

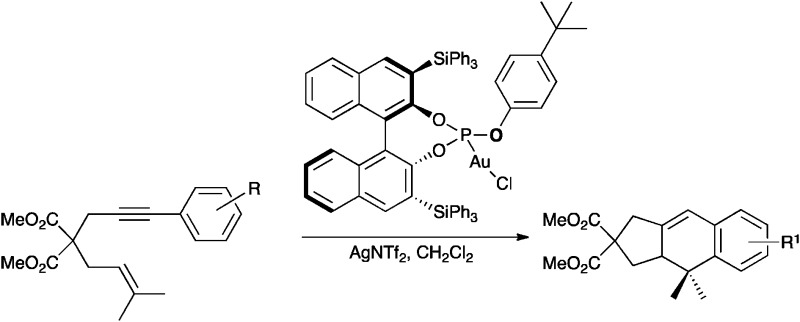
 Chiral gold(i) phosphite complexes are very reactive precatalysts for the [4+2] cycloaddition of aryl-substituted 1,6-enynes with enantiomeric ratios ranging from 86 : 14 up to 94 : 6.
Chiral gold(i) phosphite complexes are very reactive precatalysts for the [4+2] cycloaddition of aryl-substituted 1,6-enynes with enantiomeric ratios ranging from 86 : 14 up to 94 : 6.
Abstract
Chiral gold(i) phosphite complexes are readily prepared modularly from 3,3′-bis(triphenylsilyl)-1,1′-bi-2-naphthol. These chiral gold(i) phosphite complexes are very reactive precatalysts for the [4+2] cycloaddition of aryl-substituted 1,6-enynes with enantiomeric ratios ranging from 86 : 14 up to 94 : 6.
Introduction
Homogeneous gold catalysis provides efficient solutions for the construction of complex carbon skeletons under mild conditions.1–4 Much of the progress in the enantioselective C–C multiple bond activation catalysed by gold has been achieved in the last few years in intramolecular reactions.5–15 However, wide-scope enantioselective gold-catalysed transformations are still relatively scarce.
In 2005 we reported the first gold(i)-catalysed enantioselective alkoxycyclization of 1,6-enynes with a cationic catalyst generated in situ from [(R)-Tol-BINAP(AuCl)2] and AgSbF6.16 Related enantioselective cyclizations of 1,6-enynes have been carried out more recently with chiral NHC–gold(i)17 and phosphine–gold18 complexes, or using platinum catalysts.19
We have developed a general gold(i)-catalysed cycloisomerization of substrates 1 by formal [4+2] cycloaddition of arylalkynes with alkenes to form stereospecific cycloadducts 2,20 with the core structure of pycnanthuquinones (Scheme 1).21–23
Scheme 1. Gold(i)-catalyzed [4+2] cycloaddition of 1,6-enynes 1 and the structures of pycnanthuquinones A–C.

As part of a program on the development of general strategies for the synthesis of these terpenoid quinones, we examined an alternative pathway based on the gold-catalysed cyclization of benzyl-substituted 1,5-enynes.24 In parallel, we also studied the enantioselective cycloaddition of aryl-substituted 1,6-enynes 1 using a variety of gold(i) catalysts with chiral phosphineligands. Whereas we obtained modest enantioselectivities in most cases,25 the group of Genêt and Michelet reported good results in the cyclization of two substrates 1a–b in the presence of a gold(i) catalyst generated in situ from DTBM-MeOBIPHEP and AgOTf,26 although in the case of 1b the yield was significantly lower than that obtained with achiral catalysts20 (Scheme 2).
Scheme 2. Enantioselective gold(i)-catalysed [4+2] cycloaddition of 1,6-enynes 1a–b.

In an effort at developing general and practical methods for the screening of a large variety of chiral ligands in gold-catalysed reactions, we recently reported a procedure that allows performing enantioselective processes from catalysts prepared in situ from a cationic complex [Au(tmbn)2](SbF6) (tmbn = 2,4,6-trimethoxybenzonitrile) and the corresponding chiral ligand.27 As an alternative, we prepared a series of complexes bearing chiral phosphite ligands based on the BINOL motive using a relatively simple, modular approach from a commercially available 1,1′-bi-2-naphthol. We focused on phosphite ligands over phosphines because of their lower sensitivity to air and other oxidizing agents,28 and because phosphite gold(i) complexes are the most reactive catalysts for the activation of alkynes.29,30 Herein we report our efforts towards the development of chiral BINOL-derived phosphite gold(i) complexes. Chiral BINOL-derived phosphites have been used as building blocks for synthesis of chiral palladacycles, bis(phosphite) and mixed phosphite–phosphinite PCP-palladium pincer complexes.31,32 Monodentate phosphite gold(i) complexes with C 3-symmetry33 and chiral gold phosphoramidite-based catalysts have also been used in a number of gold-catalysed reactions.10–12,34
Results and discussion
We initially examined the gold(i)-catalysed cyclization of enyne1a to form adduct 2a using a wide range of complexes as precatalysts (Fig. 1). The structures of complexes L8(AuCl) (Fig. 2), L9(AuCl), L10(AuCl) (Fig. 3), L11(AuCl) (Fig. 4), and L12(AuCl)a (Fig. 5) and L12(AuCl)e were determined using X-ray diffraction.
Fig. 1. Chiral gold(i) complexes of the cyclization of 1,6-enyne 1a.

Fig. 2. X-Ray crystal structure of gold complex L8(AuCl). ORTEP plot (50% thermal ellipsoids).

Fig. 3. X-Ray crystal structures of ferrocenylphosphine gold complexes (a) L9(AuCl) and (b) L10(AuCl). ORTEP plot (50% thermal ellipsoids).

Fig. 4. X-Ray crystal structure of gold complexes L11(AuCl) and L10(AuCl). ORTEP plot (50% thermal ellipsoids). Hydrogens are omitted for clarity.

Fig. 5. X-Ray crystal structure of gold complex L12(AuCl)a. ORTEP plot (50% thermal ellipsoids). Hydrogens are omitted for clarity.

The cycloadditions were performed either at room temperature (condition A) or under microwave heating (condition B) (Table 1). Diphosphine–digold complexes L1(AuCl)2, L2(AuCl)2, and L3(AuCl)2 were investigated first (Table 1, entries 1–9). Cycloadduct 2a was obtained in all cases in good to excellent yield but only with low to moderate enantioselectivities. The best results with these diphosphine–digold complexes (56% ee) were obtained with L2(AuCl)2 in CHCl3 using AgPF6 under both conditions A and B (Table 1, entries 7 and 8). Using a 1 : 1 ratio of the digold complex to silver salt, under conditions in which the monocationic species are presumably formed, low enantioselectivities were observed. Biaryl gold–phosphine complex L4(AuCl) with the (R)-MOP ligand gave low enantiomeric excesses (Table 1, entries 11–13). BINOL-derived phosphoramidite complexes L5(AuCl) and L6(AuCl) also led to 2a in excellent yield but very poor enantioselectivities (Table 1, entries 14–19). Whereas reactions of complexes L7(AuCl)–L11(AuCl) led to poor to moderate enantioselectivities (Table 1, entries 20–27), results with phosphite gold complex L12(AuCl) were more promising (Table 1, entries 28 and 29). Although the enantiomeric excess was only marginally better than that obtained with L2(AuCl)2, phosphite gold complex L12(AuCl) was a significantly more reactive catalyst, leading to 2a in nearly quantitative yield in 12 h reaction time (vs. 24 h required with L2(AuCl)2).
Table 1. Enantioselective gold(i)-catalysed [4+2] cyclization of 1,6-enyne 1a to form 2a with complexes of Fig. 1 a .
| Entry | Au complex | AgX | Conditions | Time | Yield (%) | ee (%) |
| 1 | L1(AuCl)2 | AgSbF6 | A | 24 h | 71 | 24 |
| 2 | L1(AuCl)2 | AgSbF6 | B | 18 min | 92 | 7 |
| 3 | L1(AuCl)2 | AgPF6 | A | 24 h | 81 | 31 |
| 4 | L1(AuCl)2 | AgPF6 | B | 18 min | 90 | 39 |
| 5 | L2(AuCl)2 | AgSbF6 | A | 30 h | 90 | 25 |
| 6 | L2(AuCl)2 | AgSbF6 | A b | 18 min | 80 | 38 |
| 7 | L2(AuCl)2 | AgPF6 | A b | 24 h | 89 | 56 |
| 8 | L2(AuCl)2 | AgPF6 | B b | 15 min | 89 | 56 |
| 9 | L3(AuCl)2 | AgBF4 | A | 16 h | 91 | 25 |
| 10 | L4(AuCl) | AgSbF6 | A | 78 h | 56 | 18 |
| 11 | L4(AuCl) | AgSbF6 | B | 18 min | 78 | 20 |
| 12 | L4(AuCl) | AgPF6 | A | 78 h | 67 | 23 |
| 13 | L4(AuCl) | AgPF6 | B | 18 min | 84 | 25 |
| 14 | L5(AuCl) | AgSbF6 | A | 24 h | 91 | 8 |
| 15 | L5(AuCl) | AgSbF6 | B | 18 min | 95 | 12 |
| 16 | L5(AuCl) | AgPF6 | A | 24 h | 88 | 9 |
| 17 | L5(AuCl) | AgPF6 | B | 18 min | 94 | 14 |
| 18 | L6(AuCl) | AgSbF6 | B | 18 min | 95 | 5 |
| 19 | L6(AuCl) | AgPF6 | B | 18 min | 94 | 4 |
| 20 | L7(AuCl) | AgSbF6 | A | 12 h | 92 | 26 |
| 21 | L8(AuCl) | AgSbF6 | A | 2 h | 98 | <1 |
| 22 | L9(AuCl) | AgSbF6 | A | 24 h | >99 c | 35 |
| 23 | L9(AuCl) | OTf | A d | 24 h | >99 c | 46 |
| 24 | L9(AuCl) | NTf2 | A d | 24 h | 60 c | 50 |
| 25 | L10(AuCl) | AgSbF6 | A e | 24 h | >99 c | 50 |
| 26 | L10(AuCl) | AgSbF6 | A d | 24 h | >99 c | 39 |
| 27 | L11(AuCl) | AgSbF6 | A | 12 h | 92 | 26 |
| 28 | L12(AuCl) | AgSbF6 | A | 12 h | 99 | 57 |
| 29 | L12(AuCl) | AgBF4 | A | 16 h | 90 | 57 |
aAu complex (2.5 mol%) and AgX (2.5 or 5 mol% for mono and digold complexes, respectively). Conditions A: 23 °C, CH2Cl2. Conditions B: microwave heating at 80 °C, CH2Cl2.
bReaction in CHCl3.
cConversion determined using 1H NMR.
dReaction in benzene.
eReaction at –20 °C.
Overall, the structures of Au(i) complexes L11(AuCl) and L12(AuCl)a in the solid state are similar (Fig. 4 and 5), although the Au–P–OPh angle in L12(AuCl)a (102.90°) is significantly more acute than that of L11(AuCl) (114.98°). Complex L12(AuCl)a shows a cone-shaped binding pocket surrounding with a closest distance of 3.304 Å between the gold centre and a phenyl ring of one of the SiPh3 groups, which is within the range (3.0–3.2 Å) observed in gold(i) complexes in bulky biaryl Buchwald phosphines.35 This weak Au(i)–arene interaction is not present in complex L11(AuCl).
The preparation of a series of phosphite ligands(L12)a–n with different OR groups can be easily carried out using known methods31,32 from commercially available (R)-BINOL36 by known procedures via3,3′-bis(triphenylsilyl)-1,1′-bi-2-naphthol (3) (Scheme 3),37 which is also commercially available. Ligands(L12)a–n were routinely purified by chromatography on silica gel under an inert atmosphere and the corresponding gold(i) complexes L12(AuCl)a–n were prepared in quantitative yields by reaction with [AuCl(SMe2)].
Scheme 3. Synthesis of gold(i) phosphite complexes L12(AuCl)a–l from 3 and alcohols or phenols.

We assayed the catalytic activity of gold(i) complexes L12(AuCl)a–l (5 mol%) by mixing with AgSbF6 (5 mol%) at 0 °C in CH2Cl2, followed by addition of substrate 1a and slowly warming the reaction mixture to 23 °C over 2 h (Table 2).
Table 2. Enantioselective gold(i)-catalysed [4+2] cyclization of 1,6-enyne 1a to form 2a with complexes L12(AuCl)a–n a .
| Entry | Au complex | R | ee (%) |
| 1 | L12(AuCl)a | Ph | 70 |
| 2 | L12(AuCl)b | m-Tol | 72 |
| 3 | L12(AuCl)c | p-Tol | 80 |
| 4 b | L12(AuCl)c | p-Tol | 83 |
| 5 | L12(AuCl)d | 4-tBuC6H4 | 82 |
| 6 c | L12(AuCl)d | 4-tBuC6H4 | 88 |
| 7 | L12(AuCl)e | 4-MeOC6H4 | 60 |
| 8 | L12(AuCl)f | 2,4-Me2C6H3 | 74 |
| 9 | L12(AuCl)g | 3,5-Me2C6H3 | 81 |
| 10 | L12(AuCl)h | 2,4,6-Cl3C6H2 | 46 |
| 11 | L12(AuCl)i | 2-Napht | 70 |
| 12 | L12(AuCl)j | Me | 5 |
| 13 d | L12(AuCl)k | PhCH2 | 81 |
| 14 e | L12(AuCl)l | 3,5-tBu2C6H3CH2 | 74 |
aAu complex (5 mol%) and AgSbF6 (5 mol%), 0 to 23 °C, 2 h, CH2Cl2.
bReaction at –20 °C for 4 h.
cReaction at –20 °C for 16 h.
dReaction at –25 °C for 36 h.
eReaction at 0 °C for 7 h.
Under these conditions, L12(AuCl)a led to 2a in 70% ee (Table 2, entry 1). The enantioselectivity was raised further by using phosphite ligandsL12 derived from p-alkylsubstituted phenols (Table 2, entries 3–6). The best result (88% ee) was achieved with L12(AuCl)d derived from the tert-butylphenol group when the reaction was performed at –20 °C (Table 2, entry 6).38 Satisfactory results were also obtained with L12(AuCl)g and L12(AuCl)k (Table 2, entries 9 and 13).
The reactions with the best catalystL12(AuCl)d were slower (16–24 h) in 1,2-dichloroethane, ethyl ether, or acetone as solvent (63–82% ee), whereas no reaction was observed in toluene or 1,4-dioxane after 1–2 days. On the other hand, changing the silver salt from AgSbF6 to AgOTf or AgNTf2 did not significantly affect the reactivity and enantioselectivity, while slightly lower enantiomeric excesses were obtained with AgPF6.39
Finally, the optimized phosphite gold(i) catalyst L12(AuCl)d was applied for the cyclization of 1,6-enynes 1a–e using 2 mol% catalyst loadings (Table 3). Substrate 1b with a p-OMe group gave the corresponding cycloadduct 2b in good yield and enantioselectivity, although a longer reaction time was required (Table 3, entry 2). Good enantioselectivity was also obtained with enyne2c bearing a p-Me group (Table 3, entry 3). Sterically more demanding substrate 1d could also be cyclized in 70% yield and 79% ee (Table 3, entry 4). Finally, cyclization of 2e with a strong electron-withdrawing p-NO2 group at the phenyl ring gave cycloadduct 1d in 80% yield and 73% ee at 0 °C (Table 3, entry 5).
Table 3. Gold(i)-catalysed [4+2] cycloaddition of 1,6-enynes 1a–n with catalystL12(AuCl)d .

| ||||||
| Entry | Enyne | R | T (°C) | Time (h) | Product (yield, %) | ee (%) |
| 1 | 1a | H | –20 | 18 | 95 | 88 |
| 2 | 1b | p-MeO | –20 | 30 | 85 | 86 |
| 3 | 1c | p-Me | –20 | 15 | 98 | 87 |
| 4 | 1d | o-Me | –20 | 30 | 70 | 79 |
| 5 | 1e | p-O2N | 0 | 15 | 80 | 73 |
Conclusions
We have developed a series of chiral phosphite gold(i) complexes L12(AuCl)a–n that are easily prepared in a modular manner from BINOL. Cyclization of aryl-substituted 1,6-enynes with these complexes in the presence of a silver salt occurs with enantiomeric ratios ranging from 86 : 14 up to 94 : 6. It is also important to note that these chiral catalysts rival in reactivity with the most active catalysts for the cyclization of this more challenging class of compounds bearing a disubstituted alkyne.
Acknowledgments
We thank the MICINN (CTQ2010-16088/BQU), the AGAUR (2009 SGR 47), the European Research Council (Advanced Grant No. 321066), and the ICIQ Foundation for financial support. We also thank the ICIQ X-ray diffraction unit for the X-ray diffraction structures.
Footnotes
References
- Jiménez-Núñez E., Echavarren A. M. Chem. Rev. 2008;108:3326–3350. doi: 10.1021/cr0684319. [DOI] [PubMed] [Google Scholar]
- Gorin D. J., Sherry B. D., Toste F. D. Chem. Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Fürstner A., Davies P. W. Angew. Chem., Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]; (b) Fürstner A. Chem. Soc. Rev. 2009;38:3208–3221. doi: 10.1039/b816696j. [DOI] [PubMed] [Google Scholar]
- Michelet V., Toulleca P. Y., Genêt J.-P. Angew. Chem., Int. Ed. 2008;47:4268–4315. doi: 10.1002/anie.200701589. [DOI] [PubMed] [Google Scholar]
- (a) Huguet N. and Echavarren A. M., in Asymmetric Synthesis II, ed. M. Christmann and S. Bräse, Wiley-VCH Verlag, 2012, ch. 26, pp. 205–212. [Google Scholar]; (b) Widenhoefer R. A. Chem.–Eur. J. 2008;14:5382–5391. doi: 10.1002/chem.200800219. [DOI] [PubMed] [Google Scholar]; (c) Bongers N., Krause N. Angew. Chem., Int. Ed. 2008;47:2178–2181. doi: 10.1002/anie.200704729. [DOI] [PubMed] [Google Scholar]; (d) Sengupta S., Shi X. ChemCatChem. 2010;2:609–619. [Google Scholar]; (e) Pradal P., Toullec P. Y., Michelet V. Synthesis. 2011:1501–1514. [Google Scholar]; (f) Marinetti A., Jullien H., Voituriez A. Chem. Soc. Rev. 2012;41:4884–4908. doi: 10.1039/c2cs35020c. [DOI] [PubMed] [Google Scholar]
- (a) Johansson M. J., Gorin D. J., Staben S. T., Toste F. D. J. Am. Chem. Soc. 2005;127:18002–18003. doi: 10.1021/ja0552500. [DOI] [PubMed] [Google Scholar]; (b) Kleinbeck F., Toste F. D. J. Am. Chem. Soc. 2009;131:9178–9179. doi: 10.1021/ja904055z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hamilton G. L., Kang E. J., Mba M., Toste F. D. Science. 2007;317:496–499. doi: 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]
- (a) Liu C., Widenhoefer R. A. Org. Lett. 2007;9:1935–1938. doi: 10.1021/ol070483c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mukherjee P., Widenhoefer R. A. Angew. Chem., Int. Ed. 2012;51:1405–1407. doi: 10.1002/anie.201107877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C.-M., Beltrami D., Toullec P. Y., Michelet V. Chem. Commun. 2009:6988–6990. doi: 10.1039/b913554e. [DOI] [PubMed] [Google Scholar]
- Martínez A., García-García P., Fernández-Rodríguez M. A., Rodríguez F., Sanz R. Angew. Chem., Int. Ed. 2010;49:4633–4637. doi: 10.1002/anie.201001089. [DOI] [PubMed] [Google Scholar]
- Alonso I., Trillo B., López F., Montserrat S., Ujaque G., Castedo L., Lledós A., Mascareñas J. L. J. Am. Chem. Soc. 2009;131:13020–13030. doi: 10.1021/ja905415r. [DOI] [PubMed] [Google Scholar]
- González A. Z., Benitez D., Tkatchouk E., Goddard III W. A., Toste F. D. J. Am. Chem. Soc. 2011;133:5500–5507. doi: 10.1021/ja200084a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez-Pantiga S., Hernández-Díaz C., Rubio E., González J. M. Angew. Chem., Int. Ed. 2012;51:11552–11555. doi: 10.1002/anie.201206461. [DOI] [PubMed] [Google Scholar]
- Rodríguez L.-I., Roth T., Fillol J. L., Wadepohl H., Gade L. H. Chem.–Eur. J. 2012;18:3721–3728. doi: 10.1002/chem.201103140. [DOI] [PubMed] [Google Scholar]
- Handa S., Slaughter L. M. Angew. Chem., Int. Ed. 2012;51:2912–2915. doi: 10.1002/anie.201107789. [DOI] [PubMed] [Google Scholar]
- Sethofer S. G., Mayer T., Toste F. D. J. Am. Chem. Soc. 2010;132:8276–8277. doi: 10.1021/ja103544p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz M. P., Adrio J., Carretero J. C., Echavarren A. M. Organometallics. 2005;24:1293–1300. [Google Scholar]
- (a) Matsumoto Y., Selim K. B., Nakanishi H., Yamada K., Yamamoto Y., Tomioka K. Tetrahedron Lett. 2010;51:404–406. [Google Scholar]; (b) Wang W., Yang J., Wang F., Shi M. Organometallics. 2011;30:3859–3869. [Google Scholar]
- Chao C.-M., Genin E., Toullec P. Y., Genêt J.-P., Michelet V. J. Organomet. Chem. 2009;694:538–545. [Google Scholar]
- (a) Brissy D., Skander M., Retailleau P., Marinetti A. Organometallics. 2007;26:5782–5785. [Google Scholar]; (b) Toullec P. Y., Chao C.-M., Chen Q., Gladiali S., Genêt J.-P., Michelet V. Adv. Synth. Catal. 2008;350:2401–2408. [Google Scholar]; (c) Brissy D., Skander M., Jullien H., Retailleau P., Marinetti A. Org. Lett. 2009;11:2137–2139. doi: 10.1021/ol900724z. [DOI] [PubMed] [Google Scholar]; (d) Jullien H., Brissy D., Retailleau P., Marinetti A. Eur. J. Inorg. Chem. 2011:5083–5086. [Google Scholar]; (e) Jullien H., Brissy D., Sylvain R., Retailleau P., Naubron J.-V., Gladiali S., Marinetti A. Adv. Synth. Catal. 2011;353:1109–1124. [Google Scholar]
- (a) Nieto-Oberhuber C., López S., Echavarren A. M. J. Am. Chem. Soc. 2005;127:6178–6179. doi: 10.1021/ja042257t. [DOI] [PubMed] [Google Scholar]; (b) Nieto-Oberhuber C., Pérez-Galán P., Herrero-Gómez E., Lauterbach T., Rodríguez C., López S., Bour C., Rosellón A., Cárdenas D. J., Echavarren A. M. J. Am. Chem. Soc. 2008;130:269–279. doi: 10.1021/ja075794x. [DOI] [PubMed] [Google Scholar]
- Fort D. M., Ubillas R. P., Mendez C. D., Jolad S. D., Inman W. D., Carney J. R., Chen J. L., Ianiro T. T., Hasbun C., Bruening R. C., Luo J., Reed M. J., Iwu M., Carlson T. J., King S. R., Bierer D. E., Cooper R. J. Org. Chem. 2000;65:6534–6539. doi: 10.1021/jo000568q. [DOI] [PubMed] [Google Scholar]
- Laird D. W., Poole R., Wikström M., van Altena I. A. J. Nat. Prod. 2007;70:671–674. doi: 10.1021/np060566m. [DOI] [PubMed] [Google Scholar]
- Total synthesis of (–)-pycnanthuquinone C: Löbermann F., Mayer P., Trauner D., Angew. Chem., Int. Ed., 2010, 49 , 6199 –6202 . [DOI] [PubMed] [Google Scholar]
- López-Carrillo V., Huguet N., Mosquera Á., Echavarren A. M. Chem.–Eur. J. 2011;17:10972–10978. doi: 10.1002/chem.201101749. [DOI] [PubMed] [Google Scholar]
- Pérez-Galán P., PhD thesis, ICIQ-URV, 2005–2010. Delpont N., PhD thesis, ICIQ-URV, 2007–2011. [Google Scholar]
- Chao C.-M., Vitale M. R., Toullec P. Y., Genêt J.-P., Michelet V. Chem.–Eur. J. 2009;15:1319–1323. doi: 10.1002/chem.200802341. [DOI] [PubMed] [Google Scholar]
- Raducan M., Rodríguez-Escrich C., Cambeiro X. C., Escudero-Adán E., Pericàs M. A., Echavarren A. M. Chem. Commun. 2011;47:4893–4895. doi: 10.1039/c1cc10293a. [DOI] [PubMed] [Google Scholar]
- van Leeuwen P. W. N. M., Kamer P. C. J., Claver C., Pàmies O., Diéguez M. Chem. Rev. 2011;111:2077–2118. doi: 10.1021/cr1002497. [DOI] [PubMed] [Google Scholar]
- Amijs H. M., López-Carrillo V., Raducan M., Pérez-Galán P., Ferrer C., Echavarren A. M. J. Org. Chem. 2008;73:7721–7730. doi: 10.1021/jo8014769. [DOI] [PubMed] [Google Scholar]
- Benitez D., Shapiro N. D., Tkatchouk E., Wang Y., Goddard III W. A., Toste D. F. Nat. Chem. 2009;1:482–486. doi: 10.1038/nchem.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Bedford R. B., Chang Y.-N., Haddow M. F., McMullin C. L. Dalton Trans. 2011;40:9034–9041. doi: 10.1039/c1dt10356c. [DOI] [PubMed] [Google Scholar]; (b) Bedford R. B., Chang Y.-N., Haddow M. F., McMullin C. L. Dalton Trans. 2011;40:9042–9050. doi: 10.1039/c1dt10357a. [DOI] [PubMed] [Google Scholar]
- (a) Phopshite L11: Kawasaki M., Li P., Yamamoto H., Angew. Chem., Int. Ed., 2008, 47 , 3795 –3597 . [DOI] [PubMed] [Google Scholar]; (b) Phopshite L12a: Sakakura A., Sakuma M., Ishihara K., Org. Lett., 2011, 13 , 3130 –3797 . [DOI] [PubMed] [Google Scholar]
- González A. Z., Toste F. D. Org. Lett. 2009;12:200–203. doi: 10.1021/ol902622b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Teller H., Flügge S., Goddard R., Fürstner A. Angew. Chem., Int. Ed. 2010;49:1949–1953. doi: 10.1002/anie.200906550. [DOI] [PubMed] [Google Scholar]; (b) Teller H., Corbet M., Mantilli L., Gopakumar G., Goddard R., Thiel W., Fürstner A. J. Am. Chem. Soc. 2012;134:15331–15342. doi: 10.1021/ja303641p. [DOI] [PubMed] [Google Scholar]
- (a) Herrero-Gómez E., Nieto-Oberhuber C., López S., Benet-Buchholz J., Echavarren A. M. Angew. Chem., Int. Ed. 2006;45:5455–5459. doi: 10.1002/anie.200601688. [DOI] [PubMed] [Google Scholar]; (b) Pérez-Galán P., Delpont N., Herrero-Gómez E., Maseras F., Echavarren A. M. Chem.–Eur. J. 2010;16:5324–5332. doi: 10.1002/chem.200903507. [DOI] [PubMed] [Google Scholar]
- Brunel J. M. Chem. Rev. 2005;105:4233. doi: 10.1021/cr040079g. [DOI] [PubMed] [Google Scholar]
- Albrow V. E., Blake A. J., Fryatt R., Wilson C., Woodward S. Eur. J. Org. Chem. 2006:2549–2557. [Google Scholar]
-
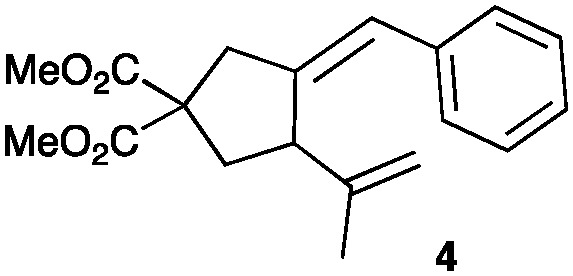
(a) (b). (a) Adduct 2a of 88% ee (determined by HPLC) has [α]20D – 25.0 ± 2.0 (c = 0.11, CHCl3). This value contrasts with that reported for 2a of 93% ee, [α]21D + 14.8, (c = 0.93, CHCl3) in ref. 26. When the cyclization of 2a with L(AuCl)2 (L = (R)-4-MeO-3,5-(tBu)2MeOBIPHEP = DTBM-MeO-BIPHEP) (3 mol%) and AgOTf (6 mol%) in Et2O, in addition to 2a (74 ee, estimated by chiral HPLC), known 4 (ref. 38b) was also obtained (72 : 28 ratio). We could not find conditions that would allow the full resolution of 4 and the enantiomers of 2a by chiral HPLC;
Porcel S., Echavarren A. M., Angew. Chem., Int. Ed., 2007, 46
, 2672
–2676
,
 . [DOI] [PubMed] [Google Scholar]
. [DOI] [PubMed] [Google Scholar] - See ESI for details
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.