

Background: There are multiple cytochrome P450 species co-localized in the endoplasmic reticulum.
Results: Membrane-bound cytochromes P450 3A4, 3A5, and 2E1 associate into heteromeric complexes, where the properties of the individual enzymes are considerably modified.
Conclusion: The properties of the P450 ensemble cannot be predicted by summation of the properties of the individual enzymes.
Significance: We disclose a mechanism of regulatory cross-talk between multiple P450 species through hetero-oligomerization.
Keywords: Allosteric Regulation, Cytochrome P450, Drug Metabolism, Membrane Protein, Protein-Protein Interaction
Abstract
The body of evidence of physiologically relevant P450-P450 interactions in microsomal membranes continues to grow. Here we probe oligomerization of human CYP3A4, CYP3A5, and CYP2E1 in microsomal membranes. Using a technique based on luminescence resonance energy transfer, we demonstrate that all three proteins are subject to a concentration-dependent equilibrium between the monomeric and oligomeric states. We also observed the formation of mixed oligomers in CYP3A4/CYP3A5, CYP3A4/CYP2E1, and CYP3A5/CYP2E1 pairs and demonstrated that the association of either CYP3A4 or CYP3A5 with CYP2E1 causes activation of the latter enzyme. Earlier we hypothesized that the intersubunit interface in CYP3A4 oligomers is similar to that observed in the crystallographic dimers of some microsomal drug-metabolizing cytochromes P450 (Davydov, D. R., Davydova, N. Y., Sineva, E. V., Kufareva, I., and Halpert, J. R. (2013) Pivotal role of P450-P450 interactions in CYP3A4 allostery: the case of α-naphthoflavone. Biochem. J. 453, 219–230). Here we report the results of intermolecular cross-linking of CYP3A4 oligomers with thiol-reactive bifunctional reagents as well as the luminescence resonance energy transfer measurements of interprobe distances in the oligomers of labeled CYP3A4 single-cysteine mutants. The results provide compelling support for the physiological relevance of the dimer-specific peripheral ligand-binding site observed in certain CYP3A4 structures. According to our interpretation, these results reveal an important general mechanism that regulates the activity and substrate specificity of the cytochrome P450 ensemble through interactions between multiple P450 species. As a result of P450-P450 cross-talk, the catalytic properties of the cytochrome P450 ensemble cannot be predicted by simple summation of the properties of the individual P450 species.
Introduction
The central role that cytochromes P450 play in drug metabolism makes these enzymes a major subject for studies of drug disposition, adverse drug effects, and drug-drug interactions. Although there has been tremendous success in delineating P450 mechanisms, the concept of the drug-metabolizing ensemble as a functionally integrated multienzyme system remains undeveloped. A common trend is to consider each particular drug-metabolizing P450 enzyme in isolation, thus disregarding the cross-talk between multiple cytochrome P450 species. However, eukaryotic cells typically possess a multitude of different cytochromes P450 that are co-localized in the membrane of the endoplasmic reticulum and share the same protein partners, NADPH-cytochrome P450 reductase (CPR)4 and cytochrome b5.
Competition of different P450 species for a limited amount of CPR, which has been observed by numerous investigators (1–6), constitutes an important element in the inter-P450 cross-talk. However, the integrative connections in microsomal monooxygenase go far beyond that and involve functional effects of physical interactions of multiple P450 molecules that result in oligomerization (7–17). Besides evidence of homo-oligomerization of various P450 species (10, 12, 15, 17–23), there are also multiple indications of interactions between dissimilar P450s in membranes of proteoliposomes (13), microsomes (14), and living cells (16). The body of evidence of profound functional effects exerted by P450-P450 interactions continues to grow (24, 25).
Typically, one of the enzymes of the interacting pair becomes activated, whereas its counterpart is inhibited or unaffected. This type of relationship is exhibited by CYP3A4/CYP1A2 (26), CYP2C19/CYP2C9 (27), CYP2D6/CYP2C9 (28), and CYP3A4/CYP2C9 (29) pairs. The most extensively studied P450-P450 pair is that of rabbit CYP1A2 and CYP2B4 enzymes (3, 6, 13, 30–34). The co-presence of CYP2B4 in mixed systems with CYP1A2 boosts the activity of the CYP1A2 (6, 30) while inhibiting CYP2B4 (3). Similar relationships were also demonstrated between CYP1A2 and the rabbit CYP2E1 (34). Analysis of these effects led Backes and co-workers (13, 30, 35) to the conclusion that the effects of CYP2B4 and CYP2E1 on CYP1A2 are exerted through the formation of mixed oligomers, where the interactions of CPR with CYP1A2 are promoted, whereas the CPR binding to the other P450 is inhibited. This conclusion is supported by our FRET-based studies of the interactions of CPR with mixtures of CYP1A2 and CYP2B4 (6).
A remarkable effect of hetero-association was demonstrated in the study of pressure-induced transitions in heterooligomers of CYP1A2 and CYP2B4 (32). Separately, these enzymes reveal quite distinct behavior upon increasing hydrostatic pressure. Whereas the CYP2B4 (Fe2+)-CO complex undergoes a P450 → P420 transition at rather low pressures, the CYP1A2 (Fe2+)-CO complex is extremely resistant to such inactivation. However, after co-incubation with CYP1A2, CYP2B4 becomes protected from pressure-induced inactivation and reveals attenuated compressibility of the heme pocket (32).
Although the above observations suggest an important effect of P450-P450 on the properties of the individual enzymes, most of these results were obtained in non-membranous in vitro systems, and the physiological relevance remains unclear. There is also a trend to consider the observations of functional effects in discrete P450 pairs in isolation. These effects are often thought to reveal the behavior of a particular P450 combination rather than being a manifestation of a general mechanism of P450-P450 cross-talk. Therefore, a general paradigm of the role of P450-P450 interactions in systems biochemistry of human drug-metabolizing ensemble is still missing.
The present study is aimed at filling this knowledge gap through a systematic exploration of the mechanisms and functional effects of P450-P450 interactions in a membrane environment. It capitalizes on the results of our recent work (23), where we developed a simple technique that produces catalytically competent membranous systems with variable content of cytochromes P450 via incorporation of the purified enzymes into insect cell microsomes containing recombinant NADPH-cytochrome P450 reductase. This innovative approach is further complemented by assessing the degree of P450 oligomerization through monitoring P450-P450 interactions with luminescence resonance energy transfer (LRET) (23).
Our initial studies with these approaches provided compelling evidence that the oligomerization of CYP3A4 is directly related to its complex allosteric behavior. In particular, we demonstrated that the stimulation by α-naphthoflavone (ANF), a prototypical activator of CYP3A4, is observed only with enzyme oligomers and becomes suppressed upon their dissociation (23). In the present work, we apply our new methods for the analysis of interactions between dissimilar P450 enzymes. We demonstrate high-affinity interactions between CYP3A4, CYP3A5, and CYP2E1 that lead to formation of mixed oligomers where the catalytic properties of the individual enzymes are modified. Furthermore, we used intermolecular cross-linking with thiol-reactive bifunctional reagents as well as the LRET measurements of interprobe distances to probe the geometry of CYP3A4 oligomers.
Taken together, our observations provide a compelling demonstration of a striking effect of P450-P450 interactions on the catalytic properties of the drug-metabolizing ensemble in microsomal membranes.
EXPERIMENTAL PROCEDURES
Materials
7-Hydroxy-4-(trifluoromethyl) coumarin, glucose-6-phosphate, glucose-6-phosphate dehydrogenase from baker's yeast, protocatechuate 3,4-dioxygenase from Pseudomonas sp., protocatechuic acid, NADPH, dl-dithiothreitol (DTT), l-α-phosphatidylcholine from egg yolk, and N,N′(o-phenylene)dimaleimide (1,2-dimaleimidobenzene; o-DMB) were the products of Sigma-Aldrich. 4,4′-Methylenebis(N-phenylmaleimide) (bis(4-maleimidophenyl) methane; bMPM) was the product of Alfa Aesar (Ward Hill, MA). l-α-Phosphatidylethanolamine from bovine liver and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (phosphatidic acid) were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL). CYP2E1 and CYP3A4 Baculosomes® Plus were obtained from Invitrogen. 7-Benzyloxyquinoline (7-BQ), 7-hydroxyquinoline, 7-methoxy-4-(trifluoromethyl)coumarin (7-MFC), CYP3A4, and CYP2E1 SupersomesTM, as well as the control SupersomesTM containing rat recombinant NADPH-cytochrome P450 reductase (catalog no. 456514) were from Corning Life Sciences (Tewksbury, MA). Erythrosine 5′-iodoacetamide (ERIA) was from AnaSpec (San Jose, CA). Dibromobimane (bBBr) and tris(2-carboxyethyl)phosphine (TCEP) were the products of Invitrogen/Molecular Probes (Eugene, OR). DY-731 maleimide (DYM) was obtained from Dyomics (MoBiTec; Göttingen, Germany). All other chemicals were of American Chemical Society grade and were used without further purification.
Site-directed Mutagenesis, Protein Purification, and Modification with Thiol-reactive Probes
The cysteine-depleted mutants CYP3A4(C58,C64) (36), CYP3A4(C468) (23) and CYP3A4(C166) were generated using the QuikChange site-directed mutagenesis kit and a template consisting of the cDNA of the N-terminally truncated (Δ3–12) S18F CYP3A4 variant with a tetrahistidine tag attached at the C terminus. This construct is referred to in this work as the “wild type CYP3A4” to distinguish it from the cysteine-depleted mutants. A similar construct was also used for expression and purification of CYP3A5. Both CYP3A proteins were expressed in Escherichia coli TOPP3 cells (37) and purified following the procedure described previously (22, 36).
The N-terminally truncated (Δ3–20) and C-terminally His-tagged variant of CYP2E1 (38) was expressed in E. coli and purified following a procedure similar to that described for CYP2B6 (39) where the ion-exchange chromatography step was omitted. Modification of the proteins with thiol-reactive probes was performed as described previously (23).
Preparation of Proteoliposomes
Proteoliposomes were obtained by the described octyl glucoside dialysis technique with some modifications (23). Specifically, we used a 2:1:0.6 mixture of phosphatidylcholine, phosphatidylethanolamine, and phosphatidic acid. Lipids (10 mg) were mixed as chloroform solutions, and the solvent was removed by evaporation. The suspension of lipids in buffer A containing 1.54% octyl glucoside was prepared using a vortex mixer and incubated for 30 min at room temperature under argon. The mixture was then diluted with the same buffer containing no detergent to a final concentration of octyl glucoside of 0.43%. After the addition of a concentrated stock (100–200 μm) of purified cytochrome P450 to the desired protein/lipid ratio, the mixture was dialyzed at 4 °C under constant gentle bubbling with argon gas against three changes of 1000 ml of buffer A, each containing 5 ml of Bio-Beads SM-2. After 72 h of dialysis (24 h with each portion of the buffer) the mixture was concentrated on Centrisart I 300,000 molecular weight cut-off concentrators (Sartorius AG, Göttingen, Germany) to a phospholipid concentration of 8–15 mm and stored at −80 °C under argon.
Incorporation of CYP3A4 into Model Microsomes
Incorporation of CYP3A4 into model microsomes was performed by incubation of an undiluted suspension (5 mg/ml protein, 2–3.5 mm phospholipid) of the CPR-containing “control” SupersomesTM (SS(R)) with purified CYP3A4, CYP3A5, CYP2E1, or their mixtures added to desired lipid/P450 molar ratios (RL/P). After incubation for 40 h at 4 °C with continuous stirring under an argon atmosphere, the suspension was diluted 8 times with 100 mm potassium phosphate buffer, pH 7.4, and centrifuged at 35,000 rpm in an Optima XL-80XP ultracentrifuge (Beckman Coulter Inc., Brea, CA) with an SW50L rotor for 90 min at 4 °C. The pellet was resuspended in the same buffer (400 μl per 500 μl of initial microsomal suspension) and briefly sonicated (2 × 10 s at 40% power) with a Biologics model 30000 sonicator with a microtip (BioLogics Inc., Manassas, VA).
Determination of P450 Concentration in Microsomal Preparations
The microsomal suspensions were diluted 1:10 to 1:20 with CO-saturated membrane solubilization buffer (100 mm potassium phosphate buffer, pH 7.3, 10% glycerol, 0.5% sodium cholate, 0.4% Igepal CO-630, 1 mm EDTA). After recording of the 340–600 nm absorbance spectrum of the ferric heme protein, the sample was reduced with sodium dithionite, and the spectrum of the CO-bound ferrous P450 state was recorded. For determination of the P450 concentration, each of the two spectra was approximated with a linear combination of appropriate prototypical spectra of P450 absorbance (40–42) and a 3rd-4th order polynomial. The polynomial term was included in the approximation to compensate for the residual turbidity of the sample. Although the value determined from the spectrum of the reduced CO-bound sample was usually slightly lower that that obtained for the ferric enzyme, the difference in the two values did not exceed 20% of their average, which was used as an estimate of the P450 content.
Determination of the Content of CYP2E1 in Its Mixtures with CYP3A Proteins
The method for selective determination of the concentration of CYP2E1 applied in this study is based on our observation that, in contrast to CYP3A proteins, CYP2E1 interacts with DTT with the formation of a hyperthiolate complex of the heme iron. This complex, where both axial ligands are represented by thiolates, has a peculiar absorbance spectrum with a characteristic split of the Soret band into two peaks positioned around 380 and 460 nm (43). In our assay procedure, we diluted 5 μl of microsomal preparations containing CYP2E1 with 100 μl of the membrane solubilization buffer (see above), placed the mixture into a quartz microcell (absorbance path length 1 cm), and performed spectrophotometric titrations with DTT. Typically, 8–10 additions of 1 m stock solution of DTT were made to reach the final concentration of 25–30 mm. After each addition of DTT, the absorbance spectrum was recorded in the region of 340–700 nm. The resulting series of spectra was subjected to the procedure of principal component analysis combined with approximation of the spectra of principal components with the prototypical spectra of absorbance of the ferric low-spin, ferric high-spin, and hyperthiolate states of CYP2E1. The concentration of the hyperthiolate complex reached at saturation with DTT was used for calculation of the concentration of CYP2E1 in the samples. This calculation was based on our observation that the steady-state concentration of the hyperthiolate complex of CYP2E1 at saturation with DTT amounts to ∼85% of the total enzyme concentration, either in solution of the purified enzyme or in CYP2E1-containing microsomes.
Cross-linking of CYP3A4
Cross-linking of CYP3A4 with the thiol-reactive bifunctional reagents bBBr, o-DMB, and bMPM was carried out at a protein concentration of 5–10 μm in buffer A. The reaction was initiated by the addition of cross-linking reagent as a 10 mm stock solution in acetone to attain the desired protein/reagent molar ratio (1:1 to 1:10). After incubation for 5–90 min with continuous stirring at room temperature, the reaction was terminated by the addition of 500 mm stock solution of reduced glutathione (GSH) to the final concentration of 5 mm. Optimal cross-linking of CYP3A4 in solution was achieved at protein/reagent ratios of 1:1 to 1:2 and prolonged incubation times (≥45 min). In the case of the membrane-incorporated proteins, the best results were obtained with a 5-min incubation time and 10-fold molar excess of bMPM. SDS-PAGE of the cross-linked proteins was performed on 8–16% gradient Ready-Gel® precast gels (Bio-Rad) at a protein load of 0.1–0.2 nmol of P450/well. The gels were stained with the SimplyBlueTM stain (Invitrogen).
Activity Measurements
The rates of 7-BQ O-debenzylation and O-demethylation of 7-MFC were measured with real-time continuous fluorometric assays (23, 44).
LRET-based Monitoring of the Formation of Mixed Oligomers of ERIA- and DYM-labeled Proteins
This was performed as described previously (23).
Data Analysis
Data analysis was performed with the use of principal component analysis as described previously (23, 45).
The equation for the equilibrium of binary association (dimerization) used in the fitting of oligomerization isotherms (dependences of LRET efficiency on the concentration of P450 in membranes) had the following form:
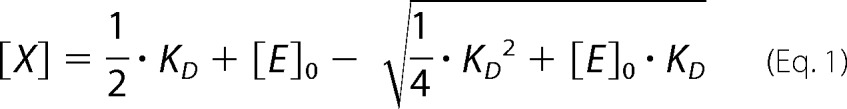 |
where [E]0, [X], and KD represent the total concentration of the associating compound (enzyme), the concentration of its dimers, and the dissociation constant, respectively.
Alternatively, we fitted the oligomerization isotherms with the equation for equilibrium of the formation of enzyme trimers (trimerization). Derivation of the analytical solution for this case results in a cubic equation, the solution of which with Vieta's trigonometric method results in the following relationship:
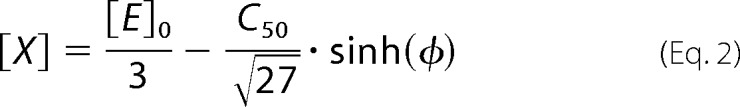 |
where
|
Here [X] is the steady-state concentration of trimers. By the analogy with the Hill equation, we introduce here the parameter C50, which defines the concentration of E at which the degree of oligomerization is equal to 50%. The relationship between C50 and KD is defined as follows:
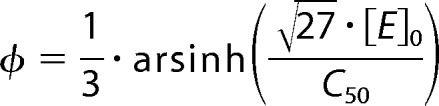 |
To fit the dependences of the relative decrease in donor fluorescence (A) on P450 concentration to Equations 1 and 2, we supplemented these relationships with the parameter Amax, which represents the amplitude of the curve extrapolated to infinite concentration of the enzyme and is determined by LRET efficiency in the oligomer,
 |
where n is the number of the enzyme molecules in the oligomer (i.e. 2 and 3 for [X] calculated according to Equation 1 and Equation 2, respectively).
Fitting of the titration isotherms to the above equations was performed with non-linear regression using a combination of Nelder-Mead and Marquardt algorithms as implemented in our SpectraLab software (41).
RESULTS
Oligomerization of CYP3A4 in Microsomes; Exploring the Architecture of CYP3A4 Oligomers with LRET and Site-directed Mutagenesis
With the use of LRET-based detection of P450-P450 interactions in the membranes of insect cell microsomes containing rat recombinant CPR (“control” Supersomes, SS(R)), we recently demonstrated that the oligomerization of CYP3A4 plays a central role in the mechanism of activation of the enzyme by ANF (23). We proposed that a structural explanation for this finding may be provided by the observation of a peripheral ligand binding site at the dimer interface in an x-ray structure of the CYP3A4 complex with progesterone (Protein Data Bank entry 1W0F (46)) and hypothesized that the intersubunit contacts observed in this dimer reproduce the interactions taking place in the oligomer of membrane-bound CYP3A4 (23). Here we probe this hypothesis by relocating LRET donor and acceptor fluorophores in the CYP3A4 molecule and examining the correlation between the observed distance changes and those expected according to the x-ray structure (Fig. 1).
FIGURE 1.
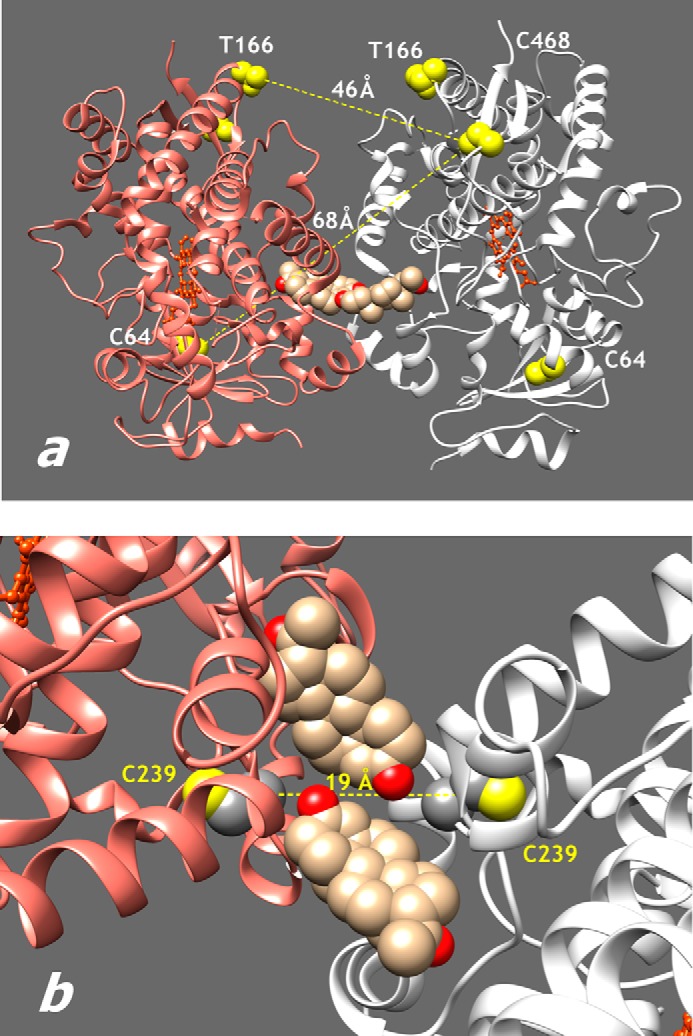
Positions of cysteine residues used in labeling and cross-linking of CYP3A4. a, structure of the crystallographic dimer of CYP3A4 in the Protein Data Bank structure 1W0F (46). Yellow space-filling models show residues 64, 166, and 468 used for the attachment of fluorescent probes. Sand color space-filling models show the two progesterone molecules bound at the intersubunit interface. b, region of intersubunit interface in the same structure showing the positions of Cys239 residues.
In our initial studies, we used the single cysteine mutants of CYP3A4 bearing the ERIA and DYM fluorophores at positions 64 and 468. The LRET efficiency specific to this pair of positions was found to be ∼10% (23). Based on this value, the interprobe distance was estimated to be 49 Å. Taking into account the large size of ERIA and DYM probes (the distances from the sulfur atom of cysteine to the most distant non-hydrogen atom of the probe are 15 and 18 Å, respectively), this value is commensurate with the distance between the sulfur atoms of Cys64 and Cys468 in the crystallographic dimer, which is equal to 67.7 Å (Fig. 1a).
We constructed the CYP3A4(C166) mutant, where a new cysteine residue was introduced by T166C mutation on a background of a construct depleted of all native cysteines except for heme-ligated Cys442. As shown in Fig. 1a, the distance between the β-carbon atom of Thr166 and the sulfur atom of Cys468 is equal to 46 Å. Therefore, relocation of one of the probes from Cys64 to Cys166 is expected to increase LRET efficiency considerably.
Fig. 2 illustrates the results of LRET experiments where we incorporated CYP3A4(C166)DYM into SS(R) containing CYP3A4(C468)ERIA. Consistent with expectations, the interactions between the two labeled proteins resulted in the appearance of LRET, the efficiency of which was considerably higher than that observed with the probes located at Cys64 and Cys468.
FIGURE 2.

Interactions of CYP3A4(C468)-ERIA with CYP3A4(C166)-DYM mutants in SS(R) microsomes studied by LRET. a, a series of spectra of delayed emission recorded during incubation of a 3 μm suspension of CYP3A4(C64)-ERIA incorporated into SS(R) at RL/P = 188 with 3 μm CYP3A4(C166)-DYM. (The RL/P ratio after incorporation of CYP3A4(C468)-DYM is equal to 94). The inset shows the time dependences of normalized intensity of donor fluorescence obtained in the experiments at an RL/P of 94, 667, and 1260. Solid lines, approximation of the kinetic curves with a biexponential equation. b, dependence of the LRET amplitude on CYP3A4 concentration in the membrane (triangles). The same dependence obtained with the CYP3A4(C64)ERIA/CYP3A4(C468)DYM pair (23) is shown in circles. Solid lines, results of the fitting of the data sets to the Hill equation. Dashed and dotted and dashed lines, approximations with Equations 1 and 2, respectively. a.u., arbitrary units.
The dependences of the LRET amplitude on P450 concentration obtained with CYP3A4(C64)ERIA/CYP3A4(C468)DYM and CYP3A4(C468)ERIA/CYP3A4(C166)DYM pairs are shown in Fig. 2b. The x axis of this plot represents the surface density of P450 in the membrane calculated based on RL/P using the value 0.95 nm2 for the area of the microsomal membrane corresponding to one molecule of phospholipid in a monolayer (47). Consistent with our earlier report, the plot obtained with CYP3A4(C64)ERIA/CYP3A4(C468)DYM was noticeably S-shaped. The sigmoidal character of the oligomerization isotherm of CYP3A4(C468)ERIA/CYP3A4(C166)DYM was less pronounced. Similar to the approach used in our previous publication (23), we employed the Hill equation in approximating these data sets. Although not based on any rigorous formalism, these approximations did not show systematic deviations from the data points (ρ2 ≥ 0.96). The parameters derived from these approximations are compared in Table 1. As seen in this table, the efficiency of LRET in CYP3A4(C468)ERIA/CYP3A4(C166)DYM reached 27.5 ± 1.7%. According to the calculation with the Förster equation, this value corresponds to the interprobe distance of ∼40 Å. Consistency of this estimate with the expected value of 46 Å corroborates the hypothesis that the orientation of the CYP3A4 molecules in the oligomer is similar to that observed in the x-ray structure 1W0F.
TABLE 1.
Oligomerization of CYP3A4, CYP3A4, and CYP2E1 in model microsomes studied by LRET
The values given in the table were obtained by fitting of the titration curves with the Hill equation (in the case of the interactions of 3A enzymes) or with the equation for the equilibrium of trimerization (Equation 2) (in the cases where at least one of the interacting proteins is represented by CYP2E1). The ± values show the confidence interval calculated for p = 0.05.
| P450 paira | [P450]50%b | (Lipid/P450)50%c | nH | LRET efficiency |
|---|---|---|---|---|
| pmol/cm2 | mol/mol | % | ||
| 3A4(C64) + 3A4(C468) | 0.47 ± 0.06 | 730 ± 95 | 3.7 ± 1.8 | 10.0 ± 1.2 |
| 3A4(C468) + 3A4(C166) | 0.56 ± 0.07 | 620 ± 80 | 2.0 ± 0.6 | 27.5 ± 1.7 |
| 3A5 + 3A5 | 0.13 ± 0.02 | 2580 ± 330 | 3.5 ± 2.0 | 10.7 ± 0.5 |
| 3A5 + 3A4(C468) | 0.19 ± 0.01 | 1830 ± 110 | 2.0 ± 0.1 | 17.3 ± 0.7 |
| 3A4(C468) + 3A5 | 0.30 ± 0.04 | 1160 ± 160 | 2.2 ± 0.6 | 9.8 ± 0.5 |
| 2E1 + 2E1 | 0.16 ± 0.05 | 2230 ± 530 | 11.4 ± 0.76 | |
| 2E1 + 3A4(C64) | 0.22 ± 0.08 | 1570 ± 460 | 5.2 ± 0.5 | |
| 2E1 + 3A5 | 0.072 ± 0.048 | 4800 ± 1920 | 8.4 ± 0.8 | |
| 3A5 + 2E1 | 0.15 ± 0.10 | 2240 ± 860 | 7.4 ± 1.1 |
a The first P450 enzyme listed in each pair was used as a bearer of the donor fluorophore (ERIA), whereas the second one had the acceptor (DYM) probe attached.
b Surface density of P450 in microsomal membranes at which the amplitude of the titration curves reaches 50% of maximal.
c Lipid/P450 ratio at which the amplitude of the titration curves reaches 50% of maximal.
Probing the Interface between CYP3A4 Subunits by Intermolecular Cross-linking
The interacting surfaces in the crystallographic dimer in the structure 1W0F include residues Cys239. The distance between the sulfur atoms of the two Cys239 residues (19.7 Å; Fig. 1b) should allow these residues to be cross-linked with thiol-reactive bifunctional reagents of appropriate size. As a control, we constructed the CYP3A4 C239S mutant. This substitution results in a stable protein with unaffected spin equilibrium and unchanged parameters of interactions with such substrates as testosterone, ANF, and 1-pyrenebutanol.
We probed cross-linking of the wild type enzyme and the C239S mutant in solution and in proteoliposomes with bBBr, o-DMB, and bMPM. As seen from the SDS-PAGE images shown in Fig. 3, incubation of the wild type enzyme in solution with any of the three reagents resulted in the appearance of cross-linked species.
FIGURE 3.
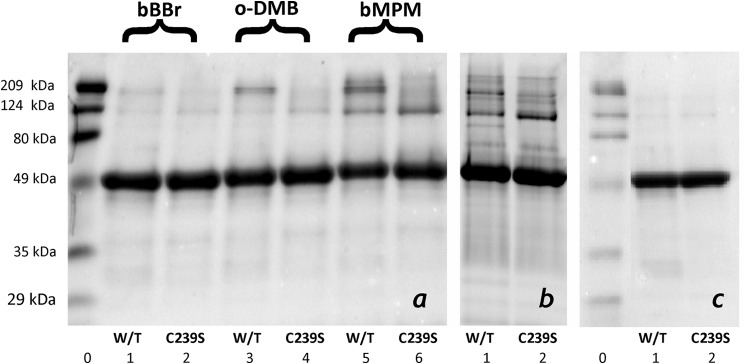
Results of SDS-PAGE of wild type CYP3A4 and its C239S mutant cross-linked with bBBr, o-DMB, and bMPM in solution (a) and bMPM in proteoliposomes (b) as compared with the intact purified proteins (c). The images on the panels show the fragments of three individual gel slabs, which were scaled according to the lanes of the molecular weight standards (lanes 0).
Comparison of the results obtained with different cross-linkers reveals a correlation between the size of the reagent and the degree of cross-linking. In the case of bBBr, which cross-links at S-S distances in the range of 3.2–6.6 Å and has maximal efficiency at a distance of 5 Å (48), the total fraction of the cross-linked protein was below 5%. Switching to the larger o-DMB (cross-linking distance of 7.7–10.5 Å with maximal efficiency at 9.6 Å (48)) and bMPM (9.4–17.3 Å and 15 Å, respectively (48)) increased the degree of cross-linking commensurate with the size of the reagent.
Despite this increase, the degree of cross-linking remained relatively low and did not exceed 30%, even in the case of bMPM. Such results are commonly observed with thiol-reactive bifunctional reagents (49–51). The ready explanation is that the reaction of two neighboring cysteines with one molecule of bifunctional reagent always competes with modification of each of the cysteines with separate molecules of the reagent. The degree of cross-linking is thus largely determined by the match of the length of the cross-linker with the average distance between the thiol groups. Therefore, the results of our cross-linking experiments should be viewed in the context of probing the protein-protein interfaces and the size of the oligomer rather than the degree of oligomerization, which is assessed quantitatively in our LRET titration experiments.
As seen from Fig. 3, cross-linking of the wild type CYP3A4 resulted in two major bands of cross-linked aggregates with molecular masses of around 120 and 190 kDa, which correspond to the cross-linked dimer and trimer, respectively. Most importantly, elimination of Cys239 caused a disappearance of the 190 kDa band, whereas the 120 kDa band became more intense, especially in the case of cross-linking with bMPM. Similar results were obtained with the use of bMPM to cross-link CYP3A4 incorporated into proteoliposomes with an RL/P ratio of 300:1 (Fig. 3b). Although the picture in this case was more complicated due to the appearance of several minor bands, the positions of the major bands of the cross-links were the same as observed in solution, and the C239S mutation resulted in a distinct redistribution of the intensities of the bands toward the band of dimers (Fig. 3).
These results suggest a trimer as the most likely size of the CYP3A4 oligomers both in solution and in the membrane. The assembly of subunits in the oligomer appears to involve two different types of interactions. Taken together, the results of our cross-linking and LRET experiments indicate that one of the subunit interfaces is similar to that observed in the structure 1W0F, where two Cys239 residues are located at a distance of 19.7 Å, which is close to the cross-linking range of bMPM (9.4–17.3 Å), the most efficient cross-linker.
Oligomerization of CYP3A5 and Its Interactions with CYP3A4
In order to incorporate the donor and acceptor fluorophores into CYP3A5, we labeled the wild type heme protein with ERIA and DYM probes and recovered the heme protein at a label/P450 ratio of 0.9–1.05. The modification did not cause any heme bleaching, P450-to-P420 conversion, or any considerable displacement of the spin equilibrium.
Incubation of CYP3A5-ERIA with SS(R)) added at the P450/lipid molar ratio (RL/P) in the range of 200:1 to 3000:1 resulted in efficient incorporation of the protein into the membranes. The recovery of the labeled heme protein in microsomes after their sedimentation and resuspension was in the range of 65–75%. Similar to CYP3A4 (23), the changes in the spectra of delayed fluorescence taken in the process of incubation of Supersomes containing CYP3A5-ERIA with CYP3A5-DYM (Fig. 4a) are indicative of the appearance of LRET due to the formation of mixed oligomers of the two labeled proteins. Similar changes were also observed in the pairs CYP3A5-ERIA/CYP3A4(C64)-DYM and CYP3A4(C64)-ERIA/CYP(3A5)-DYM.
FIGURE 4.

Oligomerization of CYP3A5 and its interactions with CYP3A4 in SS(R) microsomes studied with LRET. a, a series of spectra of delayed emission recorded during incubation of a 3 μm suspension of CYP3A5-ERIA incorporated into SS(R) at RL/P = 340 with 3 μm CYP3A5-DYM. (The RL/P ratio after incorporation of CYP3A5-DYM is equal to 170). The inset shows the time dependences of normalized intensity of donor fluorescence obtained in the experiments at the RL/P of 170 and 2400. Solid lines, approximation of the kinetic curves with a biexponential equation. b, dependence of the LRET amplitude on P450 concentration in the membrane for the interactions in the pairs CYP3A5-ERIA/CYP3A5-DYM (circles), CYP3A5-ERIA/CYP3A4(C468)-DYM (squares), and CYP3A4(C468)-ERIA/CYP3A5-DYM (triangles). Solid lines, results of the fitting of the data sets to the Hill equation (Equation 1). Dashed and dotted and dashed lines, approximations with Equations 1 and 2, respectively. a.u., arbitrary units.
The plots of the steady-state LRET efficiency versus surface density of the P450 enzymes are shown in Fig. 4b. All three titration curves reveal noticeable sigmoidal character. Our attempts to fit these data sets with equations for dimerization (Equation 1, dashed and dotted lines) or trimerization (Equation 2, dashed lines) did not produce satisfactory approximations. Therefore, analogous to the approach used in the case of CYP3A4 (Fig. 2), we approximated the oligomerization isotherms for CYP3A5 and CYP3A4/CYP3A5 mixtures with the Hill equation (Fig. 4b, solid lines). The results of this arbitrary fitting (ρ2 ≥ 0.98) did not reveal any systematic deviations from the data points. The parameters of interactions obtained in this way are summarized in Table 1. As seen from these values, the tendency of CYP3A5 to oligomerize in the membrane is higher than that observed with CYP3A4, whereas the mixed CYP3A4 + CYP3A5 system represents an intermediate case (Table 1).
Oligomerization of CYP2E1 and Its Interactions with CYP3A4 and CYP3A5
We used the unmodified truncated construct of CYP2E1 to introduce ERIA and DYM fluorophores. Similar to the labeling of CYP3A5, the modification of CYP2E1 by these probes at a 1:1 molar ratio yielded >80% of the labeled protein.
The efficiency of incorporation of CYP2E1-ERIA into SS(R) microsomes determined by the heme protein content in the membranes after their sedimentation and resuspension was as high as 95–98% of the amount taken for incorporation. Similar to CYP3A enzymes, incubation of Supersomes containing CYP2E1-ERIA with CYP2E1-DYM resulted in a considerable decrease in the donor emission accompanied by the appearance of the band of delayed fluorescence of the acceptor (Fig. 5a). The dependence of LRET amplitude on the CYP2E1 concentration in the membrane is shown in Fig. 4b (circles). In contrast to the case of CYP3A enzymes, the titration isotherm of CYP2E1 did not reveal any noticeable sigmoidal character and could be satisfactorily approximated with the steady-state equations for either dimerization (ρ2 = 0.933; dashed and dotted line) or trimerization (ρ2 = 0.946; dashed line) (Fig. 4b). However, the quality of approximation with the trimerization equilibrium equation was better (Fig. 5b).
FIGURE 5.

Oligomerization of CYP2E1 and its interactions with CYP3A4 and CYP3A5 in SS(R) microsomes studied by LRET. a, a series of spectra of delayed emission recorded during incubation of a 3 μm suspension of CYP2E1-ERIA incorporated into SS(R) at RL/P = 300 with 3 μm CYP2E1-DYM. (The RL/P ratio after incorporation of CYP2E1-DYM is equal to 150). Inset, time dependences of normalized intensity of donor fluorescence obtained in the experiments at the RL/P of 150 and 3000. Solid lines, approximation of the kinetic curves with a biexponential equation. b, dependence of the LRET amplitude on P450 concentration in the membrane for the interactions in the pairs CYP2E1-ERIA/CYP2E1-DYM (circles), CYP2E1-ERIA/CYP3A5-DYM (squares), and CYP2E1-ERIA/CYP3A4(C64)-DYM (triangles). Solid and dashed lines, results of approximation of the data sets with Equations 1 and 2, respectively. a.u., arbitrary units.
In order to probe the ability of CYP2E1 to form mixed oligomers with CYP3A4 and CYP3A5, we studied the interactions of CYP2E1-ERIA with CYP3A enzymes labeled with DYM. Whereas in the case of CYP3A5, we used the DYM-labeled wild type enzyme, the studies of CYP2E1 interactions with CYP3A4 were performed with DYM-labeled preparations of the single-cysteine mutant CYP3A4(C64). In both cases, we observed the appearance of LRET between ERIA and DYM during the incubations of Supersomes containing CYP2E1-ERIA with DYM-labeled CYP3A enzymes. A similar result was also obtained in the experiments where we incubated the Supersomes containing ERIA-labeled CYP3A5 with DYM-labeled CYP2E1. In all of these cases, the oligomerization isotherms could be satisfactorily approximated with the steady state equations of either dimerization or trimerization (Fig. 5b). The parameters of interactions obtained from the fitting of the oligomerization isotherms with the equation for the trimerization equilibrium are summarized in Table 1.
Metabolism of 7-MFC by CYP2E1-incorporated Supersomes
As we reported previously, incorporation of CYP3A4 into Supersomes containing recombinant rat CPR (SS(R)) resulted in a catalytically competent system where the parameters of CYP3A4-dependent O-debenzylation of 7-BFC were close to those observed with human liver microsomes (23). Here we report reconstitution of CYP2E1-dependent O-demethylation of 7-MFC in a similar system. In our studies, we used five individual preparations of SS(R)2E1 where the P450/lipid and CPR/P450 molar ratios (RL/P and RR/P, respectively) varied in the ranges 120:1 to 400:1 and 1:2.4 to 1:14.5, respectively. Thus, in all of our preparations, the heme protein was in molar excess over the reductase, and RR/P fell into the range typical for human liver microsomes, where the RR/P averaged over 150 human liver samples was found to be 1:7.1 (range 1:2 to 1:27) (52).
All preparations of SS(R)2E1 were active in the metabolism of 7-MFC. The dependence of the reaction rate on the concentration of 7-MFC obtained with SS(R)2E1 with RL/P = 470 and RR/P = 4.1 is exemplified in Fig. 6a (circles). The titration curves obtained in these experiments obey the Michaelis-Menten equation (ρ2 ≥ 0.98), and the values of Km were reproducible from preparation to preparation (Km = 9.5 ± 2.2 μm). At the same time, the values of Vmax vary proportionally to the value of RR/P, so that the dependence of Vmax on RR/P exhibits a strict linear correlation (Fig. 6b, circles).
FIGURE 6.
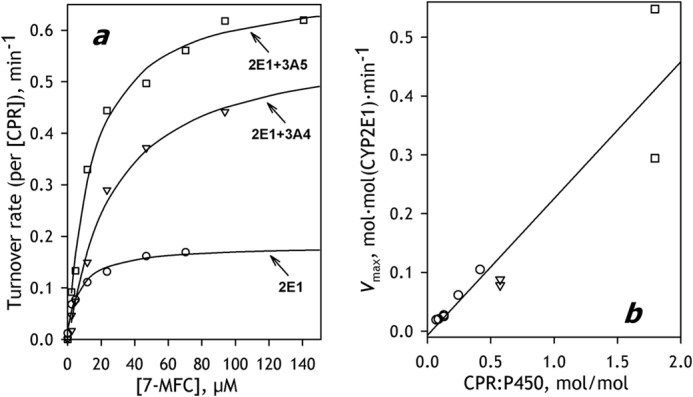
O-Demethylation of 7-MFC by CYP2E1-containing SS(R) systems and the effect of co-incorporation of CYP3A enzymes. a, dependences of the reaction rate on substrate concentrations obtained with SS(R)-2E1 (circles), SS(R)-2E1/3A5 (squares), and SS(R)-2E1/3A4 (triangles). Solid lines, approximations of the data set with the Hill equation. b, dependence of Vmax calculated per CYP2E1 concentration on the ratio of concentrations of CPR and P450 in microsomal membrane obtained with a series of SS(R)-2E1 preparations (circles) and the commercial preparations of CYP2E1 Supersomes® (squares) and CYP2E1 BaculosomesTM (triangles).
We also determined the parameters of CYP2E1-dependent O-demethylation of 7-MFC exhibited by Supersomes and Baculosome containing baculovirus-expressed recombinant human full-length CYP2E1 and human recombinant CPR. It should be noted that these preparations (SS(2E1) and BS(2E1), respectively) differ considerably from both human liver microsomes and our model system in the molar ratio of CPR to P450. In order to achieve maximal activity of P450, these commercial preparations were designed to have the RR/P ratio close to unity or even higher. According to our determination, the RR/P ratios characterizing the samples of Baculosomes and Supersomes used in our study were equal to 1.8:1 and 0.6:1, respectively.
The dependences of the rate of 7-BFC metabolism on the substrate concentration obtained with SS(2E1) and BS(2E1) are shown in Fig. 6a (triangles and bars, respectively). Fitting these curves with the Michaelis-Menten equation yields Km values of 14 ± 2 and 22 ± 4 μm for SS(2E1) and BS(2E1), respectively. As seen from Fig. 3a, the maximal velocities obtained with both preparations are higher than that exhibited by SS(R)2E1. This difference reflects the differences in RR/P ratios between these preparations. As seen from Fig. 6b, the points corresponding to SS(2E1) and BS(2E1) fall on the same linear dependence of Vmax on RR/P as the values of Vmax obtained with various preparations of SS(R)2E1 (Fig. 6b, squares and triangles, respectively). The whole data set including the values obtained with all three kinds of preparations (SS(R)2E1, SS(2E1), and BS(2E1)) is characterized by the square correlation of 0.867. The strict proportionality between Vmax and RR/P indicates that actual rate of catalysis is controlled by the concentration of CPR. Accordingly, the values of kcat calculated per concentration of CPR (which appears to be completely saturated with CYP2E1 in our conditions) are very similar for all preparations probed in this study (Table 2).
TABLE 2.
Effect of co-incorporation of CYP3A4 and CYP3A5 on CYP2E1-dependent 7-MFC O-demethylation in model microsomal systems
The values given in the table were obtained by averaging the results of 2–5 individual measurements (each with a different preparation of model microsomes), and the ± values show the confidence interval calculated for p = 0.05.
| System | S50 or Kma | nHb | Vmax | kcatc | R2E1d |
|---|---|---|---|---|---|
| μm | mol · mol(2E1) · min−1 | min−1 | |||
| SS(2E1)e | 14 ± 2 | NA | 0.42 ± 0.25 | 0.23 ± 0.14 | |
| SS(R)2E1 | 9.5 ± 2.2 | NA | 0.040 ± 0.024 | 0.24 ± 0.02 | |
| SS(R)2E1/3A4 | 20 ± 8 (0.01) | 1.23 ± 0.17 | 0.081 ± 0.031 | 0.48 ± 0.14 | 2.1 ± 0.4 (<0.01) |
| SS(R)2E1/3A5 | 15 ± 2 (0.08) | 1.09 ± 0.09 | 0.099 ± 0.049 | 0.51 ± 0.23 | 2.2 ± 0.8 (0.03) |
| BS(2E1)e | 22 ± 4 | NA | 0.083 ± 0.010 | 0.14 ± 0.02 | |
| BS(2E1)3A4f | 67 ± 26 (0.13) | NA | 0.11 ± 0.02 | 0.20 ± 0.02 | 1.4 ± 0.1 (0.03) |
| BS(2E1)3A5f | 57 ± 25 (0.12) | 1.50 ± 0.22 | 0.14 ± 0.03 | 0.23 ± 0.05 | 1.7 ± 0.4 (0.11) |
a The data were approximated with the Hill equation, except for the cases of BS(2E1) and BS(2E1) + 3A4, where the Michaelis equation was used. The values in parentheses represent the p values of Student's t test for the hypothesis of equality of the respective values of kcat measured in the microsomes containing both CYP2E1 and CYP3A enzymes with those observed in SS(R)2E1 or BS(2E1) preparations. The p values ≤0.05 are underlined to emphasize the effects with high statistical significance.
b Hill coefficient. NA designates the cases where the fitting was done with the Michaelis-Menten equation.
c Turnover rate calculated per concentration of CPR.
d Relative effect of co-incorporation of the second P450 species on Vmax. The values represent the averages of the ratios of Vmax values obtained in similar conditions, on the same date, and with the same batches of Supersomes or Baculosomes. The values in parentheses represent the p values of Student's t test for the hypothesis that the respective R2E1 ratios are equal to unity (i.e. that the co-incorporation of the CYP3A enzyme has no effect on CYP2E1 turnover). The p values ≤0.05 are underlined to emphasize the effects with high statistical significance.
e Preparation of Baculosomes (BS) or Supersomes (SS) containing human recombinant CYP2E1 and human recombinant CPR.
f Preparation of Baculosomes containing human recombinant CYP2E1 and human recombinant CPR with a co-incorporated purified CYP3A enzyme (see “Experimental Procedures”).
Effect of Interactions of CYP2E1 with CYP3A4 and CYP3A5 on CYP2E1-specific Metabolism of 7-MFC
As we demonstrated above, membrane-bound CYP2E1 forms high-affinity complexes with CYP3A4 and CYP3A5 in microsomal membranes. In order to probe how these interactions affect the functional properties of CYP2E1, we investigated the effect of co-incorporation of CYP3A4 or CYP3A5 together with CYP2E1 on CYP2E1-dependent O-demethylation of 7-MFC. In control experiments, we found that neither CYP3A4 nor CYP3A5 incorporated into SS(R) membranes exhibits any measurable activity with this substrate, so that the metabolism of 7-MFC in the mixed system can be completely attributed to the activity of CYP2E1.
Co-incorporation of CYP2E1 and CYP3A4 or CYP3A5 was achieved by incubation of a suspension of SS(R) with equimolar mixtures of CYP2E1 and a CYP3A enzyme taken at a lipid/P450 ratio of 100:1 to 300:1 as described under “Experimental Procedures.” The degree of incorporation of the heme proteins into the membrane in this case was around 50–60%, which is lower than that observed with CYP2E1 or CYP3A enzymes taken alone (70–95%).
In order to probe the concentrations of the individual P450 species in microsomal preparations containing CYP2E1/CYP3A4 or CYP2E1/CYP3A5 pairs, which we designate here as SS(R)2E1/3A4 and SS(R)2E1/3A5, respectively, we developed a spectrophotometric method for selective determination of CYP2E1. The method is based on our observation that CYP2E1 interacts with DTT with the formation of a hyperthiolate ligation complex of the heme iron. We found that the addition of DTT to purified CYP2E1 in solution resulted in a disappearance of the Soret bands of the low- and high-spin states positioned at 418 and 396 nm and a concomitant appearance of new bands at 371 and 454 nm. This “splitting” of the Soret band is indicative of the formation of the so-called hyperthiolate complex, where the sixth ligand of the heme iron is represented by a thiolate group of an external ligand (43). Formation of hyperthiolate complexes of cytochromes P450 with such organic thiols as chlorothiophenol, where the thiol group is highly ionized at neutral pH, is well documented (43). However, thiolate compounds with alkaline pKa, such as DTT, do not form this type of complexes with most P450 enzymes. The only known exception to this rule is P450 XplA from Rhodococcus rhodochrous, which has been reported to form hyperthiolate complexes with both DTT and β-mercaptoethanol (53). According to our knowledge, CYP2E1 is the first eukaryotic P450 enzyme that shares this unusual feature of P450 XplA.
Using this unique feature of CYP2E1 to determine its concentration, we found that the contents of CYP2E1 and CYP3A4/CYP3A5 enzymes in our samples were close to equimolar. The averaged molar fractions of CYP2E1 in five preparations each of SS(R)2E1/3A4 and SS(R)2E1/3A5 microsomes used in this study were estimated to be of 55 ± 9 and 49 ± 15%, respectively. The RL/P and RR/P ratios characterizing these preparations varied in the ranges of 140:1 to 280:1 and 7:1 to 14:1, respectively.
As shown in Fig. 6a, co-incorporation of either of two CYP3A enzymes results in a considerable increase in the rate of CYP2E1-dependent 7-MFC O-demethylation. The kinetic parameters in various systems are summarized in Table 2. Activation is revealed in both the values of Vmax calculated per concentration of CYP2E1 and kcat, which was calculated per concentration of CPR. In the latter case, the variability of the values obtained with different preparations of microsomes is considerably lower because the calculation takes into account the variation in the RR/P ratio between the samples.
Besides a pronounced increase in the rate of 7-MFC turnover, co-incorporation of the CYP3A enzymes also resulted in an appearance of some heterotropic cooperativity with this substrate, which is best seen in the case of SS(R)2E1/3A4, where the approximation of the titration curves with the Hill equation results in the nH value of 1.23 ± 0.17. Additionally, co-incorporation of CYP3A enzyme also produced a moderate decrease in CYP2E1 affinity for 7-MFC (Table 2).
We also evaluated the effect of co-incorporation of CYP3A enzymes with commercial preparations of Baculosomes® containing the recombinant human full-length CYP2E1 and the human recombinant CPR. Incubation of these preparations with purified CYP3A4 or CYP3A5 taken in a 1.2:1 molar ratio to CYP2E1 enzyme allowed us to obtain preparations containing CYP2E1 and CYP3A4 enzymes in a ratio close to equimolar. These preparations we designate here as BS(2E1)3A4 and BS(2E1)3A4. As seen from Table 2, the effect of CYP3A enzymes on CYP2E1-dependent metabolism of 7-MFC in Baculosomes was similar to that obtained in our SS(R) system. Here again, co-incorporation of CYP3A proteins resulted in an increase in CYP2E1 turnover and decreased substrate affinity. However, compared with the SS(R) system, the activating effect was less pronounced, the changes in the affinity were more prominent, and the emergence of homotropic cooperativity was detectable only in the case of the CYP2E1/CYP3A5 system (Table 2).
Effect of Interactions between Heterologous P450 Enzymes on CYP3A-dependent O-Debenzylation of 7-BQ
In order to probe the effect of formation of heterooligomers between CYP2E1, CYP3A4, and CYP3A5 enzymes on the catalytic properties of CYP3A enzymes, we investigated the effect of co-incorporation of CYP2E1 on CYP3A-specific activity in model microsomes. CYP2E1-containing microsomes (either SS(R)2E1, SS(2E1), or BS(2E1)) exhibit detectable activity in O-debenzylation of 7-BFC (up to 5% of the respective activity of CYP3A4). However, there was no measurable activity of any of these microsomes with 7-BQ, another CYP3A substrate.
The dependences of the rate of O-debenzylation of 7-BQ on substrate concentration obtained with SS(R)3A4, SS(R)3A5, and SS(R)3A4/3A5 are shown in Fig. 7. In all cases, the kinetic profiles of the reaction reveal prominent homotropic cooperativity and may be approximated with the Hill equation with the nH value varying from 1.4 to 3. The values of kinetic parameters obtained by averaging the results of 4–5 individual measurements are summarized in Table 3. The rate of 7-BQ metabolism by CYP3A5 is significantly higher than that observed with CYP3A4. However, although the addition of ANF increases considerably the turnover in the CYP3A4-catalyzed reaction, it has no effect on the catalysis by CYP3A5. Co-incorporation of CYP3A4 together with CYP3A5 resulted in a further increase in the rate of 7-BQ metabolism but failed to rescue the activating effect of ANF. Thus, the interaction of CYP3A5 with CYP3A4 is likely to activate the metabolism of 7-BQ by the latter enzyme at the expense of the activating effect of ANF. In contrast to the important increase of CYP2E1-specific activity in the presence of CYP3A reported above, co-incorporation of CYP2E1 elicits no statistically significant effect on O-debenzylation of 7-BQ by either of the two CYP3A enzymes.
FIGURE 7.
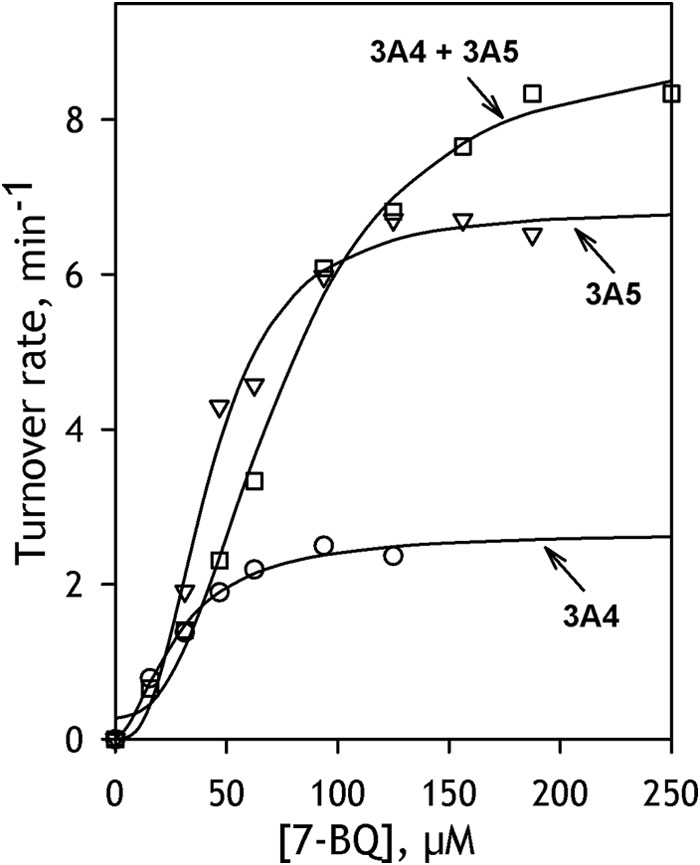
O-Debenzylation of 7-BQ by CYP3A-containing SS(R) systems. The plot shows the dependences of the reaction rate on substrate concentrations obtained with SS(R)-3A4 (circles), SS(R)-3A5 (squares), and SS(R)-3A4/3A5 (triangles). Solid lines, approximations of the data set with the Hill equation.
TABLE 3.
Effect of co-incorporation of CYP3A4, CYP3A5, and CYP2E1 on CYP3A-dependent 7-BQ O-debenzylation in model microsomal systems
The values given in the table were obtained by averaging the results of 2–5 individual measurements (each with a different preparation of model microsomes), and the ± values show the confidence interval calculated for p = 0.05.
| Systema | S50 | nHb | Vmax | kcatc | RANFd |
|---|---|---|---|---|---|
| μm | mol · mol(3A) · min−1 | min−1 | |||
| SS(R)3A4 | |||||
| −ANF | 46 ± 11 | 2.05 ± 0.49 | 0.86 ± 0.65 | 2.9 ± 0.8 | 1.7 ± 0.2 (0.02) |
| +ANF | 72 ± 22 | 1.57 ± 0.72 | 2.0 ± 1.2 | 4.8 ± 1.0 | |
| SS(R)3A5 | |||||
| −ANF | 76 ± 39 | 2.32 ± 0.65 | 1.1 ± 0.9 | 5.4 ± 3.0 (0.09) | 0.92 ± 0.1 (0.91) |
| +ANF | 62 ± 37 | 2.70 ± 0.63 | 1.3 ± 0.9 | 5.1 ± 2.3 (0.78) | |
| SS(R)3A4/3A5 | |||||
| −ANF | 61 ± 37 | 2.07 ± 0.54 | 3.1 ± 2.6 | 8.6 ± 0.1 (<0.01) | 0.85 ± 0.5 (0.62) |
| +ANF | 46 ± 11 | 2.00 ± 0.74 | 2.2 ± 0.7 | 7.3 ± 4.4 (0.18) | |
| SS(R)3A4/2E1 | |||||
| −ANF | 62 ± 34 | 1.48 ± 0.30 | 0.40 ± 0.21 | 2.2 ± 1.6 (0.64) | 2.0 ± 1.0 (0.35) |
| +ANF | 51 ± 6 | 2.31 ± 1.39 | 0.68 ± 0.10 | 3.2 ± 1.5 (0.06) | |
| SS(R)3A5/2E1 | |||||
| −ANF | 87 ± 33 | 2.26 ± 1.03 | 0.64 ± 0.24 | 2.2 ± 1.6 (0.37) | 1.8 ± 0.6 (0.44) |
| +ANF | 82 ± 49 | 2.80 ± 1.16 | 0.76 ± 0.80 | 3.7 ± 1.7 (0.45) | |
a Shown in the absence and in the presence of 25 μm ANF.
b The data were fitted with the Hill equation, and nH represents the Hill coefficient.
c Turnover rate calculated per concentration of CPR. The values in parentheses represent the p values of Student's t test for the hypothesis of equality of the respective values of kcat with the value observed for SS(R)3A4. The p values ≤0.05 are underlined to emphasize the effects with high statistical significance.
d Relative effect of the addition of 25 μm ANF on Vmax. The values represent the averages of the ratios of Vmax values obtained in the presence and in the absence of ANF with the same preparation of model microsomes. The values in parentheses represent the p values of Student's t test for the hypothesis that the respective RANF ratios are equal to unity (i.e. that ANF has no effect on the enzyme turnover). The p values ≤0.05 are underlined to emphasize the effects with high statistical significance.
DISCUSSION
This report presents the first systematic investigation of interactions between dissimilar P450 enzymes in microsomes in which the observed functional effects are correlated with the concentration of the heterologous P450-P450 complexes in the membrane. Capitalizing on our advances in developing model microsomal systems and methods for detection of P450-P450 interactions, we demonstrated the high propensity of CYP3A4, CYP3A5, and CYP2E1 to oligomerize. Furthermore, we observed the formation of high-affinity P450-P450 complexes in the pairs CYP3A4/CYP3A5, CYP2E1/CYP3A4, and CYP2E1/CYP3A5 and demonstrated a profound alteration of the catalytic properties of the individual P450 enzymes involved in these interactions.
The model system with purified recombinant P450 proteins incorporated into SS(R) microsomes introduced in our recent study (23) and further developed here provides unparalleled opportunities for studying P450-P450 interactions in the microsomal membrane. It allows obtaining the membranes with variable concentrations and isoform composition of the P450 ensemble. The use of this system in combination with labeling of P450 proteins with appropriate fluorescent and phosphorescent probes allows utilization of a wide variety of fluorescence-based techniques of detecting P450-P450 interactions, probing the degree of oligomerization, and determining the size of the oligomers.
Our comparison of P450-incorporated microsomes with commercial P450-containing insect cell microsomes (Baculosomes® and SupersomesTM) suggests that the SS(R) system provides an adequate model of the membrane-incorporated system, even when it is used with the N-terminally truncated P450 constructs. The comparison of the N-terminal sequences of CYP3A4, CYP3A5, and CYP2E1 with that of CYP51 from Saccharomyces cerevisiae, the first full-length membranous P450 enzyme with resolved structure of the N-terminal segment (Protein Data Bank entry 4LXJ (54)), suggests that the transmembrane helices of CYP3A and CYP2E1 are constituted by residues 13–33 and 8–30, respectively. Therefore, although the whole transmembrane segment appears to be retained in our Δ3–12 CYP3A constructs, the Δ3–20 variant of CYP2E1 retains only 11 of the 23 transmembrane residues. Despite this partial loss of the N-terminal anchor, the truncated variant of CYP2E1 retains its ability to be efficiently incorporated into microsomal membranes. Moreover, comparison of the SS(R)2E1 containing the truncated enzyme with the commercial preparations of Supersomes® and BaculosomesTM containing the full-length recombinant enzyme does not reveal any major functional differences between the two systems (Table 2) in terms of turnover rates and Km values in the metabolism of 7-MFC and effects of co-incorporation of CYP3A4 and CYP3A5 on the CYP2E1 activity.
These findings suggest that the truncation of CYP2E1 does not change the mode of the enzyme interactions with the membrane or considerably modify its functional properties. This conclusion is in agreement with the results of Baylon et al. (55), who concluded that the membrane binding of P450 is largely independent of the presence of the N-terminal hydrophobic anchor. According to the current concepts, the interactions of the microsomal P450 enzymes with the membrane are in large part determined by the regions between the N-terminal anchor and α-helix A (54, 56, 57) as well as by the hydrophobic surface in the region of α-helices F′ and G′(55, 58, 59). Furthermore, according to the x-ray structure of the full-length CYP51, the constrained orientation of the P450 catalytic domain relative to the membrane is dictated by a network of interactions of polar residues in the C-terminal part of the transmembrane helix and the following proline-rich region (54), which are completely retained in our constructs.
Importantly, our studies revealed a high degree of oligomerization of all three studied P450 proteins at concentrations similar to those in the endoplasmic reticulum of liver cells. According to our measurements, the concentrations (surface densities) at which 50% of the individual heme proteins are oligomerized vary from 0.16 pmol/cm2 for CYP2E1 to 0.47 pmol/cm2 for CYP3A4. The respective values for the pairs of heterologous P450 enzymes range from 0.07 to 0.30 pmol/cm2 for CYP3A4/CYP3A5 (Table 1). For comparison, the measurements of Watanabe et al. (60) suggest that the surface density of cytochromes P450 in the endoplasmic reticulum of mouse hepatocytes lies in the range of 0.6–2.8 pmol/cm2. We may infer therefore that at physiological concentrations in the endoplasmic reticulum membrane, all three heme proteins are heavily oligomerized, and an important part of their pool is involved in hetero-association between different P450 species.
Results of our cross-linking experiments with the wild type CYP3A4 and its C239S mutant suggest that the assembly of subunits in CYP3A4 oligomers is likely to involve two different types of interactions, and, most probably, the number of subunits in the oligomer is a multiple of 3. Taken together, the results of cross-linking and LRET studies indicate that one of the subunit interfaces is similar to that observed in the CYP3A4 x-ray structure 1W0F, where Cys239 residues of two subunits are located in close proximity to each other. These findings strengthen our prior inference that the peripheral steroid-binding site in the vicinity of the F′ and G′ helices of two interacting CYP3A4 molecules observed in the x-ray structure 1W0F is physiologically relevant (23, 61). The interactions at the second type of the intersubunit interface are also likely to bring some cysteine residues other than Cys239 of two CYP3A4 subunits into close proximity to each other.
Our conclusion that the minimal building block of the P450 oligomer is a trimer is in a good agreement with the results of the measurements of the rate of rotational diffusion of membrane-bound cytochromes P450 by different techniques, which suggest a hexamer as the preferential size of the P450 oligomers (18, 19). Interestingly, the predominating size of the oligomers of purified CYP2B4 (63) and CYP3A4 (64) in solution is also a hexamer. Furthermore, according to the results of electron microscopy, the aggregates of purified CYP2B4 in solution are represented by hexamers organized as a two-layer dimer of trimers (65). The close similarity of the results of our cross-linking experiments in solution and in the membrane (Fig. 3) indicates that the trimeric building blocks of the oligomers in solution and in the membrane may share the same structure.
An important insight into the mechanisms of the functional effects of P450-P450 interactions may be derived from the results of the present study together with the prior observations of “persistent conformational heterogeneity.” We originated this term to designate unusual “non-equilibrating” divergence of the pool of membrane-bound P450 proteins into functionally distinct fractions, which has been observed in numerous studies with various techniques (see Ref. 66 for review). In particular, an oligomerization-related heterogeneity was demonstrated in our study of dithionite-dependent reduction of CYP3A4. Whereas the kinetics of reduction in CYP3A4 oligomers in solution or in proteoliposomes obeys a triexponential equation, the monomerization of the enzyme by its incorporation into Nanodiscs or into liposomes with high RL/P, renders the process monophasic (67). The most remarkable finding is that, whereas the fraction of oligomeric P450 reduced in the fast phase is almost completely low-spin, the high-spin state strongly predominates in the slowly reducible fraction (67).
A similar contrast between the high- and low-spin states of P450 was also observed in the studies of NADPH-dependent reduction. Opposite to the case of dithionite, the fast phase of the NADPH-dependent reduction is preferential for the high-spin heme protein. Selective reduction of the high-spin P450 in the fast phase of the NADPH-dependent process has been demonstrated with CYP2C11 (68), CYP2B4 (69), and most recently with CYP3A4 (22, 40).
The contrast in the kinetics of reduction between the high- and the low-spin states seemingly contradicts the very high rate of P450 spin transitions (62, 70–72), which implies that the position of equilibrium between the two states must remain unaffected throughout the reduction. This contradiction suggests that the spin equilibrium in the oligomers cannot be considered as being applied to the whole P450 pool. The enzyme is rather distributed between non-interconverting subpopulations that differ in the position of spin equilibrium, affinity for substrates, and ability to form electron transfer complexes with the flavoprotein partner. The latter difference was best revealed in our studies with the use of the flavin domain of cytochrome P450-BM3 (BMR) as a soluble substitute for CPR, where we found that only about 50% of oligomeric CYP3A4 was able to be reduced by BMR, either in solution or in proteoliposomes. Here again, the monomerization of CYP3A4 resulted in its complete reducibility (22, 40).
The relevance of the functional effects of hetero-association to differential modulation of the affinities of interacting P450 species for CPR was demonstrated in our studies with the CYP1A2/CYP2B4 pair in a soluble reconstituted system, where we probed the formation of P450-CPR complexes by FRET from the CPM-labeled CPR to the heme of P450 (6). We demonstrated that, whereas CPR has similar affinities for CYP1A2 and CYP2B4, titration with CYP1A2/CYP2B4 mixtures in the presence of 7-ethoxyresorufin, a substrate of CYP1A2, reveals a dependence of KD of the P450-CPR complex on the CYP1A2/CYP2B4 ratio that is described by an asymmetric bell-shaped curve (6). A multifold decrease in the apparent affinity for CPR observed in the mixtures with a high molar fraction of 2B4 suggests that the association of 2B4 with 1A2 in the presence of 7-ethoxyresorufin “hides” 2B4 from the interactions with CPR (6). This conclusion is in good agreement with the studies of Backes and co-workers (13, 31).
The present study provides a plausible structural explanation for the “persistent heterogeneity” and its relevance to the functional consequences of hetero-association of dissimilar P450 species. According to our results, the oligomers of CYP3A4 in the membrane are presumably organized as trimers or their multiples, where the interactions between the constituting subunits involve two different types of protein-protein contacts. We hypothesize that this architecture gives rise to orientational and/or conformational dissimilarity of the subunits in the trimer, which is likely to be organized as a dimer with an associated third subunit. As a consequence, the formation of mixed oligomers of multiple P450 species may result in selective activation of one enzyme and/or suppression of the activity of its counterpart.
According to our hypothesis illustrated in Fig. 8, the two types of subunits in P450 oligomer differ in both the propensity to form productive electron transfer complexes with CPR and the ability to interact with substrates. Due to this difference, the presence of a substrate specific to one of the interacting P450 species would cause a redistribution of the multiple enzymes between the active and “hindered” locations, so that the P450 complexed with substrate will be activated. A beneficial result of this mechanism would be to maintain a balance between the oxygenation of P450 substrates and the uncoupled production of reactive oxygen species by the P450 ensemble and ensure rapid adaptation to any changes in cellular exposure to xenobiotics.
FIGURE 8.
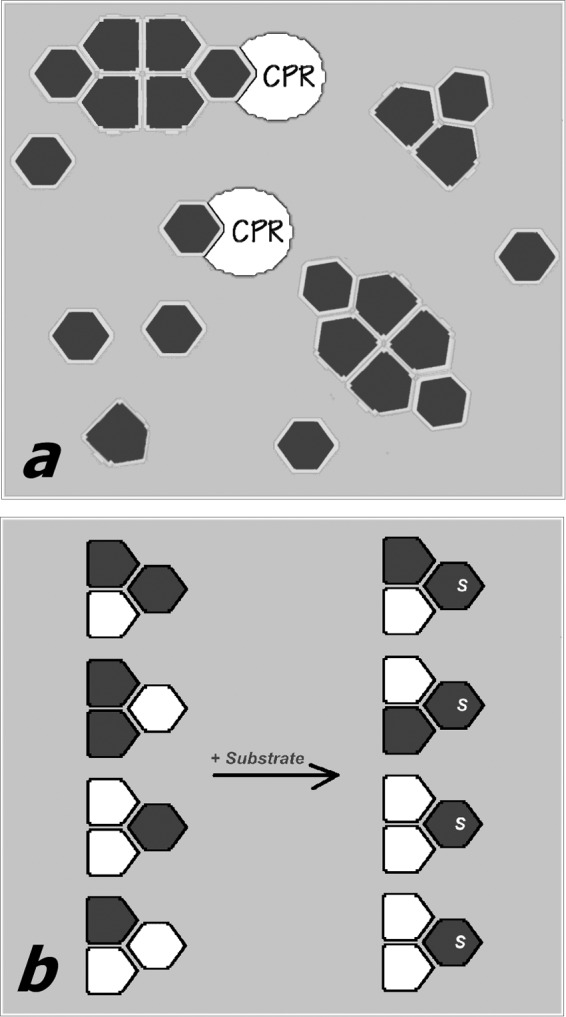
Schematic illustrating our hypothesis about the mechanism of the functional effects of P450 oligomerization. In a, a P450 heme protein (dark gray-filled polygons) exists in equilibrium between monomers and oligomers. The subunits of the oligomer attain two different conformations (hexagons and pentagons), depending on their position in the trimer, which is the minimal building block of the oligomers. One of the two conformations (hexagons) preferentially forms electron transfer complexes with the reductase (white-filled partial ovals) and interacts with the P450 substrates. The “pentagon-shaped” conformation is therefore excluded (either completely or partially) from electron transfer and catalysis. b, illustrates the hypothesis of substrate-induced reorganization of mixed oligomers of two different P450 enzymes (white- and dark gray-filled polygons). In the absence of substrate (left) the distribution of the two species between two P450 conformations (hexagons and pentagons) is random. The addition of a substrate specific to one of the two P450 species (dark gray) causes a redistribution of the two species in the oligomers, so that the gray protein now occupies the positions that are open for the interactions with substrate (hexagon-shaped subunits).
An important feature of this mechanism is that the degree of activation caused by hetero-association is determined by the disparity between the interacting enzymes in their affinities for each particular P450 substrate. The most pronounced must be the activating effect observed with a highly selective substrate of one of the two interacting species, as we observed in the case of 7-MFC and CYP2E1/CYP3A pairs. Admittedly, there is a finite possibility that the formation of the mixed oligomers with CYP2E1 changes the substrate selectivity of CYP3A enzymes, so that they become capable of metabolizing 7-MFC. However, this seems very unlikely in view of the lack of any effect of CYP2E1 on the metabolism of 7-BQ by CYP3A enzymes. It is difficult to imagine a situation where a stimulus that makes the enzyme capable of metabolizing a new substrate (7-MFC) would not affect the metabolism of other substrates (such as 7-BQ). Therefore, the activating effect of CYP2E1-CYP3A interactions on the metabolism of 7-MFC is better explained with the model of substrate-dependent reorganization of oligomers (Fig. 8b), assuming that the affinity of CYP2E1 for 7-MFC is much higher than that of the CYP3A enzymes. In contrast, the effect of interactions of two closely similar P450 species, such as CYP3A4 and CYP3A5, may be difficult to detect due to their nearly matching substrate specificities.
The proposed mechanism may be further complicated by different propensities of the interacting enzymes to occupy active and hindered positions even in the substrate-free state. Furthermore, oligomerization of certain P450 enzymes (such as CYP3A4) may result in emergence of oligomer-specific effector-binding sites that may be involved in regulation of P450-P450 interactions and control the catalytic properties of the P450 ensemble. Although this hypothetical model requires further rigorous examination, it provides a plausible explanation for all known data on the functional effects of interactions between dissimilar P450 enzymes.
Our results demonstrate a critical importance of physical interactions between multiple cytochrome P450 species as a major determinant of the functional properties of the human drug-metabolizing ensemble. Further elucidation of the mechanistic basis of complex, non-additive behavior of P450 enzymes revealed in this study is critical for establishing a systems biology approach to human drug-metabolizing ensemble and in depth understanding of the physiological effects of the developmental, age-dependent, and temporal changes in the P450 expression profile.
This work was supported, in whole or in part, by National Institutes of Health Grant GM054995 (to J. R. H.).
- CPR
- NADPH-cytochrome P450 reductase
- LRET
- luminescence resonance energy transfer
- o-DMB
- N,N′(o-phenylene)dimaleimide, 1,2-dimaleimidobenzene
- bMPM
- 4,4′-methylenebis(N-phenylmaleimide)
- 7-BQ
- 7-benzyloxyquinoline
- 7-MFC
- 7-methoxy-4-(trifluoromethyl)coumarin
- 7-BFC
- 7-benzyloxy-4-(trifluoromethyl)coumarin
- ANF
- α-naphthoflavone
- ERIA
- erythrosine 5′-iodoacetamide
- bBBr
- dibromobimane
- DYM
- DY-731 maleimide
- SS(R)
- “control” SupersomesTM containing rat recombinant NADPH-cytochrome P450 reductase manufactured by Corning Life Sciences (catalog no. 456514).
REFERENCES
- 1. Kaminsky L. S., Guengerich F. P. (1985) Cytochrome P-450 isozyme/isozyme functional interactions and NADPH-cytochrome P-450 reductase concentrations as factors in microsomal metabolism of warfarin. Eur. J. Biochem. 149, 479–489 [DOI] [PubMed] [Google Scholar]
- 2. Chen G. F., Ronis M. J., Ingelman-Sundberg M., Badger T. M. (1999) Hormonal regulation of microsomal cytochrome P4502E1 and P450 reductase in rat liver and kidney. Xenobiotica 29, 437–451 [DOI] [PubMed] [Google Scholar]
- 3. Cawley G. F., Batie C. J., Backes W. L. (1995) Substrate-dependent competition of different P450 isozymes for limiting NADPH-cytochrome P450 reductase. Biochemistry 34, 1244–1247 [DOI] [PubMed] [Google Scholar]
- 4. Yamazaki H., Inoue K., Mimura M., Oda Y., Guengerich F. P., Shimada T. (1996) 7-Ethoxycoumarin O-deethylation catalyzed by cytochromes P450 1A2 and 2E1 in human liver microsomes. Biochem. Pharmacol. 51, 313–319 [DOI] [PubMed] [Google Scholar]
- 5. Tan Y., Patten C. J., Smith T., Yang C. S. (1997) Competitive interactions between cytochromes P450 2A6 and 2E1 for NADPH-cytochrome P450 oxidoreductase in the microsomal membranes produced by a baculovirus expression system. Arch. Biochem. Biophys. 342, 82–91 [DOI] [PubMed] [Google Scholar]
- 6. Davydov D. R., Petushkova N. A., Bobrovnikova E. V., Knyushko T. V., Dansette P. (2001) Association of cytochromes P450 1A2 and 2B4: are the interactions between different P450 species involved in the control of the monooxygenase activity and coupling? Adv. Exp. Med. Biol. 500, 335–338 [DOI] [PubMed] [Google Scholar]
- 7. Yamada M., Ohta Y., Bachmanova G. I., Archakov A. I., Hatta I., Kawato S. (2001) Effect of microsome-liposome fusion on the rotational mobility of cytochrome P450IIB4 in rabbit liver microsomes. J. Inorg. Biochem. 83, 261–268 [DOI] [PubMed] [Google Scholar]
- 8. Greinert R., Finch S. A., Stier A. (1982) Conformation and rotational diffusion of cytochrome P-450 changed by substrate binding. Biosci. Rep. 2, 991–994 [DOI] [PubMed] [Google Scholar]
- 9. Hildebrandt P., Garda H., Stier A., Bachmanova G. I., Kanaeva I. P., Archakov A. I. (1989) Protein-protein interactions in microsomal cytochrome P-450 isozyme LM2 and their effect on substrate binding. Eur. J. Biochem. 186, 383–388 [DOI] [PubMed] [Google Scholar]
- 10. Schwarz D., Pirrwitz J., Meyer H. W., Coon M. J., Ruckpaul K. (1990) Membrane topology of microsomal cytochrome P-450: saturation transfer EPR and freeze-fracture electron microscopy studies. Biochem. Biophys. Res. Commun. 171, 175–181 [DOI] [PubMed] [Google Scholar]
- 11. McIntosh P. R., Kawato S., Freedman R. B., Cherry R. J. (1980) Evidence from cross-linking and rotational diffusion studies that cytochrome P450 can from molecular aggregates in rabbit-liver microsomal membranes. FEBS Lett. 122, 54–58 [DOI] [PubMed] [Google Scholar]
- 12. Myasoedova K. N., Magretova N. N. (2001) Cross-linking study of cytochrome P450 1A2 in proteoliposomes. Biosci. Rep. 21, 63–72 [DOI] [PubMed] [Google Scholar]
- 13. Reed J. R., Eyer M., Backes W. L. (2010) Functional interactions between cytochromes P450 1A2 and 2B4 require both enzymes to reside in the same phospholipid vesicle: evidence for physical complex formation. J. Biol. Chem. 285, 8942–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alston K., Robinson R. C., Park S. S., Gelboin H. V., Friedman F. K. (1991) Interactions among cytochromes P-450 in the endoplasmic reticulum: detection of chemically cross-linked complexes with monoclonal antibodies. J. Biol. Chem. 266, 735–739 [PubMed] [Google Scholar]
- 15. Praporski S., Ng S. M., Nguyen A. D., Corbin C. J., Mechler A., Zheng J., Conley A. J., Martin L. L. (2009) Organization of cytochrome P450 enzymes involved in sex steroid synthesis: protein-protein interactions in lipid membranes. J. Biol. Chem. 284, 33224–33232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ozalp C., Szczesna-Skorupa E., Kemper B. (2005) Bimolecular fluorescence complementation analysis of cytochrome P4502C2, 2E1, and NADPH-cytochrome P450 reductase molecular interactions in living cells. Drug Metab. Disp. 33, 1382–1390 [DOI] [PubMed] [Google Scholar]
- 17. Reed J. R., Connick J. P., Cheng D., Cawley G. F., Backes W. L. (2012) Effect of homomeric P450-P450 complexes on P450 function. Biochem. J. 446, 489–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greinert R., Finch S. A., Stier A. (1982) Cytochrome P-450 rotamers control mixed-function oxygenation in reconstituted membranes: rotational diffusion studied by delayed fluorescence depolarization. Xenobiotica 12, 717–726 [DOI] [PubMed] [Google Scholar]
- 19. Kawato S., Gut J., Cherry R. J., Winterhalter K. H., Richter C. (1982) Rotation of cytochrome P-450. I. Investigations of protein-protein interactions of cytochrome P-450 in phospholipid vesicles and liver microsomes. J. Biol. Chem. 257, 7023–7029 [PubMed] [Google Scholar]
- 20. Myasoedova K. N., Berndt P. (1990) Cytochrome P-450LM2 oligomers in proteoliposomes. FEBS Lett. 275, 235–238 [DOI] [PubMed] [Google Scholar]
- 21. Hu G., Johnson E. F., Kemper B. (2010) Cytochrome P450 2C8 exists as a dimer in natural membranes. Drug Metab. Dispos. 38, 1976–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davydov D. R., Sineva E. V., Sistla S., Davydova N. Y., Frank D. J., Sligar S. G., Halpert J. R. (2010) Electron transfer in the complex of membrane-bound human cytochrome P450 3A4 with the flavin domain of P450BM-3: the effect of oligomerization of the heme protein and intermittent modulation of the spin equilibrium. Biochim. Biophys. Acta 1797, 378–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Davydov D. R., Davydova N. Y., Sineva E. V., Kufareva I., Halpert J. R. (2013) Pivotal role of P450-P450 interactions in CYP3A4 allostery: the case of α-naphthoflavone. Biochem. J. 453, 219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davydov D. R. (2011) Microsomal monooxygenase as a multienzyme system: the role of P450-P450 interactions. Expert Opin. Drug Metab. Toxicol. 7, 543–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reed J. R., Backes W. L. (2012) Formation of P450·P450 complexes and their effect on P450 function. Pharmacol. Ther. 133, 299–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamazaki H., Gillam E. M., Dong M. S., Johnson W. W., Guengerich F. P., Shimada T. (1997) Reconstitution of recombinant cytochrome P450 2C10(2C9) and comparison with cytochrome P450 3A4 and other forms: effects of cytochrome P450- P450 and cytochrome P450-b5 interactions. Arch. Biochem. Biophys. 342, 329–337 [DOI] [PubMed] [Google Scholar]
- 27. Hazai E., Kupfer D. (2005) Interactions between CYP2C9 and CYP2C19 in reconstituted binary systems influence their catalytic activity: possible rationale for the inability of CYP2C19 to catalyze methoxychlor demethylation in human liver microsomes. Drug Metab. Dispos. 33, 157–164 [DOI] [PubMed] [Google Scholar]
- 28. Subramanian M., Low M., Locuson C. W., Tracy T. S. (2009) CYP2D6-CYP2C9 protein-protein interactions and isoform-selective effects on substrate binding and catalysis. Drug Metab. Dispos. 37, 1682–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Subramanian M., Tam H., Zheng H., Tracy T. S. (2010) CYP2C9-CYP3A4 protein-protein interactions: role of the hydrophobic N-terminus. Drug Metab. Dispos. 38, 1003–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Backes W. L., Batie C. J., Cawley G. F. (1998) Interactions among P450 enzymes when combined in reconstituted systems: formation of a 2B4–1A2 complex with a high affinity for NADPH-cytochrome P450 reductase. Biochemistry 37, 12852–12859 [DOI] [PubMed] [Google Scholar]
- 31. Cawley G. F., Zhang S., Kelley R. W., Backes W. L. (2001) Evidence supporting the interaction of CYP2B4 and CYP1A2 in microsomal preparations. Drug Metab. Dispos. 29, 1529–1534 [PubMed] [Google Scholar]
- 32. Davydov D. R., Petushkova N. A., Archakov A. I., Hoa G. H. (2000) Stabilization of P450 2B4 by its association with P450 1A2 revealed by high-pressure spectroscopy. Biochem. Biophys. Res. Commun. 276, 1005–1012 [DOI] [PubMed] [Google Scholar]
- 33. Kelley R. W., Reed J. R., Backes W. L. (2005) Effect of ionic strength on the functional interactions between CYP2B4 and CYP1A2. Biochemistry 44, 2632–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kelley R. W., Cheng D., Backes W. L. (2006) Heteromeric complex formation between CYP2E1 and CYP1A2: evidence for the involvement of electrostatic interactions. Biochemistry 45, 15807–15816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Backes W. L., Kelley R. W. (2003) Organization of multiple cytochrome P450s with NADPH-cytochrome P450 reductase in membranes. Pharmacol. Ther. 98, 221–233 [DOI] [PubMed] [Google Scholar]
- 36. Tsalkova T. N., Davydova N. Y., Halpert J. R., Davydov D. R. (2007) Mechanism of interactions of α-naphthoflavone with cytochrome P450 3A4 explored with an engineered enzyme bearing a fluorescent probe. Biochemistry 46, 106–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harlow G. R., Halpert J. R. (1998) Analysis of human cytochrome P450 3A4 cooperativity: construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics. Proc. Natl. Acad. Sci. U.S.A. 95, 6636–6641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spatzenegger M., Liu H., Wang Q., Debarber A., Koop D. R., Halpert J. R. (2003) Analysis of differential substrate selectivities of CYP2B6 and CYP2E1 by site-directed mutagenesis and molecular modeling. J. Pharmacol. Exp. Ther. 304, 477–487 [DOI] [PubMed] [Google Scholar]
- 39. Jang H.-H., Dayydov D. R., Lee G.-Y., Yun C.-H., Halpert J. R. (2014) The role of cytochrome P450 2B6 and 2B4 substrate access channel residues predicted based on crystal structures of the amlodipine complexes. Arch. Biochem. Biophys. 545, 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fernando H., Halpert J. R., Davydov D. R. (2008) Kinetics of electron transfer in the complex of cytochrome P450 3A4 with the flavin domain of cytochrome P450BM-3 as evidence of functional heterogeneity of the heme protein. Arch. Biochem. Biophys. 471, 20–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Davydov D. R., Deprez E., Hoa G. H., Knyushko T. V., Kuznetsova G. P., Koen Y. M., Archakov A. I. (1995) High-pressure-induced transitions in microsomal cytochrome P450 2B4 in solution: evidence for conformational inhomogeneity in the oligomers. Arch. Biochem. Biophys. 320, 330–344 [DOI] [PubMed] [Google Scholar]
- 42. Renaud J. P., Davydov D. R., Heirwegh K. P. M., Mansuy D., Hui Bon Hoa G. (1996) Thermodynamic studies of substrate binding and spin transitions in human cytochrome P-450 3A4 expressed in yeast microsomes. Biochem. J. 319, 675–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sono M., Andersson L. A., Dawson J. H. (1982) Sulfur donor ligand-binding to ferric cytochrome P-450-cam and myoglobin: ultraviolet-visible absorption, magnetic circular-dichroism, and electron-paramagnetic resonance spectroscopic investigation of the complexes. J. Biol. Chem. 257, 8308–8320 [PubMed] [Google Scholar]
- 44. Davydov D. R., Davydova N. Y., Tsalkova T. N., Halpert J. R. (2008) Effect of glutathione on homo- and heterotropic cooperativity in cytochrome P450 3A4. Arch. Biochem. Biophys. 471, 134–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sineva E. V., Rumfeldt J. A. O., Halpert J. R., Davydov D. R. (2013) A large-scale allosteric transition in cytochrome P450 3A4 revealed by luminescence resonance energy transfer (LRET). PLoS One 8, e83898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Williams P. A., Cosme J., Vinkovic D. M., Ward A., Angove H. C., Day P. J., Vonrhein C., Tickle I. J., Jhoti H. (2004) Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 305, 683–686 [DOI] [PubMed] [Google Scholar]
- 47. Wibo M., Amar-Costesec A., Berthet J., Beaufay H. (1971) Electron microscope examination of subcellular fractions III: quantitative analysis of the microsomal fraction isolated from rat liver. J. Cell Biol. 51, 52–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Green N. S., Reisler E., Houk K. N. (2001) Quantitative evaluation of the lengths of homobifunctional protein cross-linking reagents used as molecular rulers. Protein Sci. 10, 1293–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nagy J. K., Lau F. W., Bowie J. U., Sanders C. R. (2000) Mapping the oligomeric interface of diacylglycerol kinase by engineered thiol cross-linking: homologous sites in the transmembrane domain. Biochemistry 39, 4154–4164 [DOI] [PubMed] [Google Scholar]
- 50. Paumard P., Arselin G., Vaillier J., Chaignepain S., Bathany K., Schmitter J. M., Brèthes D., Velours J. (2002) Two ATP synthases can be linked through subunits i in the inner mitochondrial membrane of Saccharomyces cerevisiae. Biochemistry 41, 10390–10396 [DOI] [PubMed] [Google Scholar]
- 51. Prahlad J., Hauser D. N., Milkovic N. M., Cookson M. R., Wilson M. A. (2014) Use of cysteine-reactive cross-linkers to probe conformational flexibility of human DJ-1 demonstrates that Glu18 mutations are dimers. J. Neurochem. 130, 839–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gomes A. M., Winter S., Klein K., Turpeinen M., Schaeffeler E., Schwab M., Zanger U. M. (2009) Pharmacogenomics of human liver cytochrome P450 oxidoreductase: multifactorial analysis and impact on microsomal drug oxidation. Pharmacogenomics 10, 579–599 [DOI] [PubMed] [Google Scholar]
- 53. Bui S. H., McLean K. J., Cheesman M. R., Bradley J. M., Rigby S. E. J., Levy C. W., Leys D., Munro A. W. (2012) Unusual spectroscopic and ligand binding properties of the cytochrome P450-flavodoxin fusion enzyme XplA. J. Biol. Chem. 287, 19699–19714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Monk B. C., Tomasiak T. M., Keniya M. V., Huschmann F. U., Tyndall J. D. A., O'Connell J. D., 3rd, Cannon R. D., McDonald J. G., Rodriguez A., Finer-Moore J. S., Stroud R. M. (2014) Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc. Natl. Acad. Sci. U.S.A. 111, 3865–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Baylon J. L., Lenov I. L., Sligar S. G., Tajkhorshid E. (2013) Characterizing the membrane-bound state of Cytochrome P450 3A4: structure, depth of insertion, and orientation. J. Am. Chem. Soc. 135, 8542–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Williams P. A., Cosme J., Sridhar V., Johnson E. F., McRee D. E. (2000) Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol. Cell 5, 121–131 [DOI] [PubMed] [Google Scholar]
- 57. Liu J., Tawa G. J., Wallqvist A. (2013) Identifying cytochrome P450 functional networks and their allosteric regulatory elements. PLoS One 8, e81980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Berka K., Paloncýová M., Anzenbacher P., Otyepka M. (2013) Behavior of human cytochromes P450 on lipid membranes. J. Phys. Chem. B 117, 11556–11564 [DOI] [PubMed] [Google Scholar]
- 59. Cojocaru V., Balali-Mood K., Sansom M. S. P., Wade R. C. (2011) Structure and dynamics of the membrane-bound cytochrome P450 2C9. PLoS Comput. Biol. 7, e1002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Watanabe J., Asaka Y., Kanai K., Kanamura S. (1992) Relation between cytochrome P-450 increase and endoplasmic-reticulum proliferation in hepatocytes of mice treated with phenobarbital: a microphotometric and morphometric study. J. Histochem. Cytochem. 40, 353–357 [DOI] [PubMed] [Google Scholar]
- 61. Davydov D. R., Rumfeldt J. A. O., Sineva E. V., Fernando H., Davydova N. Y., Halpert J. R. (2012) Peripheral ligand-binding site in cytochrome P450 3A4 located with fluorescence resonance energy transfer (FRET). J. Biol. Chem. 287, 6797–6809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ziegler M., Blanck J., Ruckpaul K. (1982) Spin equilibrium relaxation kinetics of cytochrome P450 LM2. FEBS Lett. 150, 219–222 [Google Scholar]
- 63. Berndt P., Magretova N. N., Miasoedova K. N., Cherniak V. Y. (1989) Cytochrome P-450 LM2 hexamer dissociation in the presence of a nonionic detergent Emulgene 913. Biokhimiia 54, 338–341 [PubMed] [Google Scholar]
- 64. Fernando H., Davydov D. R., Chin C. C., Halpert J. R. (2007) Role of subunit interactions in P450 oligomers in the loss of homotropic cooperativity in the cytochrome P450 3A4 mutant L211F/D214E/F304W. Arch. Biochem. Biophys. 460, 129–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tsuprun V. L., Myasoedova K. N., Berndt P., Sograf O. N., Orlova E. V., Chernyak V. Y., Archakov A. I., Skulachev V. P. (1986) Quaternary structure of the liver microsomal cytochrome P-450. FEBS Lett. 205, 35–40 [DOI] [PubMed] [Google Scholar]
- 66. Davydov D. R., Halpert J. R. (2008) Allosteric P450 mechanisms: multiple binding sites, multiple conformers or both? Expert Opin. Drug Metab. Toxicol. 4, 1523–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Davydov D. R., Fernando H., Baas B. J., Sligar S. G., Halpert J. R. (2005) Kinetics of dithionite-dependent reduction of cytochrome P450 3A4: heterogeneity of the enzyme caused by its oligomerization. Biochemistry 44, 13902–13913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Backes W. L., Tamburini P. P., Jansson I., Gibson G. G., Sligar S. G., Schenkman J. B. (1985) Kinetics of cytochrome P-450 reduction: evidence for faster reduction of the high-spin ferric state. Biochemistry 24, 5130–5136 [DOI] [PubMed] [Google Scholar]
- 69. Karyakin A. V., Davydov D. R. (1988) Kinetics of electron-transfer reactions in monooxygenase system. Vestnik Akad. Med. Nauk SSSR 1988, 53–62 [PubMed] [Google Scholar]
- 70. Tsong T. Y., Yang C. S. (1978) Rapid conformational changes of cytochrome P-450: effect of dimyristoyl lecithin. Proc. Natl. Acad. Sci. U.S.A. 75, 5955–5959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fisher M. T., Sligar S. G. (1987) Temperature jump relaxation kinetics of the P-450cam spin equilibrium. Biochemistry 26, 4797–4803 [DOI] [PubMed] [Google Scholar]
- 72. Brenner S., Hay S., Girvan H. M., Munro A. W., Scrutton N. S. (2007) Conformational dynamics of the cytochrome P450BM3/N-palmitoylglycine complex: the proposed “proximal-distal” transition probed by temperature-jump spectroscopy. J. Phys. Chem. B 111, 7879–7886 [DOI] [PubMed] [Google Scholar]