcat, but with an overall decreased catalytic efficiency relative to the wild-type (16).
Biochemical studies with a number of compounds have failed to identify an alternate substrate for the oxidation reaction (17), and other members of the D-hydroxy acid dehydrogenase family are incapable of carrying out phosphite oxidation. These and other studies lend credence to the biological role of phosphite dehydrogenase as a bona fide phosphite-oxidizing enzyme (18). The only known effective inhibitor of the enzyme is sulfite (SO32-), a competitive inhibitor with phosphite (Ki = 16 μM) thought to act by forming a covalent adduct with NAD that resembles the transition state of the hydride transfer reaction (16).
Phosphite dehydrogenase has also drawn considerable attention for use as a cofactor regeneration system for use during the enzymatic preparation of enantiomerically enriched products (19). The reduction of NAD+ to NADH by phosphite dehydrogenase has applications in cofactor recycling in enzymatic processes that utilize NADH or NADPH as hydride donors (20–22). The equilibrium constant for the oxidation of phosphite by PTDH can be estimated to be 1011, indicating that the reaction is essentially irreversible (8). The strong thermodynamic driving force for catalysis and the relatively inexpensive substrate phosphite have made PTDH an attractive candidate for use in the enzymatic regeneration of NADH from NAD+ (23, 24).
Although phosphite dehydrogenase can efficiently utilize NAD+ to regenerate NADH, the wild-type enzyme is incapable of utilizing NADP+ as a cofactor. In order to expand the utility of PTDH as a universal nicotinamide cofactor regeneration enzyme, site-specific variants at the active site were generated based on analysis of a homology model structure (25). Two putative active site residues, Glu-175 and Ala-176, were identified as possible determinants of cofactor specificity. These modeling studies suggested that Glu-175 may interact with the adenine hydroxyl groups of NAD+, and thus would sterically clash with the 2′ phosphate of NADP+, while replacement of Ala-176 by a basic residue would favorably stabilize the 2′ phosphate of NADP+. Kinetic analyses of the Glu-175→Ala/Ala-176→Arg double variant demonstrated a 1000-fold improvement in the catalytic efficiency with NADP+ as a substrate, relative to the wild-type (25). This double variant also demonstrated an unexpected four-fold increase in catalytic efficiency with NAD+ as a substrate, although the structural foundations for this improvement have not been borne out.
In order to further aid in the mechanistic elucidations of this biologically unusual catalyst, and to provide a molecular basis for further engineering efforts for improved NAD(P)H regeneration, we have elucidated five different crystallographic structures of a thermostable variant of PTDH (26), as well as those of several active-site mutants, bound to various cofactors. This thermostable PTDH variant (hereafter, TS-PTDH to reflect the use of the thermostable variant) was generated by directed evolution/random mutagenesis and contains 16 mutations (see Experimental Procedures for details) that result in increased thermostability and increased activity but with kinetic isotope effects and pre-steady state kinetic parameters similar to those for the wild-type enzyme (27). The crystal structures presented here consist of the binary complex of TS-PTDH with NAD+ at 1.7 Å resolution, the TS-PTDH-NAD+-sulfite ternary complex at 2.2 Å resolution, the relaxed cofactor specificity variant Glu-175→Ala TS-PTDH-NAD+ binary complex at 2.3 Å resolution, the cofactor specificity double variant Glu-175→Ala/Ala-176→Arg TS-PTDH-NAD+ binary complex at 1.9 Å resolution, and the Glu-175→Ala/Ala-176→Arg TS-PTDH-NADP+ binary complex at 1.85 Å resolution. The crystallographically independent copies observed in crystals of the Glu-175→Ala TS-PTDH-NAD+ complex contain both ligand-bound and apo forms of the enzyme. Hence, these five structures provide six distinct chemical views of TS-PTDH representing the “wild-type” and variant enzymes interacting with either NAD+ or NADP+ cofactors, and with the competitive inhibitor sulfite.
EXPERIMENTAL PROCEDURES
Protein expression and purification
The expression constructs for the thermostable PTDH, as well as those for the Glu-175→Ala single, and Glu-175→Ala/Ala-176→Arg double mutant, have been described previously (16, 25, 26). The TS-PTDH variant contains mutations at the following 16 residues, from wt-PTDH from P. stutzeri: Asp13→Glu, Met26→Ile, Val71→Ile, Glu130→Lys, Gln132→Arg, Gln137→Arg, Ile150→Phe, Gln215→Leu, Arg275→Gln, Leu276→Gln, Ile313→Leu, Val315→Ala, Ala319→Glu, Ala325→Val, Glu332→Asn, and Cys336→Asp. The expression vectors were introduced into E. coli BL21 (DE3) competent cells, subject to antibiotic selection (ampicillin at a concentration of 100 μg/mL), and individual colonies were grown in 10 mL of LB medium supplemented with ampicillin (100 mg/mL) at 37 °C overnight. Each culture was transferred to a fresh LB medium with the same antibiotics, and grown until the optical density at 600 nm reached approximately 0.4. The temperature was then decreased to 16 °C, protein production was induced with the addition of 0.4 mM IPTG and the cells were grown for an additional 18 hours. For growth of selenomethionine labeled TS-PTDH, a 200 mL starter culture was inoculated in minimal media (6 g/L Na2HPO4, 3 g/L KH2PO4, 1 g/L NH4Cl, 0.5 g/L NaCl) supplemented with 2 mM MgSO4, 0.5 % glucose, and 0.05 % glycerol, and grown at 37 °C overnight. The starter culture (8 mL) was then used to inoculate 800 mL of the same supplemented minimal media additionally containing adenosine monophosphate (1 μg/mL) and thiamine (1 μg/ml). After growth at 37 °C to an OD600 of 1.0, the temperature was reduced to 16 °C and 5 mL of amino acid mix (14 mg/mL each of lysine, threonine, and phenylalanine; 7 mg/mL each of leucine, isoleucine, and valine; and 8.4 mg/mL selenomethionine) was added. Twenty min later, protein expression was induced with 0.4 mM IPTG and the cells were grown for an additional 18 hours.
Cells were harvested by centrifugation (5,000 rpm, 4 °C, 15 min), and re-suspended in 50 mL of lysis buffer (50 mM Tris-HCl, pH 7.5, 300 mM KCl) supplemented with a cocktail of protease inhibitors (2 mM 2-aminoethyl benzenesulfonyl fluoride, 0.3 μM aprotinin, 130 μM bestatin, 1 μM leupeptin). Cells were disrupted using an Emulsiflex C5 French press cell (Avestin Inc., Ottawa, Canada), and the lysate was clarified by centrifugation (15,000 rpm, 4 °C, 30 min). The supernatant was filtered through a 0.22 μm pore size Durapore membrane (Millipore, Bedford, MA). The filtrate was applied to HisTrap FF 5 mL (GE Healthcare, Piscataway, NJ) column, and unbound proteins were washed with 20 column volumes of lysis buffer. The bound proteins were eluted with elution buffer (50 mM Tris-HCl, pH 7.5, 300 mM KCl, 250 mM imidazole), and dialyzed overnight against a solution of 50 mM Tris-HCl, pH 8.5, 100 mM KCl.
Protein crystallization
Prior to crystallization, the poly-histidine affinity tag was cleaved using thrombin and the resultant sample was further purified using anion exchange chromatography (5 mL HiTrap Q, G. E. Healthcare) using a gradient of increasing KCl concentration, and by size-exclusion chromatography (Sephacryl 200, GE Healthcare) in a buffer consisting of 20 mM Na+-HEPES (pH 7.5) and 100 mM KCl. Protein concentrations were determined using the Bradford method prior to crystallization. Initial crystallization conditions were established with commercial screens from Hampton Research (Aliso Viejo, CA) and Emerald Biosystems (Bainbridge Island, WA). Refinement of promising conditions yielded large crystals suitable for diffraction analysis. Typically, 2 μL of variant TS-PTDH at 10 mg/mL (in 100 mM KCl, 10 mM Na+-HEPES, pH 7.5) was added to 2 μL of precipitant (0.2M KCl, 25–30% polyethylene glycol 3350) using buffers with a pH range of 6.5 to 8.0 (typically 50 mM Na+-MES monohydrate, pH 6.5, 50 mM Na+-HEPES, pH 7.5, or 50 mM Tris-HCl, pH 8.0) and equilibrated over a well containing the precipitant solution at 8 °C. For co-crystallization experiments, the appropriate ligand was added to the protein sample at a final concentration of 5 mM and kept on ice for 30 min prior to crystallization. Crystals grew over a period of three weeks, and after transient immersion into a precipitant solution supplemented with 25% glycerol, were vitrified by direct immersion into liquid nitrogen.
Structure determination and refinement
Crystallographic phases were determined by single wavelength anomalous diffraction using crystals of selenomethionine (SeMet) labeled Glu-175→Ala TS-PDTH, as crystals of this variant could be grown reproducibly. Using crystals of SeMet Glu-175→Ala TS-PDTH, a four-fold redundant anomalous diffraction data set was collected to 2.3 Å resolution using a Mar 300 CCD detector, at an insertion device synchrotron beam line (LS-CAT Sector 21 ID-D, Advanced Photon Source, Argonne, IL), and integrated and scaled using the HKL2000 package (28). Initial heavy atom positions were determined using HySS (36 SeMet sites), and subjected to maximum likelihood refinement using SHARP (figure of merit = 0.313) (29). The resultant map was of excellent quality and allowed for independent manual modeling of all six copies of the polypeptide in the crystallographic asymmetric unit using XtalView (30). Despite incubation with a stoichiometric excess of NAD+ prior to crystallization, only four of the molecules in the asymmetric unit show density corresponding to a well-bound nucleotide molecule. As a result, there are considerable differences in the relative orientations of the two domains amongst the six independent molecules in the asymmetric unit. Non-crystallographic symmetry restraints were not used at any point during refinement. The final model was further improved by crystallographic refinement using REFMAC5, interspersed with rounds of manual model building (31, 32).
The structures of all other variants of TS-PDTH and respective ligand complexes were solved by molecular replacement using the final refined coordinates of one monomer of Glu-175→Ala TS-PDTH as a search probe. Successful molecular replacement solutions could only be determined by conducting independent searches using the two individual domains of the molecule. After rigid body refinement of the initial molecular replacement solution, the atomic model was subject to automatic rebuilding using ARP/wARP (33), resulting in near complete traces of each of the structures. Cycles of manual rebuilding followed by crystallographic refinement were carried out for each of the co-crystal structures. The respective ligands were manually built into the difference Fourier maps after the free R factors dropped below 30%. Cross-validation, using 5% of the data for the calculation of the free R factor, was utilized throughout model building process in order to monitor building bias (34). The stereochemistry of the models was routinely monitored throughout the course of refinement using PROCHECK (35). The refined coordinates have been deposited in the PDB.
RESULTS AND DISCUSSION
Structure Determination
The crystal structure of the thermostable variant of PTDH, with an additional relaxed cofactor specificity mutation Glu-175→Ala, in complex with NAD+, was determined to a resolution of 2.3 Å by single wavelength anomalous diffraction. The structures of all other active site variants and ligand complexes were determined by molecular replacement. Crystal parameters, data collection parameters and refinement statistics for each of the structures are summarized in Table 1.
Table 1.
Data collection, phasing and refinement statistics
| PTDH NAD+ |
PTDH NAD+ and Sulfite |
SeMet Glu-175→Ala PTDH-NAD+ |
Glu-175→Ala/Ala-176→Arg PTDH-NAD+ |
Glu-175→Ala/Ala-176→Arg PTDH-NADP+ |
|
|---|---|---|---|---|---|
| PDB code | 4E5N | 4E5K | 4EBF | 4E5P | 4E5M |
| Data collection | |||||
| Space group | P21 | P21 | P21 | P21 | P21 |
| a, b, c (Å), β (°) | 90.7, 113.5, 130.2 100.2 |
73.1, 114.2, 88.3 112.3 |
70.2, 122.6, 135.0 96.4 |
70.3, 122.3, 134.3 96.4 |
48.2, 114.0, 58.8 94.1 |
| Resolution (Å)1 | 80–1.7 (1.8–1.7) | 82–1.95 (2.0–1.95) | 50–2.3 (2.38–2.3) | 50–1.9 (1.97–1.9) | 50–1.85 (1.92–1.85) |
| Rsym (%) | 5.4 (43.6) | 6.8 (41.0) | 8.2 (37.3) | 7.4 (33.9) | 7.1 (18.9) |
| I/σ(I) | 19.2 (3.0) | 16.27 (3.2) | 19.4 (3.9) | 17.8 (3.5) | 19.8 (5.3) |
| Completeness(%) | 95.0 (74.1) | 97.8 (84.9) | 98.9 (98.3) | 97.6 (89.6) | 96.6 (79.3) |
| Redundancy | 6.0 (3.8) | 4.6 (3.5) | 4.1 (4.1) | 4.2 (3.9) | 4.3 (3.0) |
| Refinement | |||||
| Resolution (Å) | 25.0–1.7 | 25.0–1.95 | 25.0–2.3 | 25.0–1.9 | 25.0–1.85 |
| No. reflections | 256,745 | 90,507 | 91,875 | 163,207 | 49,262 |
| Rwork/Rfree2 | 21.0/24.7 | 21.9/26.5 | 20.8/27.5 | 20.3/24.3 | 18.7/21.9 |
| Number of atoms | |||||
| Protein | 20,133 | 10,066 | 15,030 | 15,076 | 5,035 |
| NAD+/NADP+ | 352 | 176 | 176 | 264 | 96 |
| Sulfite | - | 16 | - | - | - |
| Water | 2,458 | 570 | 754 | 1,338 | 516 |
| B-factors | |||||
| Protein | 29.0 | 11.5 | 40.2 | 29.5 | 14.9 |
| NAD+/NADP+ | 23.7 | 26.1 | 40.6 | 33.6 | 21.8 |
| Sulfite | - | 33.4 | - | - | - |
| Water | 38.3 | 14.3 | 38.6 | 36.4 | 24.3 |
| R.m.s deviations | |||||
| Bond lengths (Å) | 0.007 | 0.013 | 0.012 | 0.009 | 0.007 |
| Bond angles (°) | 1.16 | 1.55 | 1.55 | 1.29 | 1.14 |
Highest resolution shell is shown in parenthesis.
R-factor = Σ(|Fobs|-k|Fcalc|)/Σ |Fobs|and R-free is the R value for a test set of reflections consisting of a random 5% of the diffraction data not used in refinement.
Overall structure
Our structural studies utilized a thermostable variant of PDTH, and the 1.7 Å resolution co-crystal structure of this variant will be described as the “wild-type” structure. As expected from the low, but significant, similarity in primary sequence to members of the D-2-hydroxyacid dehydrogenases family, the core architecture of TS-PTDH is similar to those of other enzymes within this group (Figure 1A). Notable structural homologs include Pyrococcus horikoshii glyoxylate reductase (PDB Code: 2DBQ; Z score = 41; RMSD of 1.6 Å over 319 aligned Cα atoms; 34% sequence identity), Hyphomicrobium methylovorum glycerate dehydrogenase (PDB Code: 1GDH; Z score = 35.6; RMSD of 2.4 Å over 317 aligned Cα atoms; 29% sequence identity), and Aquifex aeolicus lactate dehydrogenase (PDB Code: 3KB6; Z score = 34.5; RMSD of 2.2 Å over 312 aligned Cα atoms; 25% sequence identity). Similar to other members within the D-2-hydroxyacid dehydrogenases superfamily, TS-PTDH exists both in solution and in the crystal as a dimer. The multiple copies in the crystallographic asymmetric unit for the various complexes pack as sets of equivalent homodimers.
Figure 1. Overall structure of PTDH.

A. Ribbon diagram derived from the 1.75 Å resolution crystal structure of the thermostable PTDH variant showing the disposition of the substrate-binding domain (in pink) and the NAD-binding domain (in blue). The bound NAD+ cofactor is shown in yellow ball-and-stick. B. The structure of the TS-PTDH homodimer with one monomer in the same orientation and color scheme as in A and the other monomer colored in gray. C. Close-up view of the NAD+ binding site. The cofactor is shown in yellow ball-and-stick and active site residues are colored in green. Superimposed is a difference Fourier electron density map contoured at 3σ over background (in blue) and 8σ over background (in yellow), calculated with coefficients |Fobs| - |Fcalc| and phases from the final refined model with the coordinates of NAD+ deleted prior to one round of refinement.
The TS-PTDH monomer may be divided into a large and a small domain, separated by a flexible hinge region. The large domain (NAD+ binding domain) is composed of a contiguous stretch of residues spanning Leu-100 to Pro-291, and consists of six parallel β strands, sandwiched between seven α helices. The small domain (substrate binding domain) is composed from secondary structural elements from two non-contiguous regions of the polypeptide: Met-1 through Val-96 form a domain containing five parallel β strands flanked by four α helices, and residues Ala-299 through Pro-329 contains an additional helix (Figure 1A and 1B).
The NAD+ cofactor is housed at the junction between the two domains, where residues from the large subunit engage in interactions with the ligand (Figure 1C). The adenine ring is nestled in between two loops composed of His-174 through Ala-178 and Ala-207 through Thr-214, and makes van der Waals contacts with Leu-151, Ala-176, and Leu-208. Lys-76 engages in favorable electrostatic interactions with the diphosphate oxygens of the cofactor. The only other consequential interaction with the cofactor occurs through the interaction of Glu-175 with the 2′- and 3′-hydroxyls of the adenine ribose (Oε1 – 2′-OH distance of 2.6 Å and Oε2 – 3′-OH distance of 2.5 Å). There is a constellation of polar residues near the nicotinamide, in the vicinity of the putative substrate-binding region, and these include the canonical His/Glu/Arg triad (Arg-237, Glu-266, and His-292 in PTDH) found in all D-2-hydroxyacid dehydrogenases. The proximity of these residues at the active site had been previously predicted by homology modeling studies and their importance has been established through mutational analyses (16, 25).
As noted, the use of a thermostable variant of PTDH, which proved amenable to crystallization, is justified by the fact that pre-steady state kinetics for this variant are similar to those for the wild-type enzyme (27). Mapping of the sites of the mutations onto the crystal structure reveals that all of the changes occur at positions that are distal relative to the active site (Figure 2).
Figure 2. Thermostabilizing mutations in PTDH.

Ribbon diagram of the thermostable PTDH variant that was used for structural studies showing the 16 thermostabilizing mutations. Two of the mutations (Glu-332→Asn and Cys-336→Asp) occur within the six carboxy-terminal residues that are not visible in the structures. Note that all of these mutations are distal to the active site.
The crystal structure of the TS-PTDH-NAD+ complex also identifies a number of additional polar residues in the vicinity of the active site that may play a role in catalysis. Notable among these is Asp-79 that interacts with Arg-237 and may serve to orient the side chain of the latter to position substrate phosphite for hydride transfer. Prior mutational analysis at Asp-79 demonstrates that alteration of this side chain results in significant compromises in catalytic efficiency (up to a 2600-fold decrease in kcat/Km for the Asp-79→Ala mutant) (27). Another potentially catalytically relevant residue is Arg-301, which has a side chain that is oriented directly towards the putative substrate-binding site, but is located at a distance of over 5 Å from this site. Kinetic analysis of site-specific variants at this residue is described in the accompanying study (36).
Co-crystal structure with the competitive inhibitor sulfite
In order to elucidate the determinants for substrate recognition, we determined the 1.95 Å resolution co-crystal structure of TS-PTDH in complex with NAD+ and the competitive inhibitor sulfite (Ki = 16 μM). The co-crystal structure contains four copies of the TS-PDTH monomer in the crystallographic asymmetric unit, allowing for independent views of the enzyme-inhibitor complex. The sulfite anion is situated proximal to the nicotinamide of the cofactor, where it is engaged through interactions with the side chains of Arg-237 (Nη1-O distance of 2.6 Å, and Nη2-O distance of 3.0 Å), His-292 (Nε2-O distance of 2.8 Å), and the backbone amides of Lys-76 (2.8 Å) and Gly-77 (2.7 Å) (Figure 3A). In PTDH, Arg-237, Glu-266 and His-292 constitute the canonical His/Glu/Arg triad found in all D-2-hydroxyacid dehydrogenases, and Asp-79 is proposed to orient the side chain of Arg-237. Our TS-PTDH-sulfite co-crystal structure suggests that these residues are likely involved in positioning the substrate phosphite to facilitate hydride transfer. The co-crystal structure also shows that Arg-301 is oriented towards the sulfite but is located at a distance greater than 5 Å away from the sulfite oxygen atoms. Kinetic analysis of site-specific mutations at this residue (presented in the accompanying manuscript) demonstrate that it is important for catalysis (36). Lastly, local movements near the active site upon binding of sulfite include modest movement of the loop encompassing Ala-74 through Phe-78, which results in favorable interactions with the backbone amides, and torsional displacement of the side chain of Met-53 (Sγ-O distance of 3.1 Å), which may facilitate movement of the catalytic hydroxide for attack on the phosphorus atom of substrate phosphite (Figure 3B).
Figure 3. Active site of PTDH in complex with the competitive inhibitor sulfite.
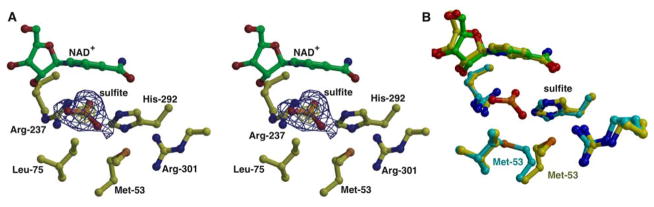
A. The inhibitor is colored in orange, the cofactor is shown in green ball-and-stick and active site residues are colored in yellow. Superimposed is a difference Fourier electron density map contoured at 3σ over background (in blue) and 8σ over background (in yellow), calculated with coefficients |Fobs | - |Fcalc | and phases from the final refined model with the coordinates of sulfite deleted prior to one round of refinement. B. A superposition of the active sites of unliganded PTDH (in cyan) with the sulfite complex (in yellow). Note the movement of Met-53 that results in the formation of a defined substrate-binding pocket.
Prior spectroscopic analysis suggests that, at the active site, sulfite forms a covalent adduct with NAD+ that may resemble the transition state for hydride transfer (17). This adduct was presumed to occur through the covalent linkage of the sulfur to nicotinamide C4. Within the active site of the sulfite co-crystal structure, sulfite is not bound as an adduct but rather as a distinct species. Furthermore, the distance between the sulfur and the nicotinamide C4 is 3.9 Å, which is not indicative of a covalently linked adduct. It is currently unclear why an adduct can not be visualized in the co-crystal structure or even whether the spectroscopic data are truly indicative of an adduct. However, given the long distance of 3.9 Å between the sulfur and the nicotinamide C4, modest movements of the enzyme or the substrate and/or cofactor must occur in order to achieve a transition-state configuration that would optimally place (donor-acceptor distance of 2.7–3.1 Å) the hydrogen of the substrate phosphite over the re-face of the nicotinamide. Structural studies, molecular dynamics and cross-correlation analysis are consistent with a ~0.5 Å movement of the hydride donor to the nicotinamide C4 in near-attack ground state conformers of related enzymes of the D-2-hydroxyacid dehydrogenase superfamily (37).
Crystal structures of relaxed cofactor specificity mutants
Attempts to expand the utility of PTDH as a universal nicotinamide cofactor regeneration enzyme capable of also utilizing NADP+ have identified two residues, Glu-175 and Ala-176, as determinants of cofactor specificity (16). In the wild-type structure, Glu-175 engages both the 2′ and 3′ hydroxyl of the NAD+ adenine ribose, and this residue would result in steric and electrostatic preclusion of the 2′-phosphate of NADP+ (Figure 1C). Intriguingly, although the Glu-175→Ala mutant increased the kcat/KM,NADP+ (which may approximate the ability of the enzyme to bind the co-factor) by a factor of about 30, relative to the wild-type, this variant also demonstrated an unexpected 4-fold increase in the kcat/KM,NAD+, despite the anticipated loss of the favorable hydrogen bonds provided by Glu-175 (25). The 2.3 Å resolution co-crystal structure of Glu-175→Ala TS-PTDH in complex with NAD+ provides a rationale for the observed increase in kcat/KM,NAD+. Although the overall co-crystal structures of the wild-type and Glu-175→Ala TS-PTDH with NAD+ are nearly identical, the Glu-175→Ala mutation results in local compensatory changes near the vicinity of the bound cofactor (Figure 4A). First, the loop containing residues His-174 through Leu-179 moves inward towards the adenine ring of NAD+, resulting in more favorable van der Waals contact. Second, the movement of this loop results in orientation of the backbone amide of Ala-175 within hydrogen-bonding distance of N3 of adenine (NH - N3 distance of 3.1 Å in Glu-175→Ala and 3.6 Å in the wild-type TS-PTDH). And finally, the side chain of Leu-208, located on the other side of the adenine ring, also reorients to provide more favorable van der Waals contact with the cofactor. Although these changes are local, and are the result of small movements in the variant polypeptide, the identical changes can be observed in the multiple, independent copies of the Glu-175→Ala TS-PTDH that are in the crystallographic asymmetric unit. The net result of these movements appears to be better interaction with the NAD+ cofactor, providing an explanation for the improved kcat/KM,NAD+.
Figure 4. Structures of PTDH variants with relaxed cofactor specificity.
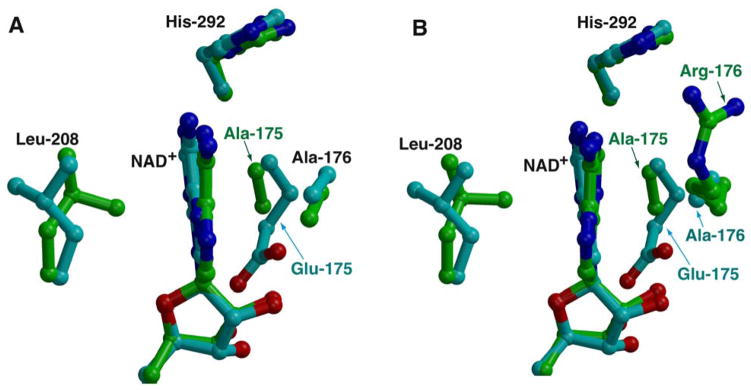
A. Superposition of the structures of complexes of NAD+ with TS-PTDH (in green) and with the Glu-175→Ala variant (in cyan). Note the movements of Ala-195, Ala-196 and Leu-208 towards the adenine ring of the NAD+ cofactor. B. Superposition structures of the complexes of NAD+ with TS-PTDH (in green) and with the Glu-175→Ala/Ala-176→Arg double variant that can also utilize NADPH as a cofactor (in cyan).
Cofactor specificity has been shown to be further expanded towards NADP+ in the Glu-175→Ala/Ala-176→Arg double variant, which increases the kcat/KM,NADP+ by 1000-fold relative to the wild-type. The 1.9 Å resolution co-crystal structure of Glu-175→Ala/Ala-176→Arg TS-PTDH in complex with NAD+ also demonstrates the compensatory movement of the His-174 through Leu-179 loop, relative to the wild-type, observed in the single mutant (Figure 4B). The Arg-176 side chain points away from the active site, towards bulk solvent and is positioned parallel to the adenine ring of NAD+. As no additional contacts occur between Arg-176 and NAD+, this structure explains why the kcat/KM,NAD+ is not significantly lowered in this double variant (11.8 μM−1 min−1), in comparison with the Glu-175→Ala single mutant (13.1 μM−1 min−1).
In order to understand the basis for the expanded preference observed only towards NADP+ in this double variant, we solved the 1.85 Å resolution structure of Glu-175→Ala/Ala-176→Arg TS-PTDH in complex with NADP+ (Figure 5A). A comparison of the co-crystal structures of the double variants in complex with the two different cofactors reveals that the side chain of Arg-176 rotates away from solvent and faces inward towards the NADP+ phosphate (Figure 5B). Although only one of the phosphate oxygen atoms is within hydrogen bonding distance (Nη2 - O distance of 2.6 Å), the other oxygen atoms are within 4 Å of the Nε of Arg-176 and are also stabilized by several ordered solvent molecules. The planar stacking of the guanidinium group with the adenine ring of the cofactor further stabilizes the movement of the Arg-176 side chain towards the NADP+ phosphate. Such arginine-aromatic stacking interactions have been previously observed in the structures of the periplasmic ribose binding protein, and P2 myelin, and in T. foetus IMP dehydrogenase (38, 39) and are suggested to orient the Arg side chain without interfering with the ability to form hydrogen bonds (40).
Figure 5. Structure of the relaxed cofactor specificity variant with NADP+.
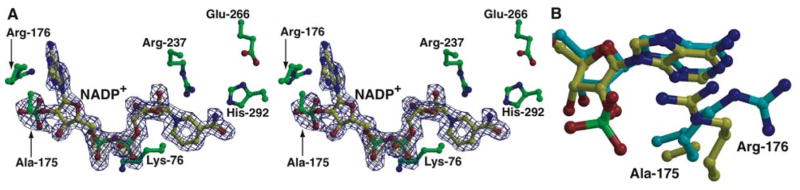
A. Close-up view of the active site of Glu-175→Ala/Ala-176→Arg TS-PTDH with bound NADP+. The cofactor is shown in yellow ball-and-stick and active site residues are colored in green. Superimposed is a difference Fourier electron density map contoured at 3σ over background (in blue) and 8σ over background (in yellow), calculated with coefficients |Fobs| - |Fcalc| and phases from the final refined model with the coordinates of NADP+ deleted prior to one round of refinement. B. Superposition of the structure of Glu-175→Ala/Ala-176→Arg TS-PTDH with bound NAD+ (in cyan) and with bound NADP+ (in yellow). Note the movements of Arg-176 towards the phosphate in the NADP+-bound structure.
Cofactor induced conformational movement
In the Glu-175→Ala TS-PDTH structure, only three of the molecules in the asymmetric unit show density corresponding to a well-bound nucleotide molecule, despite incubation of the protein with a 20-fold stoichiometric excess of NAD+ prior to crystallization. Weak, but convincing density, corresponding to a poorly occupied ligand, can be observed in two of the remaining molecules, while the final molecule does not contain any ligand. Consequently, a comparison of the six independent molecules in the crystallographic asymmetric unit of Glu-175→Ala TS-PDTH allows visualization of the enzyme in conformations with and without a bound cofactor.
A comparison of ligand free Glu-175→Ala TS-PDTH with that of its NAD+ complex reveals the rigid body rotation characteristic of the family of NAD+-dependent dehydrogenases (41). Binding of the cofactor causes the movement of the smaller substrate-binding domain towards the larger domain, resulting in a narrowing of the active site cleft. The rigid body movement corresponds to a rotation of about 6° around the hinge region that separates the two domains. As with other enzymes in this same superfamily, the ligand-free conformation can be considered as the “open” conformation and the NAD+-bound form as the “closed” conformation. In addition to a narrowing of the active site cleft, the transition from the open to closed conformation results in placement of Lys-76 into a position where it can favorably interact with the diphosphates of NAD+. In addition, the side chains of Met-53, Leu-75 and Leu-100 close off the active site to shield the bound cofactor away from solvent.
CONCLUSION
Phosphite dehydrogenase has drawn considerable attention for both its role as a catalyst in a biologically unusual reaction and for its use in cofactor recycling in bioprocesses that utilize NADH or NADPH as hydride donors. The co-crystal structures of the thermostable variant of PTDH, and various complexes of active site mutants presented here provide a three-dimensional framework for further engineering efforts aimed at improving the catalytic efficiency, as well as expanded cofactor repertoire. These co-crystal structures also identify a number of active site residues that may function in catalysis and additional structure-based functional studies aimed at validating the functions of these residues is presented in the accompanying manuscript (36).
Acknowledgments
This work was supported in part by the National Science Foundation (NSF 0822536 to W.A.V.) and the Biotechnology Research and Development Consortium (Project 2-4-121 to H.Z.).
We thank Keith Brister at LS-CAT (21-ID at APS) for facilitating data collection.
ABBREVIATIONS
- PTDH
phosphite dehydrogenase
- ptxD
phosphite dehydrogenase encoding gene
- NAD+ and NADH
nicotinamide adenine dinucleotide
References
- 1.White AK, Metcalf WW. Microbial metabolism of reduced phosphorus compounds. Annu Rev Microbiol. 2007;61:379–400. doi: 10.1146/annurev.micro.61.080706.093357. [DOI] [PubMed] [Google Scholar]
- 2.Malacinski G, Konetzka WA. Bacterial oxidation of orthophosphate. J Bacteriol. 1966;91:578–582. doi: 10.1128/jb.91.2.578-582.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malacinski GM, Konetzka WA. Orthophosphite-nicotinamide adenine dinucleotide oxidoreductase from Pseudomonas fluorescens. J Bacteriol. 1967;93:1906–1910. doi: 10.1128/jb.93.6.1906-1910.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lauwers AM, Heinen W. Bio-degradation and utilization of silica and quartz. Arch Mikrobiol. 1974;95:67–78. doi: 10.1007/BF02451749. [DOI] [PubMed] [Google Scholar]
- 5.Metcalf WW, Wolfe RS. Molecular genetic analysis of phosphite and hypophosphite oxidation by Pseudomonas stutzeri WM88. J Bacteriol. 1998;180:5547–5558. doi: 10.1128/jb.180.21.5547-5558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White AK, Metcalf WW. The htx and ptx operons of Pseudomonas stutzeri WM88 are new members of the pho regulon. J Bacteriol. 2004;186:5876–5882. doi: 10.1128/JB.186.17.5876-5882.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White AK, Metcalf WW. Isolation and biochemical characterization of hypophosphite/2-oxoglutarate dioxygenase. A novel phosphorus-oxidizing enzyme from Psuedomonas stutzeri WM88. J Biol Chem. 2002;277:38262–38271. doi: 10.1074/jbc.M204605200. [DOI] [PubMed] [Google Scholar]
- 8.Costas AM, White AK, Metcalf WW. Purification and characterization of a novel phosphorus-oxidizing enzyme from Pseudomonas stutzeri WM88. J Biol Chem. 2001;276:17429–17436. doi: 10.1074/jbc.M011764200. [DOI] [PubMed] [Google Scholar]
- 9.Grant GA. A new family of 2-hydroxyacid dehydrogenases. Biochem Biophys Res Commun. 1989;165:1371–1374. doi: 10.1016/0006-291x(89)92755-1. [DOI] [PubMed] [Google Scholar]
- 10.Dengler U, Niefind K, Kiess M, Schomburg D. Crystal structure of a ternary complex of D-2-hydroxyisocaproate dehydrogenase from Lactobacillus casei, NAD+ and 2-oxoisocaproate at 1.9 A resolution. J Mol Biol. 1997;267:640–660. doi: 10.1006/jmbi.1996.0864. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg JD, Yoshida T, Brick P. Crystal structure of a NAD-dependent D-glycerate dehydrogenase at 2.4 A resolution. J Mol Biol. 1994;236:1123–1140. doi: 10.1016/0022-2836(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 12.Lamzin VS, Aleshin AE, Strokopytov BV, Yukhnevich MG, Popov VO, Harutyunyan EH, Wilson KS. Crystal structure of NAD-dependent formate dehydrogenase. Eur J Biochem. 1992;206:441–452. doi: 10.1111/j.1432-1033.1992.tb16945.x. [DOI] [PubMed] [Google Scholar]
- 13.Lamzin VS, Dauter Z, Popov VO, Harutyunyan EH, Wilson KS. High resolution structures of holo and apo formate dehydrogenase. J Mol Biol. 1994;236:759–785. doi: 10.1006/jmbi.1994.1188. [DOI] [PubMed] [Google Scholar]
- 14.Schuller DJ, Grant GA, Banaszak LJ. The allosteric ligand site in the Vmax-type cooperative enzyme phosphoglycerate dehydrogenase. Nat Struct Biol. 1995;2:69–76. doi: 10.1038/nsb0195-69. [DOI] [PubMed] [Google Scholar]
- 15.Relyea HA, van der Donk WA. Mechanism and applications of phosphite dehydrogenase. Bioorg Chem. 2005;33:171–189. doi: 10.1016/j.bioorg.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 16.Woodyer R, Wheatley JL, Relyea HA, Rimkus S, van der Donk WA. Site-directed mutagenesis of active site residues of phosphite dehydrogenase. Biochemistry. 2005;44:4765–4774. doi: 10.1021/bi047868c. [DOI] [PubMed] [Google Scholar]
- 17.Relyea HA, Vrtis JM, Woodyer R, Rimkus SA, van der Donk WA. Inhibition and pH dependence of phosphite dehydrogenase. Biochemistry. 2005;44:6640–6649. doi: 10.1021/bi047640p. [DOI] [PubMed] [Google Scholar]
- 18.Vrtis JM, White AK, Metcalf WW, van der Donk WA. Phosphite dehydrogenase: an unusual phosphoryl transfer reaction. J Am Chem Soc. 2001;123:2672–2673. doi: 10.1021/ja004301k. [DOI] [PubMed] [Google Scholar]
- 19.Vrtis JM, White AK, Metcalf WW, van der Donk WA. Phosphite dehydrogenase: a versatile cofactor-regeneration enzyme. Angew Chem Int Ed Engl. 2002;41:3257–3259. doi: 10.1002/1521-3773(20020902)41:17<3257::AID-ANIE3257>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 20.Chenault HK, Whitesides GM. Regeneration of nicotinamide cofactors for use in organic synthesis. Appl Biochem Biotechnol. 1987;14:147–197. doi: 10.1007/BF02798431. [DOI] [PubMed] [Google Scholar]
- 21.van der Donk WA, Zhao H. Recent developments in pyridine nucleotide regeneration. Curr Opin Biotechnol. 2003;14:421–426. doi: 10.1016/s0958-1669(03)00094-6. [DOI] [PubMed] [Google Scholar]
- 22.Zhao H, van der Donk WA. Regeneration of cofactors for use in biocatalysis. Curr Opin Biotechnol. 2003;14:583–589. doi: 10.1016/j.copbio.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Johannes TW, Woodyer RD, Zhao H. Efficient regeneration of NADPH using an engineered phosphite dehydrogenase. Biotechnol Bioeng. 2007;96:18–26. doi: 10.1002/bit.21168. [DOI] [PubMed] [Google Scholar]
- 24.Woodyer R, van der Donk WA, Zhao H. Optimizing a biocatalyst for improved NAD(P)H regeneration: directed evolution of phosphite dehydrogenase. Comb Chem High Throughput Screen. 2006;9:237–245. doi: 10.2174/138620706776843246. [DOI] [PubMed] [Google Scholar]
- 25.Woodyer R, van der Donk WA, Zhao H. Relaxing the nicotinamide cofactor specificity of phosphite dehydrogenase by rational design. Biochemistry. 2003;42:11604–11614. doi: 10.1021/bi035018b. [DOI] [PubMed] [Google Scholar]
- 26.Johannes TW, Woodyer RD, Zhao H. Directed evolution of a thermostable phosphite dehydrogenase for NAD(P)H regeneration. Appl Environ Microbiol. 2005;71:5728–5734. doi: 10.1128/AEM.71.10.5728-5734.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fogle EJ, van der Donk WA. Pre-steady-state studies of phosphite dehydrogenase demonstrate that hydride transfer is fully rate limiting. Biochemistry. 2007;46:13101–13108. doi: 10.1021/bi701550c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Borek D, Majewski W, Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- 29.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 30.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 31.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 33.Perrakis A, Sixma TK, Wilson KS, Lamzin VS. wARP: improvement and extension of crystallographic phases by weighted averaging of multiple-refined dummy atomic models. Acta Crystallogr D Biol Crystallogr. 1997;53:448–455. doi: 10.1107/S0907444997005696. [DOI] [PubMed] [Google Scholar]
- 34.Kleywegt GJ, Brunger AT. Checking your imagination: applications of the free R value. Structure. 1996;4:897–904. doi: 10.1016/s0969-2126(96)00097-4. [DOI] [PubMed] [Google Scholar]
- 35.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 36.Hung JE, Fogle EJ, Relyea HA, Christman HD, Johannes TW, Zhao H, Metcalf WW, van der Donk WA. Accompanying paper. 2011. [Google Scholar]
- 37.Luo J, Bruice TC. Anticorrelated motions as a driving force in enzyme catalysis: the dehydrogenase reaction. Proc Natl Acad Sci U S A. 2004;101:13152–13156. doi: 10.1073/pnas.0405502101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gan L, Petsko GA, Hedstrom L. Crystal structure of a ternary complex of Tritrichomonas foetus inosine 5′-monophosphate dehydrogenase: NAD+ orients the active site loop for catalysis. Biochemistry. 2002;41:13309–13317. doi: 10.1021/bi0203785. [DOI] [PubMed] [Google Scholar]
- 39.Prosise GL, Lueke H. Crystal structures of Tritrichomonasfoetus inosine monophosphate dehydrogenase in complex with substrate, cofactor and analogs: a structural basis for the random-in ordered-out kinetic mechanism. Journal of Molecular Biology. 2003;326:517–527. doi: 10.1016/s0022-2836(02)01383-9. [DOI] [PubMed] [Google Scholar]
- 40.Flocco MM, Mowbray SL. Planar stacking interactions of arginine and aromatic side-chains in proteins. J Mol Biol. 1994;235:709–717. doi: 10.1006/jmbi.1994.1022. [DOI] [PubMed] [Google Scholar]
- 41.Eklund H, Branden CI. Crystal Structure, Coenzyme Conformations, and Protein Interactions. In: Dolphin D, Poulson R, Avramovic O, editors. Pyridine Nucleotide Coenzyme: Chemical, Biochemical and Medical Aspects. Wiley and Sons; 1987. pp. 51–98. [Google Scholar]