

Abstract
Nonribosomal peptide synthetases (NRPS) are large modular macromolecular machines that produce small peptide molecules with wide-ranging biological activities, such as antibiotics and green chemicals. The condensation (C) domain is responsible for amide bond formation, the central chemical step in nonribosomal peptide (NRP) synthesis. Here we present two crystal structures of the first condensation domain of the calcium-dependent antibiotic (CDA) synthetase (CDA-C1) from Streptomyces coelicolor, determined at resolutions of 1.8 Å and 2.4 Å. The conformations adopted by CDA-C1 are quite similar in these two structures, yet distinct from those seen in other NRPS C domain structures. High pressure liquid chromatography based reaction assays show that this CDA-C1 construct is catalytically active, and small angle X-ray scattering experiments suggest that the conformation observed in these crystal structures could faithfully represent the conformation in solution. We have performed targeted molecular dynamics simulations, normal mode analyses and energy minimized linear interpolation to investigate the conformational changes required to transition between the observed structures. We discuss the implications of these conformational changes in the synthetic cycle, and of the observation that the “latch” that covers the active site is consistently formed in all studied C domains.
Introduction
Nonribosomal peptide synthetases (NRPSs) are large macromolecular machines that catalyze the assembly of monomer substrates into biologically active secondary metabolites 1–3. As the name implies, NRPS substrates are often amino acids, but over four hundred monomers are known to be used as substrates, including D-amino acids, aryl acids, keto acids, hydroxy acids, and fatty acids 4. Nonribosomal peptides have important and diverse biological activity and include anti-fungals, anti-bacterials, antivirals, anti-tumours, siderophores, and Immunosuppressants 3, including well-known compounds such as penicillin 5, daptomycin 6, and cyclosporin 7.
NRPSs are organized into modules of >110 kDa, with each module responsible for the addition of one specific monomer. Modules contain multiple domains, each performing specific functions in product synthesis 8. A basic elongation module contains a condensation (C) domain, an adenylation (A) domain and a peptide carrier protein (PCP) domain. The A domain selects the cognate amino acid and adenylates it, then attaches it to a phosphopantetheinyl (PPE) group on the PCP domain. The PCP domain transports the amino acid to the C domain, which catalyzes peptide bond formation between this amino acid and the peptide attached to the PCP domain of the preceding module, thus elongating the peptide chain. Next, the PCP domain brings the elongated peptide chain to the downstream module, where it is passed off and further elongated in the next peptidyl transferase reaction.
In this reaction cycle, numerous conformational changes are known, or have been proposed, to occur. As described above, the PCP domain must completely translocate to interact with different partner domains 9, 10, and is also known to change conformation, depending on its functional state 11. Large scale-movements are known to occur in the A domain between the conformation which catalyzes the adenylation reaction and that which catalyzes the thiolation reaction (attachment of substrate to PCP domain) 12–15. Thus, conformational cycling is absolutely essential for NRPS function.
The C domain catalyzes the key catalytic event of NRPS function, peptide (amide) bond formation 16, 17. Three structures which include NRPS C domains have been determined by X-ray crystallography: a stand-alone C domain 18, a C–PCP didomain complex 19 and a C-A-PCP-Te termination module 10. The C-domain comprises ~450 amino acids and has a pseudo-dimer configuration, with both N and C-terminal subdomains having cores with folds in the CoA dependent acyltransferase (CAT) superfamily. The active site sits at the bottom of a “canyon” 19 or “V” 10 formed by the two subdomains of the C domain, and is covered by a “latch” that crosses over from C to N subdomain. The catalytic center includes an HHxxDG sequence motif 16, 17, 20 and must have binding sites for its donor and acceptor substrates. The conformations of the C domain visualized in these three structures vary somewhat, though it is unclear whether these differences stem from the fact that different proteins were used in the three studies or from the possibility that they are in different functional states. Although there are hundreds of different C domains in NRPS biosynthetic clusters, they likely all share a common mode of action.
In this study we focus on the first C domain of the calcium dependent antibiotic (CDA) synthetase, CDA-C1 (Figure 1) 21, 22. The calcium dependent antibiotic is part of a class of acidic lipopeptides which includes the last resort antibiotic daptomycin 23. These antibiotics work by binding to and disrupting cytosolic membranes in bacteria 24. The CDA biosynthetic cluster in Streptomyces coelicolor includes an 11-module canonical NRPS spread over 3 proteins, which adds the 11 amino acids and cyclizes the product (Figure 1a) 22. The first monomer is not an amino acid, but a fatty acid, a 2,3-epoxyhexanoyl group which is synthesized as a hexanoyl fatty acid on an acyl carrier protein (ACP) by a fatty acid synthase, then modified to the epoxy form by epoxidation enzymes HxcO and/or HcmO (Figure 1b) 25, 26. The first C domain of CDA synthetase catalyses the transfer of the 2,3-epoxyhexanoyl group from 2,3-epoxyhexanoyl-ACP to the serinyl-PCP domain of the first module. Although normally present as part of the 799 kDa CDA PS1, excised CDA-C1 is active in catalysis 26.
Figure 1. Schematic diagrams for CDA components.

a) A schematic diagram of the proteins CDA peptide synthetase 1, 2 and 3 which make up the NRPS system for calcium dependent antibiotic synthesis in Streptomyces coelicolor. b) A schematic diagram illustrating the reaction catalyzed by CDA-C1. c) A schematic diagram of the chemical structure of CDA-4b, a representative CDA peptide synthesized in Streptomyces coelicolor 4, 26.
Here we present two structures of the first condensation domain of the calcium-dependent antibiotic (CDA) synthetase (CDA-C1), determined by X-ray crystallography at resolutions of 1.8 Å and 2.4 Å, and accompanying small angle X-ray scattering (SAXS), activity assays and computational analyses.
Results and discussion
Purification, crystallization and structure determination of CDA-C1
CDA-C1 is the N-terminal domain of CdaPS1, a 20 domain, 7463 residue NRPS protein 26. To study CDA-C1, we designed a gene construct by aligning the sequences of CDA-C1 to C domains with known structure to enable C-terminal boundary definition. The resulting construct was heterologously expressed in E. coli with extremely high yields and purified to homogeneity using a four-column chromatographic protocol. CDA-C1 was subjected to high-throughput crystallization trials, which readily yielded crystals in several crystallization conditions. Two crystallization conditions were optimized and could be used reproducibly to produce crystals in P212121 and P21 space groups which diffract to high resolution. Data collection at a rotating anode “home” source gave complete data sets with good statistics (Table S1). These were subjected to phasing trials using molecular replacement with many different search models derived from one or both subdomains of the known C domains, with no success. Selenomethionine-derivatized protein was then produced, which could be purified and crystallized by the same protocols. Multi-wavelength data sets were collected from P212121 crystals at the National Synchrotron Light Source, and multiwavelength anomalous dispersion (MAD) phasing techniques were used to determine the structure to 1.8 Å resolution (Figure 2, Supplementary Figure S2, Table S1). Electron density maps showed that there are two molecules in asymmetric unit with very similar conformation, and allowed building of residues 4 – 449 in molecule A and 2 – 448 in molecule B. The use of this structure as a search model readily gave a molecular replacement solution for the P21 crystal form using home source data with resolution solved to 2.4 Å (Figure S2b, Table S1). This final model included residues 4 – 222 and 234 – 450 in molecule A and 5 – 230, 241 – 410 and 420 – 448 in molecule B.
Figure 2. Structure of CDA-C1.
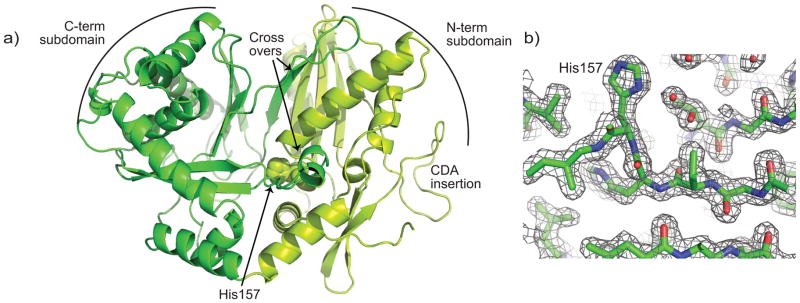
a) Ribbons representation of CDA-C1 determined in the P212121 space group. The N-terminal subdomain is chartreuse, the C-terminal subdomain is green, and the domain crossovers, CDA-specific insertion and active site H157 are all indicated. b) A 2FO-Fc electron density map contoured at 1σ.
The structure of CDA-C1
CDA-C1 adopts the classical C domain conformation 10, 18, 19, with two subdomains, each having a core CAT fold, two points of crossover from the C-terminal subdomain to the N-terminal subdomain, and the active site in the center of the “V” formed by the subdomains (Figure 2). Superimposition of all four independent molecules determined here (two molecules from the P212121 crystal form and two from the P21 crystal form) show that in all these molecules, the subdomains adopt approximately the same relative orientation to one another (Supplementary Figure S3). The only major difference between these structures is in the conformation of the loop 82–96. Loop 82–96 is in the linker between two beta sheets of the N-terminus. Multiple alignment of over 500 C domains 27 shows that this loop is almost unique to CDA-C1, with only a protein 930752.1 from Photorhabdus luminescens also having an insertion in the same location (albeit with no conservation between the two sequences) (Supplementary Figure S1). Since CDA-C1 and Plu 930752 are initiator C domains, this face may have less stringent conservation requirements: the C domain does not have to pack with a full upstream module, rather it needs to interact only with the small ACP proteins.
Comparison of the structures presented here with the structures of the three previously determined structures of C domains reveals significant conformational differences. When the C-terminal subdomains of the characterized C domains from tyrocidine synthetase III (TycC) 19, vibriobactin synthase (VibH) 18 and surfactin A synthetase C (SrfAC) 10 are superimposed upon CDA-C1, the N-terminal subdomains of these proteins are in strikingly different positions (Figure 3, Supplementary Figure S4). CDA-C1 is in a much more “closed” conformation, with its N and C-terminal subdomains closer together. The N-terminal subdomain would require a shift and large rotation of ~15°, ~22°, and ~25° respectively to assume the conformations seen with VibH, TycC and SrfAC. This is a large movement – the alpha carbon positions in the N-terminal domain move by as much as 19, 20 and 23 Å and on average differ by 6, 8 and 11 Å.
Figure 3. Comparison of CDA-C1 with other C domains.

Alignment of the C-terminal subdomain shows different relative subdomain–subdomain orientation in the structures of C domains. CDA-C1 displays the most “closed” conformation. CDA-C1 is in green, the C domain from SrfAC is yellow, from VibH is brown and from TycC is orange. See supplementary figure S4 for individual comparisons.
SAXS analysis of CDA-C1
This “closed” conformation of CDA-C1 seemed unlikely to be artificially forced by crystal packing. All CDA-C1 monomers are observed in very similar conformations, even though the packing contacts between the two molecules in each asymmetric unit (crystallographic dimers) are solely through the C-terminal subdomain, and many of the other crystal contacts are different for the two space groups (Supplementary Figure S3d). To address whether this “closed” conformation is indeed adopted in solution, we undertook a SAXS analysis of CDA-C1.
SAXS data was collected on CDA-C1 at multiple concentrations with each experiment giving consistent results (a typical scattering result is shown in Figure 4). Molecular weight of CDA-C1 in solution was found to be ~97 kDa using the Kratky plot method (comparing to BSA and lysozyme) which agrees well with calculated molecular weight of CDA-C1 dimer (96.8 kDa) (Figure S5). We calculated theoretical scattering curves from the structures of both a single CDA-C1 and the dimer of CDA-C1 observed in both P212121 and P21 crystal forms. Comparison of the scattering data and the theoretical curves obviously indicates that CDA-C1 is a dimer in solution, and that the dimer is in a conformation very similar to that of the crystallographic dimer (Figure 4a). The envelope calculation also shows a good consistency between our crystallographic dimer and the solution conformations as assayed by SAXS (Figure 4b). Although CDA-C1 appears to exist as a dimer in solution and in the crystal, and this dimer interface is accessible in the SrfaC termination module 10, the dimer interface does not show any sequence conservation among C domains so may not have biological significance in the intact NRPS.
Figure 4. SAXS analyses of CDA-C1.

a) Fitting of calculated scattering curves to merged experimental data indicating that CDA-C1 is a dimer in solution b) superposition of averaged filtered envelope on crystallographic CDA-C1 dimer c) quality of fit of various models to experimental scattering data suggesting that CDA-C1 in solution is best described by crystallographic dimer in it’s observed conformation.
To test whether the SAXS data could differentiate between the C domain conformation observed in the crystal structure reported here and those conformations seen in the previously reported structures (Figure 3, Supplementary Figure S4), we created models of CDA-C1 in the conformations of VibH, TycC and SrfAC. These models were used to calculate theoretical scattering curves which were fitted to the experimental SAXS scattering data, with goodness of fit assessed by calculating χ2 values. CDA-C1 had the lowest χ2 to the SAXS data (Figure 4c), although the values for VibH and SrfAC were only slightly higher. TycC had a substantially higher χ2. Thus, the conformation we observe in both P212121 and P21 crystal forms is at least consistent with solution studies.
CDA-C1 is catalytically active
We next asked whether the construct of CDA-C1 we made, which adopts this “closed” conformation, is catalytically active. We first attempted to show activity of our construct using small molecule acyl-N-acetylcysteamine thioester analogues (acyl-SNACs) 28 of the two substrates (2,3-epoxy-hexanoyl-SNAC and serinyl-SNAC), but we were unable to detect product (2,3-epoxy-hexanoyl-serinyl-SNAC) formation. While we were performing our studies, Marahiel and co-workers published experiments with a similar construct of CDA-C1, which showed their construct to be active in an assay where the two substrates are delivered by carrier proteins and the product 2,3-epoxy-hexanoyl-serinyl-PCP is separated by HPLC and confirmed by mass spectrometry 26. We could demonstrate catalytic activity of our CDA-C1 construct using a version of this assay (Figure 5). The assay is complicated by the fact that serinyl-PCP (and, in our experience, 2,3-epoxy-hexanoyl-ACP) is liable to hydrolysis, and that several PCP species migrate very similarly on reverse phase columns. Nonetheless, we were able to clearly identify product formation when CDA-C1 was in the reaction mix, but not when it was replaced by CDA-C1 harboring an H157A active site mutation (Figure 5) 26.
Figure 5. CDA-C1 is catalytically active.

a) A schematic diagram explaining the C domain activity assay. b) HPLC traces and c) mass spectra of reaction assays show that product epoxyl-hexanoyl-serinyl-PCP1 is formed only when wild type CDA-C1 (blue) is present in the reaction mixture. Reactions with CDA-C1 harboring the active-site mutation H157A (red) do not lead to product formation. Expected mass of epoxyl-hexanoyl-serinyl-PCP1 is 12618.3 Da. Expected and observed masses for other carrier protein states are listed in Table S2.
Thus the CDA-C1 construct we crystallized is catalytically active, and the conformation we observe in the crystal structures is consistent with SAXS results probing its solution conformation.
Computational analyses of C domain movement
Although each structurally characterized C domains is seen in a different conformation, there appears to be sufficient space at the active site to bind substrate in each form. One could imagine that progressively more open C domain conformations would occur in C domains from later modules of NRPS to accommodate larger substrates, but that is unlikely to be the cause of the different conformations: VibH is an initiator C domain like CDA-C1, but found in an open conformation, and SrfAC is more open than TycC despite coming from an earlier module in their respective NRPS (Figure 3, Figure S4, Movie S7). It is unclear if all these conformations are catalytically active, or if a C domain would sample each of the observed conformations during an NRPS catalytic cycle. There is extensive precedent for using crystal structures of homologous proteins to investigate conformational changes that a protein may undergo in solution 29–32. Therefore, we undertook analyses of the types of movement required to transition between observed conformations.
We subjected CDA-C1 to normal mode analysis to observe whether a transition from “closed” to “open” form would be replicated by normal modes, using the programs WEBnm@ 33 and NOMAD-Ref 34. Both programs reveal that such a movement is reasonable. WEBnm@ replicates this movement very well (Movie S1), although in this simulation, the amplitude is not quite enough to bring it to a fully “open” state. Likewise, several NOMAD-Ref modes appear to replicate similar movements (Movie S2–S3).
To study and visualize the full transition between closed and open states as well as between the various open states, we carried out two further computational exercises. First, we performed linear interpolation with energy minimization refinement (“morphing”) to interconvert the conformations. The resulting animations show the transition from the conformation seen in this study to those of previously published structures (Movie S4–6), as well as the movements required to transition between these states (Movie S7). Secondly, we performed targeted molecular dynamics simulations on the same transitions (Movie S8–S11). The targeted molecular dynamics simulations transitions appear smooth and do not seem to have to pass through any obviously unfavorable conformations. The root mean square deviations between progressive models in the simulations and the target models in SrfAC, TycC and VibH conformation smoothly decrease for residues in the N terminal subdomain and crossover strands (Figure S5).
Together, the normal mode analyses, the morphing and targeted molecular dynamics simulations suggest that the conformation changes which would be required to interconvert the four conformations observed in NRPS C domains are reasonable.
The putative conformational changes describe here involve relative movement between the two CAT fold containing subdomains, while the conformation of each CAT core fold remains unchanged. There are numerous proteins which contain CAT folds, three of which have been characterized structurally and shown to also contain two “pseudodimeric” CAT folds: murine carnitine acetyltransferase (CrAT) 35, human choline acetyltransferase (CHAT) 36 and polyketide-associated protein A5 (PapA5; which has a similar overall structure to C domains) 37. We asked whether similar opening has been seen in these three proteins. CrAT does not undergo significant conformational changes upon binding substrate 35; CHAT undergoes small but significant conformational change (~1.5° opening) upon substrate binding 36 and PapA5 is hypothesized to undergo some kind of opening of the active site to allow substrate to bind, as an α helix blocks the active site in the apo conformation, but this opening has yet to be observed 37. Therefore, the opening of two CAT fold subdomains is a feature in some, but not all proteins which contain them.
Though some C domains are expressed at stand-alone proteins, they are more usually part of a much larger protein, so the conformational changes must be considered in the context of a full NRPS. Within a module, an outside face of the C-terminal subdomain of the C domain forms a large binding interface with the A domain 10. The open or closing of the C domain would not abolish this C – A interface, though subtle changes within the contact area might occur. We speculate that conformational changes such as the opening of the C domain may be part of a communication network in NRPSs by which the functional state of one domain is conveyed to other domains. This may include sensing of the Asub domain position, which forms part of the CA interface in the adenylation state but rotates away in the thiolation state 10, 12–15, or PCP domain position, which has been observed bound at the acceptor site of an open C domain 10. Such a communication network could to contribute to appropriate timing and coordination of the many reactions in the NRPS synthetic cycle, increasing the efficiency and rate of small molecule synthesis 38, 39. Confirmation of this theoretical network would require multiple structures of intact NRPSs and accompanying biochemical analyses.
The “latch” of the C domain
There are two points of crossover where a segment from the C-terminal subdomain crosses over to form part of the N-terminal subdomain. Residues ~295–309 cross over and form a small alpha helix, which backs against N-terminal subdomain helices h3 and h4. Residues ~367–388 cross and donate a beta strand to the major beta sheet in the N-terminal subdomain (formed of strands s1, s4, s5, s6) (Figure 2 and Figure S7a). This segment has been described as a “latch”, forming a “roof” of the active site, and has been proposed to disengage from the N-terminal subdomain during the reaction cycle 19. However, in all structures of C-domains, the crossover “latch” interaction is intact (Figure S7). Furthermore, the interaction remains intact throughout every NMA, MD and morph simulation performed here (Movies S1–11). Buried surface area calculations suggest that it is possible that the latch, at least in some C domains, could remain intact: In CDA-C1, SrfAC, TycC and VibH the latch buries 954 Å2, 903 Å2, 641 Å2 and 927 Å2 of surface area respectively, where interface on the order of 700 Å2 is known to support heterodimer formation 40,41. Finally, Samel et al. 19 argue that high B factors in the loop suggest that it could be a mobile element, but the latch residues in our structures display slightly lower than average B factors (average B factor for latch: 20.8 Å2; average B factor for structure: 24.1 Å2). Conceptually, it seems reasonable that the latch would open, because if it did not, the growing peptide chain would need to be threaded through each C domain active site. However, we do not feel that there is at this time compelling evidence to support latch opening.
An active site tunnel and transition state model
As no structure of a C domain with substrates has been determined, the precise binding site and approach of the PPE-bound substrates are not definitively known, but can be reasonably guessed 10, 18, 19. The donor and acceptor substrates must approach and bind from opposite faces and meet at the active site H157. If the latch is not open, as is the case in all structures and models to date, the active site can be described as being at the center of a tunnel in the middle of the C domain (Figure 6). We identified this tunnel visually and also by using the program Caver 42. This tunnel, formed partially by the latch, stays intact through the morphing and MD simulations, and in all crystal structures. The tunnel is ~30 Å long and wide enough to accommodate substrates. Indeed, the tunnel entrances correspond to what is known about the binding sites for the upstream and downstream carrier protein domains. The SrfAC module has a substrate-less PCP domain positioned with its PPE attachment site at a reasonable distance from the C domain’s catalytic histidine, and positioned at the tunnel entrance shown in Figure 6b 19. The structure of the TycC didomain has an upstream PCP domain in an unproductive conformation, but it is generally positioned near the tunnel entrance shown in Figure 6c, showing that the upstream carrier protein domain can reach this site. To give a holistic view of the C domain at the point of NRPS peptide bond formation, we constructed a model of the transition state of the reaction, including the C domain, 2,3-epoxy-hexenoyl-ACP and serinyl-PCP domain (Figure 6). The starting position of the PCP domain was taken from the SrfAC model, while the ACP was docked, using the program HADDOCK, taking into account the residues on the carrier proteins required for productive donor binding data 43–45. The pantetheinyl substrates were roughly positioned along the tunnel, and the whole complex was subject to multiple rounds of Cartesian coordinate energy minimization in the program CNS.
Figure 6. A tunnel to the active site of CDA-C1.

a,b) Surface representations of CDA-C1 show a tunnel through CDA-C1, with the active site in its center. c) A representation of the tunnel from CAVER 42 output. d,e) A model of the transition state of the CDA-C1 catalyzed reaction including CDA-C1, ACP, PCP1 and covalently-attached transition state. Note that the binding sites of upstream and downstream carrier proteins are not related by the same symmetry operation that relates the CAT core of the N- and C-terminal subdomains. The CAT cores are related by a ~120° rotation about an axis approximately parallel to the acceptor portion of the tunnel. Since the tunnel has a carrier protein at each end, rotation of the entire modeled complex about this axis does not interchange the positions of the carrier proteins.
The model fits exactly into the described tunnel, with the nucleophilic alpha amino group of the serinyl-PCP domain modeled at 3 Å distance from the putative catalytic residue, as would be expected for the intermediate state of the reaction. The fatty acid 2,3-epoxy hexanoyl side chain fits nicely into a pocket lined by α helix 4 and the major β sheet in the N-terminal subdomain, whereas the serine faces the C-terminal subdomain. Although precise analysis of substrate-C domain interactions awaits successful co-complex determination, this model will be useful in efforts to dissect the substrate specificity shown by C domains 26, 28, 46–49.
In summary, we have presented a structure of an active, previously underdetermined NRPS condensation domain, which adopts a novel conformation. Conformational changes in C domains such as those modeled in our computational analyses are likely to occur in the catalytic cycle of NRPS C domains and may be important for peptide synthesis.
Methods
Cloning and expression of CDA-C1
The CDA-C1 construct was designed by aligning the sequence of first C domain of CDA peptide synthetase I of Streptomyces coelicolor A3(2) (NCBI NP_627443.1) to C domains of known structure (Figure S1). The CDA-C1 construct was synthesized by DNA 2.0, Inc. (Menlo Park, CA, USA), featuring an N-terminal octahistidine tag and tobacco etch virus (TEV) protease cleavage site of sequence MHHHHHHHHENLYFQG and CDA residues Met1-Thr449. Protein production was performed in BL21 (DE3) E coli cells grown in LB medium supplemented by 300 μg ml−1 kanamycin (LB-kan) at 37°C. CDA-C1 expression was induced at an OD600 ~0.5–0.6 with the addition of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and expression continued overnight at 16°C. Selenomethionine derivatized (SeMet) CDA-C1 was expressed in the same cell strain grown in kanamycin-containing M9 minimal medium supplemented with 0.2% glucose, 2 μM MgSO4, and 0.1 μM CaCl2. After reaching an OD600 of 0.5, the medium was further supplemented with the amino acids K, F, T (100 mg l−1 each), I, L, V (50 mg ml−1 each) and seleno-L-methionine (60 mg l−1) 50. Fifteen minutes after supplementation, cultures were induced with 1 mM IPTG and grown overnight at 25°C.
Protein purification of CDA-C1
CDA-C1 cell pellets were resuspended in a buffer of 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM β-mercaptoethanol (βME), 1 mM imidazole pH 8.0, and 1 mM phenylmethanesulfonyl fluoride (PMSF). Cells were lysed by sonication, and centrifuged for 30 min at 40 000 × g and 4°C. The supernatant was pooled and applied onto a 5 ml HiTrap IMAC FF column (GE Healthcare) charged with Ni2+. CDA-C1 was eluted with 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM βME, 250 mM imidazole pH 8.0, and 1 mM PMSF; purity was assessed using SDS-PAGE. The fractions containing CDA-C1 were diluted 5-fold with a buffer of 50 mM Tris-HCl pH 7.5 and 2 mM β-mercaptoethanol, and applied to a MonoQ 5/50 GL column (GE Healthcare) and eluted with a gradient of 0–1 M NaCl. Relevant fractions were pooled and digested overnight at 4°C with N-His-TEV protease, using a ratio of 1 mg N-His-TEV protease per 40 mg CDA-C1. The cleaved sample was reapplied onto the 5 ml HiTrap IMAC FF column charged with Ni2+ and the flow through collected. The sample was brought to 1 M (NH4)2SO4, applied to 2 × 1 ml HiTrap phenyl HP column (GE Healthcare) and eluted with a gradient of 1–0 M (NH4)2SO4. After dialysis into 50 mM Tris-HCl pH 7.5, 100 mM NaCl and 2 mM dithiothreitol (DTT), CDA-C1 was concentrated to 10 mg ml−1, flash-frozen and stored in liquid nitrogen.
Crystallization and data collection
Sparse-matrix crystallization trials of CDA-C1 and subsequent optimization identified two crystallization solutions that allowed growth of diffraction-quality crystals in sitting drop format using 2 μl of 10 mg ml−1 protein sample and 2 μl of crystallization solution in the drop and a 400 μl in the reservoir: (a) 25–27% PEG 3000, 0.2–0.25 M lithium sulfate, 0.1 M HEPES, pH 7.5, and (b) 9–13% PEG 10000 and 0.1 M HEPES pH 7.1–7.3. Crystals in condition (a) were directly mounted in cryoloops and flash-cooled in liquid nitrogen, while crystals in (b) were cryoprotected in a solution containing 20% PEG 10000 prior to mounting. SeMet-CDA-C1 crystals were grown in 26–31% PEG 3000, 0.2–0.26 M lithium sulfate and 0.1 M HEPES pH 7.5.
Data from SeMet-CDA-C1 crystals were collected at Se peak, inflection and remote wavelengths at X6A beamline at the National Synchrotron Light Source (NSLS), Brookhaven National Laboratory (BNL), Brookhaven, NY with a 0.5° frame width (Table S1). Native data sets were collected using a Rigaku RUH-3R rotating copper-anode source equipped with a R-AXIS IV++ image plate detector at McGill University, Montreal, Canada (GRASP) with a 0.1° frame width.
Structure determination
Data were indexed and scaled using the program HKL-2000 51 The structure of SeMet-CDA-C1 was determined by a three-wavelength multiwavelength anomalous dispersion (MAD) phasing experiment using the program Phenix 52 (Table S1). The structure of CDA-C1 in the crystal form grown in the PEG 10000 – containing condition was determined by molecular replacement with SeMet-CDA-C1 as the reference model, using the program Phaser 53. Structures were subjected to iterative rounds of modeling and refinement to give the final model (Table S1).
SAXS analyses
Purified CDA-C1 was subjected to gel filtration on Superdex200 10/300 GL column equilibrated with 25 mM Bis-TRIS pH 7.0 150 mM NaCl 10% glycerol. The most concentrated fraction (0.843 mg/ml) was used unaltered for SAXS data collection. The remaining fractions were pooled and concentrated on an Amicon(R) Ultra concentrator with 10kDa cut-off membrane to produce samples at 3.48, 9.82 and 23.04 mg/ml. Samples were filtered through a Millipore Ultrafree-MC VV 0.1 um filter prior to loading into SAXS cell. SAXS data was collected on an Anton Paar SAXSess MC2 CCD system on a PANalytical PW3830 generator with a Cu LFF tube (GRASP). Beam was collimated to 8mm length. Data points from 0.35 to 0.4 1/nm were covered by a CCD blemish and thus omitted. Primary data was processed and desmeared (Lake algorithm) using SAXSquant software. Scattering curves were merged using the program PRIMUS 54. Theoretical scattering was calculated and fitted to experimental data using the program CRYSOL 55. P(r) plots were calculated with the program GNOM 55. For ab initio shape reconstruction 50 models generated with the program DAMMIF 56 were averaged with the program DAMAVER 57 assuming 2-fold symmetry. The resulting averaged and filtered envelope was superimposed onto crystal structure using the program SUPCOMB 58.
Expression and purification of enzymes required for reaction assay
ACP SCO3249 (ACP) (NCBI NP_627461.1, residues Met1-Ala89), DptA-PCP1 (PCP1) (NCBI WP_006122820.1, residues Asn929-Thr1030) and HxcO (NCBI NP_627459.1, residues Thr2-Pro600) constructs were designed based on the sequences reported by Marahiel and colleagues 25, 26 and synthesized by DNA 2.0, Inc. (Menlo Park, CA, USA). ACP and HxcO both featured an N-terminal tag sequence MHHHHHHHHENLYFQG, and PCP1 featured a C-terminal tag sequence ENLYFQGHHHHHH. Protein production of HxcO, DptA and ACP was done as described for CDA-C1. Sfp from Bacillus subtilis was expressed and purified based on the protocol of Walsh and colleagues 59.
The first step of purification of ACP, PCP1 and HxcO was nickel affinity chromatography as described for CDA-C1. HxcO was further purified using a Q Sepharose column (GE Healthcare) with a gradient of 0–1 M NaCl in a buffer of 50 mM Tris-HCl pH 7.5 and 1 mM tris(2-carboxyethyl)phosphine (TCEP). PCP1 and ACP were cleaved with TEV protease and reapplied to the 5 ml HiTrap IMAC FF column charged with Ni2+, then subjected to anion exchange chromatography using a 5 ml HiTrap Q Sepharose column (GE Healthcare) and a gradient of 0–1 M NaCl in a buffer of 50 mM Tris-HCl pH 7.5, 1 M NaCl and 2 mM βME. PCP1 was placed into a buffer of 25 mM HEPES pH 7.0, 50 mM NaCl, 1 mM TCEP, then concentrated and frozen. ACP was applied to a HiPrep 16/60 Sephacryl S-200 HR in buffer containing 25 mM HEPES pH 7.0 and 50 mM NaCl, then concentrated and frozen.
Assay for CDA-C1 activity
Assay for CDA-C1 activity was adapted from Marahiel and colleagues 26. ACP (200 μM) was incubated for 60 minutes at room temperature with hexanoyl-coenzyme A (800 μM) (Sigma-Aldrich, Oakville, Canada) and Sfp (40.2 μM), in buffer containing 25 mM HEPES pH 7.0, 50 mM NaCl and 1 mM TCEP. HxcO (5 μM), FAD (25 μM) and HCl (3.75 mM) were added; the reaction was incubated for a further 60 minutes. In parallel, PCP1 was incubated at room temperature with serinyl-coenzyme A (800 μM) (Zamboni Chem Solutions, Montreal, Canada) and Sfp (40.2 μM) in buffer containing 25 mM HEPES pH 7.0, 50 mM NaCl and 1 mM TCEP for 20 minutes. At the completion of loading of the carrier domains, the reactions were mixed, CDA-C1 (6.2 μM) was added, and the solution was further incubated for one hour.
Separation of differentially loaded carrier proteins was performed using reverse phase chromatography (Varian, C4 4.6 × 250 mm Microsorb 300Å, 5 μM) at a flow rate of 0.5 ml/min using gradient elution from 40–50% B over 25 min, followed by a gradient up to 55% B over another 25 min (buffer A: 0.2% TFA with H2O and buffer B: 0.2% TFA with acetonitrile). Protein masses were determined offline using ESI-MS (Esquire HCT Ultra; Bruker Daltonics) in positive-ion mode with an ESI nebulizer (Mass Spectrometry Core Facility, Bellini Life Science Complex, Montreal, Canada). The mass spectrometer was set to acquire spectra in the mass range 900–3000 m/z for an average of 3 minutes and the protein was infused through a syringe pump at 240 microliters per hour. Acquired spectra were averaged and the charge states of the protein was determined with the aid of a charge state ruler (Esquire Data Analysis; Bruker Daltonics).
Computational analyses
Models of CDA-C1 in the conformation observed in the structures SrfAC, TycC and VibH were produced to allow energy minimized refined linear interpolation, targeted molecular dynamics simulations and analysis of SAXS data using the SWISS-MODEL server 60 in alignment mode. Energy minimized refined linear interpolations (“morphings”) were performed using the rigimol and refine functions in the program iPyMOL (Incentive PyMOL 2006 release, DeLano Scientific). Targeted molecular dynamics simulations were performed with the program NAMD 61 using a hydrated sphere of 47.2 Å radius, a temperature of 310 K and an elastic constant TMDk of 200 kcal/mol/Å2 for between 40 ps and 1 ns. Morphing and TMD was performed between the CDA-C1 structure and models with sequence of CDA-C1 in the conformations of SrfAC, TycC and VibH, but note that the reciprocal transition using the structure of the more “open” C domains and a model with its sequence in the CDA-C1 conformation works equally well. Normal mode analyses were undertaken using the web-based programs WEBnm@ 33 and NOMAD-Ref 34. Buried surface areas were calculated using the Lee & Richards buried surface accessibility calculation 62 implemented in the program CNS 63. The active site tunnel was identified with the program CAVER 47, using the coordinates of H157 as the search seed.
For the model of the transition state of the C domain – catalyzed reaction, the server SWISS-MODEL was used to produce homology models of upstream ACP starting from the structure of ACP from Staphylococcus aureus (PDB ID 4DXE; Center for Structural Genomics of Infectious Diseases, unpublished), and the downstream PCP from the structure of the SrfAC module 10. The initial position of ACP was derived from molecular docking using the program HADDOCK 64, with outputs vetted by whether the resulting model agreed with experimental protein-protein contact data 43, 44. The initial position of PCP1 was achieved by superimposition of the SrfAC C domain on CDA-C1. Restraints parameters for the acyl-PPE transition state (TS) were generated using the program PRODRG 65. Atoms of the TS were arbitrarily placed along the tunnel, and the entire system was subjected to conjugate gradient minimization with no experimental energy terms in the program CNS, to yield the final holo ACP–C–PCP1 model.
Supplementary Material
An alignment of CDA-C1 with its orthologue from Streptomyces lividans, Plu3535, the three C domains with known structures (SrfAC, VibH, TycC), and three of the more homologous C domains as returned by BLAST 66 search (Dhbf, Entf, Gram).
a) An FO electron density map from the P212121 crystal form contoured at 1σ. b) A 2FO – FC electron density map from the P21 crystal form, contoured at 1σ.
a) Alignment of all four monomers in the asymmetric units of the P212121 crystal form and the P21 crystal form. b) The conformations of a unique insert in the CDA-C1 sequence shows variable conformations in the four monomers. c) The dimers which are present in one asymmetric unit of the P212121 crystal form and the P21 crystal form are in similar conformations. d) The packing of the dimers in the crystals are different for the P212121 crystal form and the P21 crystal form.
Alignment of the C-terminal subdomain shows different relative subdomain–subdomain orientation in the structures of CDA-C1 (green) and those of a) VibH (brown), b) TycC (orange) and c) SrfAC (yellow).
Additional SAXS data for CDA-C1. a) Normalized scattering curves of CDA-C1 at different concentrations showing little concentration dependence. b) Kratky plots of CDA-C1 and BSA indicating that CDA-C1 is a dimer. c) P(r) plot generated from merged data and used for envelope reconstruction.
The root mean square deviation (rmsd) between Cα atoms of the model and target during targeted molecular dynamics simulations using a) SrfAC, b) TycC and c) VibH C domains as models. Simulations were 1 ns in length and rmsd values for every 100th model are shown. Residues ~295–309 and ~367–388 cross over to the N-terminal subdomain.
The subdomain crossover “latch” interaction is present in (a) CDA-C1 (b) VibH (c) TycC and (d) SrfaC.
Movie S1-S3. Normal mode analyses. Normal modes calculated using WEBNM@ (Movie S1) and NOMAD-Ref (Movie S2–S3) show conformational changes similar to what would be required to interconvert “closed” and “open” forms of the C domain.
Movie S4-S7. Morphing between observed C domain conformations. Energy minimized linear interpolation (“morphing”) of CDA-C1 between conformations observed in the crystal structures of CDA-C1 and SrfAC (Movie S4), CDA-C1 and TycC (Movie S5), CDA-C1 and TycC (Movie S6), and between all four conformations (Movie S7). The CDA-C1 insert is removed for these computations. The active site is colored red.
Movie S8-S11. Targeted molecular dynamics simulations between observed C domain conformations. Targeted molecular dynamics simulations of CDA-C1 between conformations observed in the crystal structures of CDA-C1 and SrfAC (Movie S8), CDA-C1 and TycC (Movie S9), CDA-C1 and TycC (Movie S10), and between all four conformations (Movie S11). The CDA-C1 insert is removed for these computations. The active site is colored red.
Table S1. Data collection and refinement statistics for the P212121 and P21 crystal forms of CDA-C1.
Deconvoluted masses of differentially-loaded carrier proteins identified from the HPLC assay.
Acknowledgments
We are indebted to Vivian Stojanoff and Edwinto Lazo for data collection at beamline X6A of NSLS, Brookhaven National labs, and to Kurt Dejgaard for advice and training in mass spectrometry, as well as Janice Reimer for purification of Sfp protein, Michael Tarry, Diego Alonzo and Fabien Bergeret for advice and discussion, and Albert Berghuis for advice, critical reading of the manuscript and support of DR. The plasmid containing Sfp was a kind gift of Nathan Magarvey (McMaster University). We thank Alex Deiters, Qingyang Liu and Yan Zou (North Carolina State University) for preparation of SNAC analogues. We are indebted to John Colucci, Helmi Zaghdane and Robert Zamboni (Zamboni Chem Solutions) for an extremely generous gift of serinyl-CoA. This research was supported by the Canadian Institutes of Health Research (CIHR) Operating Grant No. MOP 106615 awarded to TMS, a Human Frontiers Science Program Organization Career Development Award to TMS, a Tier 2 Canada Research Chair in Macromolecular Machines held by TMS and a CIHR Strategic Training Initiative in Chemical Biology studentship held by KB.
Footnotes
Accession numbers
The coordinates and structure factors for SeMet-CDA-C1 in P212121 space group and CDA-C1 in P21 space group have been deposited in the PDB with accession codes 4JN3 and 4JN5.
References
- 1.Schwarzer D, Finking R, Marahiel MA. Nonribosomal peptides: from genes to products. Nat Prod Rep. 2003;20:275–287. doi: 10.1039/b111145k. [DOI] [PubMed] [Google Scholar]
- 2.Felnagle EA, Jackson EE, Chan YA, Podevels AM, Berti AD, McMahon MD, Thomas MG. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol Pharm. 2008;5:191–211. doi: 10.1021/mp700137g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konz D, Marahiel MA. How do peptide synthetases generate structural diversity? Chem Biol. 1999;6:R39–48. doi: 10.1016/S1074-5521(99)80002-7. [DOI] [PubMed] [Google Scholar]
- 4.Caboche S, Pupin M, Leclere V, Fontaine A, Jacques P, Kucherov G. NORINE: a database of nonribosomal peptides. Nucleic Acids Res. 2008;36:D326–31. doi: 10.1093/nar/gkm792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Liempt H, von Dohren H, Kleinkauf H. delta-(L-alpha-aminoadipyl)-L-cysteinyl-D-valine synthetase from Aspergillus nidulans. The first enzyme in penicillin biosynthesis is a multifunctional peptide synthetase. J Biol Chem. 1989;264:3680–3684. [PubMed] [Google Scholar]
- 6.Wessels P, von Dohren H, Kleinkauf H. Biosynthesis of acylpeptidolactones of the daptomycin type. A comparative analysis of peptide synthetases forming A21978C and A54145. Eur J Biochem. 1996;242:665–673. doi: 10.1111/j.1432-1033.1996.0665r.x. [DOI] [PubMed] [Google Scholar]
- 7.Zocher R, Nihira T, Paul E, Madry N, Peeters H, Kleinkauf H, Keller U. Biosynthesis of cyclosporin A: partial purification and properties of a multifunctional enzyme from Tolypocladium inflatum. Biochemistry. 1986;25:550–553. doi: 10.1021/bi00351a005. [DOI] [PubMed] [Google Scholar]
- 8.Weber T, Marahiel MA. Exploring the domain structure of modular nonribosomal peptide synthetases. Structure. 2001;9:R3–9. doi: 10.1016/s0969-2126(00)00560-8. [DOI] [PubMed] [Google Scholar]
- 9.Frueh DP, Arthanari H, Koglin A, Vosburg DA, Bennett AE, Walsh CT, Wagner G. Dynamic thiolation-thioesterase structure of a non-ribosomal peptide synthetase. Nature. 2008;454:903–906. doi: 10.1038/nature07162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanovic A, Samel SA, Essen LO, Marahiel MA. Crystal structure of the termination module of a nonribosomal peptide synthetase. Science. 2008;321:659–663. doi: 10.1126/science.1159850. [DOI] [PubMed] [Google Scholar]
- 11.Koglin A, Mofid MR, Lohr F, Schafer B, Rogov VV, Blum MM, Mittag T, Marahiel MA, Bernhard F, Dotsch V. Conformational switches modulate protein interactions in peptide antibiotic synthetases. Science. 2006;312:273–276. doi: 10.1126/science.1122928. [DOI] [PubMed] [Google Scholar]
- 12.Gulick AM, Starai VJ, Horswill AR, Homick KM, Escalante-Semerena JC. The 1.75 Å crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry. 2003;42:2866–2873. doi: 10.1021/bi0271603. [DOI] [PubMed] [Google Scholar]
- 13.Reger AS, Wu R, Dunaway-Mariano D, Gulick AM. Structural characterization of a 140 degrees domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry. 2008;47:8016–8025. doi: 10.1021/bi800696y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sundlov JA, Shi C, Wilson DJ, Aldrich CC, Gulick AM. Structural and Functional Investigation of the Intermolecular Interaction between NRPS Adenylation and Carrier Protein Domains. Chem Biol. 2012;19:188–198. doi: 10.1016/j.chembiol.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell CA, Shi C, Aldrich CC, Gulick AM. The Structure of PA1221, a Non-Ribosomal Peptide Synthetase containing Adenylation and Peptidyl Carrier Protein Domains. Biochemistry. 2012;51:3252–3263. doi: 10.1021/bi300112e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Crecy-Lagard V, Marliere P, Saurin W. Multienzymatic non ribosomal peptide biosynthesis: identification of the functional domains catalysing peptide elongation and epimerisation. C R Acad Sci III. 1995;318:927–936. [PubMed] [Google Scholar]
- 17.Stachelhaus T, Mootz HD, Bergendahl V, Marahiel MA. Peptide bond formation in nonribosomal peptide biosynthesis. Catalytic role of the condensation domain. J Biol Chem. 1998;273:22773–22781. doi: 10.1074/jbc.273.35.22773. [DOI] [PubMed] [Google Scholar]
- 18.Keating TA, Marshall CG, Walsh CT, Keating AE. The structure of VibH represents nonribosomal peptide synthetase condensation, cyclization and epimerization domains. Nat Struct Biol. 2002;9:522–526. doi: 10.1038/nsb810. [DOI] [PubMed] [Google Scholar]
- 19.Samel SA, Schoenafinger G, Knappe TA, Marahiel MA, Essen LO. Structural and functional insights into a peptide bond-forming bidomain from a nonribosomal peptide synthetase. Structure. 2007;15:781–792. doi: 10.1016/j.str.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 20.Bergendahl V, Linne U, Marahiel MA. Mutational analysis of the C-domain in nonribosomal peptide synthesis. Eur J Biochem. 2002;269:620–629. doi: 10.1046/j.0014-2956.2001.02691.x. [DOI] [PubMed] [Google Scholar]
- 21.Chong PP, Podmore SM, Kieser HM, Redenbach M, Turgay K, Marahiel M, Hopwood DA, Smith CP. Physical identification of a chromosomal locus encoding biosynthetic genes for the lipopeptide calcium-dependent antibiotic (CDA) of Streptomyces coelicolor A3(2) Microbiology. 1998;144:193–199. doi: 10.1099/00221287-144-1-193. [DOI] [PubMed] [Google Scholar]
- 22.Hojati Z, Milne C, Harvey B, Gordon L, Borg M, Flett F, Wilkinson B, Sidebottom PJ, Rudd BA, Hayes MA, Smith CP, Micklefield J. Structure, biosynthetic origin, and engineered biosynthesis of calcium-dependent antibiotics from Streptomyces coelicolor. Chem Biol. 2002;9:1175–1187. doi: 10.1016/s1074-5521(02)00252-1. [DOI] [PubMed] [Google Scholar]
- 23.Strieker M, Marahiel MA. The structural diversity of acidic lipopeptides antibiotics. Chembiochem. 2009;10:607–616. doi: 10.1002/cbic.200800546. [DOI] [PubMed] [Google Scholar]
- 24.Lakey JH, Lea EJ, Rudd BA, Wright HM, Hopwood DA. A new channel-forming antibiotic from Streptomyces coelicolor A3(2) which requires calcium for its activity. J Gen Microbiol. 1983;129:3565–3573. doi: 10.1099/00221287-129-12-3565. [DOI] [PubMed] [Google Scholar]
- 25.Kopp F, Linne U, Oberthür M, Marahiel MA. Harnessing the chemical activation inherent to carrier protein-bound thioesters for the characterization of lipopeptides fatty acid tailoring enzymes. Journal of the American Chemical Society. 2008;130:2656–2666. doi: 10.1021/ja078081n. [DOI] [PubMed] [Google Scholar]
- 26.Kraas FI, Giessen TW, Marahiel MA. Exploring the mechanism of lipid transfer during biosynthesis of the acidic lipopeptide antibiotic CDA. FEBS Lett. 2012;586:283–288. doi: 10.1016/j.febslet.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 27.Rausch C, Hoof I, Weber T, Wohlleben W, Huson DH. Phylogenetic analysis of condensation domains in NRPS sheds light on their functional evolution. BMC Evol Biol. 2007;7:78. doi: 10.1186/1471-2148-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehmann DE, Trauger JW, Stachelhaus T, Walsh CT. Aminoacyl-SNACs as small-molecule substrates for the condensation domains of nonribosomal peptide synthetases. Chem Biol. 2000;7:765–772. doi: 10.1016/s1074-5521(00)00022-3. [DOI] [PubMed] [Google Scholar]
- 29.Vonrhein C, Schlauderer GJ, Schulz GE. Movie of the structural changes during a catalytic cycle of nucleoside monophosphate kinases. Structure. 1995;3:483–490. doi: 10.1016/s0969-2126(01)00181-2. [DOI] [PubMed] [Google Scholar]
- 30.Yonus H, Neumann P, Zimmermann S, May JJ, Marahiel MA, Stubbs MT. Crystal structure of DltA. Implications for the reaction mechanism of nonribosomal peptide synthetase adenylation domains. J Biol Chem. 2008;283:32484–32491. doi: 10.1074/jbc.M800557200. [DOI] [PubMed] [Google Scholar]
- 31.Van Wynsberghe A, Li G, Cui Q. Normal-mode analysis suggests protein flexibility modulation throughout RNA polymerase’s functional cycle. Biochemistry. 2004;43:13083–13096. doi: 10.1021/bi049738+. [DOI] [PubMed] [Google Scholar]
- 32.Marechal JD, Perahia D. Use of normal modes for structural modeling of proteins: the case study of rat heme oxygenase 1. Eur Biophys J. 2008;37:1157–1165. doi: 10.1007/s00249-008-0279-y. [DOI] [PubMed] [Google Scholar]
- 33.Hollup SM, Salensminde G, Reuter N. WEBnm@: a web application for normal mode analyses of proteins. BMC Bioinformatics. 2005;6:52. doi: 10.1186/1471-2105-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindahl E, Azuara C, Koehl P, Delarue M. NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res. 2006;34:W52–6. doi: 10.1093/nar/gkl082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsiao YS, Jogl G, Tong L. Crystal structures of murine carnitine acetyltransferase in ternary complexes with its substrates. J Biol Chem. 2006;281:28480–28487. doi: 10.1074/jbc.M602622200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim AR, Rylett RJ, Shilton BH. Substrate binding and catalytic mechanism of human choline acetyltransferase. Biochemistry. 2006;45:14621–14631. doi: 10.1021/bi061536l. [DOI] [PubMed] [Google Scholar]
- 37.Buglino J, Onwueme KC, Ferreras JA, Quadri LE, Lima CD. Crystal structure of PapA5, a phthiocerol dimycocerosyl transferase from Mycobacterium tuberculosis. J Biol Chem. 2004;279:30634–30642. doi: 10.1074/jbc.M404011200. [DOI] [PubMed] [Google Scholar]
- 38.Hahn M, Stachelhaus T. Harnessing the potential of communication-mediating domains for the biocombinatorial synthesis of nonribosomal peptides. Proc Natl Acad Sci U S A. 2006;103:275–280. doi: 10.1073/pnas.0508409103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hur GH, Meier JL, Baskin J, Codelli JA, Bertozzi CR, Marahiel MA, Burkart MD. Crosslinking studies of protein-protein interactions in nonribosomal peptide biosynthesis. Chem Biol. 2009;16:372–381. doi: 10.1016/j.chembiol.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janin J, Chothia C. Stability and specificity of protein-protein interactions: the case of the trypsin-trypsin inhibitor complexes. J Mol Biol. 1976;100:197–211. doi: 10.1016/s0022-2836(76)80148-9. [DOI] [PubMed] [Google Scholar]
- 41.Amit AG, Mariuzza RA, Phillips SE, Poljak RJ. Three-dimensional structure of an antigen-antibody complex at 2.8 A resolution. Science. 1986;233:747–753. doi: 10.1126/science.2426778. [DOI] [PubMed] [Google Scholar]
- 42.Chovancova E, Pavelka A, Benes P, Strnad O, Brezovsky J, Kozlikova B, Gora A, Sustr V, Klvana M, Medek P, Biedermannova L, Sochor J, Damborsky J. CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput Biol. 2012;8:e1002708. doi: 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai JR, Fischbach MA, Liu DR, Walsh CT. A protein interaction surface in nonribosomal peptide synthesis mapped by combinatorial mutagenesis and selection. Proc Natl Acad Sci U S A. 2006;103:5314–5319. doi: 10.1073/pnas.0601038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lai JR, Fischbach MA, Liu DR, Walsh CT. Localized protein interaction surfaces on the EntB carrier protein revealed by combinatorial mutagenesis and selection. J Am Chem Soc. 2006;128:11002–11003. doi: 10.1021/ja063238h. [DOI] [PubMed] [Google Scholar]
- 45.Trivedi OA, Arora P, Vats A, Ansari MZ, Tickoo R, Sridharan V, Mohanty D, Gokhale RS. Dissecting the mechanism and assembly of a complex virulence mycobacterial lipid. Mol Cell. 2005;17:631–643. doi: 10.1016/j.molcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 46.Belshaw PJ, Walsh CT, Stachelhaus T. Aminoacyl-CoAs as probes of condensation domain selectivity in nonribosomal peptide synthesis. Science. 1999;284:486–489. doi: 10.1126/science.284.5413.486. [DOI] [PubMed] [Google Scholar]
- 47.Stachelhaus T, Marahiel MA. Modular structure of peptide synthetases revealed by dissection of the multifunctional enzyme GrsA. J Biol Chem. 1995;270:6163–6169. doi: 10.1074/jbc.270.11.6163. [DOI] [PubMed] [Google Scholar]
- 48.Linne U, Marahiel MA. Control of directionality in nonribosomal peptide synthesis: role of the condensation domain in preventing misinitiation and timing of epimerization. Biochemistry. 2000;39:10439–10447. doi: 10.1021/bi000768w. [DOI] [PubMed] [Google Scholar]
- 49.Clugston SL, Sieber SA, Marahiel MA, Walsh CT. Chirality of peptide bond-forming condensation domains in nonribosomal peptide synthetases: the C5 domain of tyrocidine synthetase is a (D)C(L) catalyst. Biochemistry. 2003;42:12095–12104. doi: 10.1021/bi035090+. [DOI] [PubMed] [Google Scholar]
- 50.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 51.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 52.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konarev Petr V, Volkov Vladimir V, Sokolova Anna V, Koch Michel HJ, Svergun Dmitri I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography. 2003;36:1277–1282. [Google Scholar]
- 55.Svergun D, Barberato C, Koch MHJ. CRYSOL - a Program to Evaluate Xray Solution Scattering of Biological Macromolecules from Atomic Coordinates. Journal of Applied Crystallography. 1992;25:495–503. [Google Scholar]
- 56.Franke D, Svergun D. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of Applied Crystallography. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Volkov V, Svergun DI. Uniqueness of ab initio shape determination in small angle scattering. Journal of Applied Crystallography. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. Journal of Applied Crystallography. 2001;34:33–41. [Google Scholar]
- 59.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 60.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 61.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 63.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 64.de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data driven biomolecular docking. Nat Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 65.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 66.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
An alignment of CDA-C1 with its orthologue from Streptomyces lividans, Plu3535, the three C domains with known structures (SrfAC, VibH, TycC), and three of the more homologous C domains as returned by BLAST 66 search (Dhbf, Entf, Gram).
a) An FO electron density map from the P212121 crystal form contoured at 1σ. b) A 2FO – FC electron density map from the P21 crystal form, contoured at 1σ.
a) Alignment of all four monomers in the asymmetric units of the P212121 crystal form and the P21 crystal form. b) The conformations of a unique insert in the CDA-C1 sequence shows variable conformations in the four monomers. c) The dimers which are present in one asymmetric unit of the P212121 crystal form and the P21 crystal form are in similar conformations. d) The packing of the dimers in the crystals are different for the P212121 crystal form and the P21 crystal form.
Alignment of the C-terminal subdomain shows different relative subdomain–subdomain orientation in the structures of CDA-C1 (green) and those of a) VibH (brown), b) TycC (orange) and c) SrfAC (yellow).
Additional SAXS data for CDA-C1. a) Normalized scattering curves of CDA-C1 at different concentrations showing little concentration dependence. b) Kratky plots of CDA-C1 and BSA indicating that CDA-C1 is a dimer. c) P(r) plot generated from merged data and used for envelope reconstruction.
The root mean square deviation (rmsd) between Cα atoms of the model and target during targeted molecular dynamics simulations using a) SrfAC, b) TycC and c) VibH C domains as models. Simulations were 1 ns in length and rmsd values for every 100th model are shown. Residues ~295–309 and ~367–388 cross over to the N-terminal subdomain.
The subdomain crossover “latch” interaction is present in (a) CDA-C1 (b) VibH (c) TycC and (d) SrfaC.
Movie S1-S3. Normal mode analyses. Normal modes calculated using WEBNM@ (Movie S1) and NOMAD-Ref (Movie S2–S3) show conformational changes similar to what would be required to interconvert “closed” and “open” forms of the C domain.
Movie S4-S7. Morphing between observed C domain conformations. Energy minimized linear interpolation (“morphing”) of CDA-C1 between conformations observed in the crystal structures of CDA-C1 and SrfAC (Movie S4), CDA-C1 and TycC (Movie S5), CDA-C1 and TycC (Movie S6), and between all four conformations (Movie S7). The CDA-C1 insert is removed for these computations. The active site is colored red.
Movie S8-S11. Targeted molecular dynamics simulations between observed C domain conformations. Targeted molecular dynamics simulations of CDA-C1 between conformations observed in the crystal structures of CDA-C1 and SrfAC (Movie S8), CDA-C1 and TycC (Movie S9), CDA-C1 and TycC (Movie S10), and between all four conformations (Movie S11). The CDA-C1 insert is removed for these computations. The active site is colored red.
Table S1. Data collection and refinement statistics for the P212121 and P21 crystal forms of CDA-C1.
Deconvoluted masses of differentially-loaded carrier proteins identified from the HPLC assay.