

Abstract
The necessary infrastructure to carry out genomics-driven oncology is now widely available and has resulted in the exponential increase in characterized cancer genomes. While a subset of genomic alterations is clinically actionable, the majority of somatic events remain classified as variants of unknown significance and will require functional characterization. A careful cataloging of the genomic alterations and their response to therapeutic intervention should allow the compilation of an “actionability atlas” and the creation of a genomic taxonomy stratified by tumor type and oncogenic pathway activation. The next phase of genomic medicine will therefore require talented bioinformaticians, genomic navigators, and multidisciplinary approaches to decode complex cancer genomes and guide potential therapy. Equally important will be the ethical and interpretable return of results to practicing oncologists. Finally, the integration of genomics into clinical trials is likely to speed the development of predictive biomarkers of response to targeted therapy as well as define pathways to acquired resistance.
Introduction
The dawn of the 21st century brought about a critical turning point in our understanding of the molecular basis of cancer and the potential to translate cancer genetics discoveries into therapeutic advances. The dramatic efficacy of imatinib in the treatment of Philadelphia chromosome–positive chronic myelogenous leukemia (CML) (1, 2), as well as the recognition that activating mutations in EGFR predict the therapeutic response of patients with non–small-cell lung cancer (NSCLC) to EGFR inhibitors (3, 4), were thought to be harbingers of a time when targeted therapy could be paired with genomic information to identify responsive patients and, effectively, personalize medicine. While additional success stories exist, cancer is a complex disease, and the envisioned tsunami of the genomic revolution has taken time to reach shore.
Cancer researchers have benefited tremendously from the advent of high-throughput sequencing. The genomes of thousands of tumors have now been sequenced both by individual groups and through collective efforts such as the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA) (1, 2, 5). At the same time, there has been an explosion in the development and clinical testing of targeted agents (6, 7). Therefore, two of the fundamental building blocks of an effective genomics-driven oncology program are now available. While conceptually the integration of genomics-driven oncology into clinical practice would seem straightforward, its implementation has proven to have real-world complexities such as the unintended discovery of germline variants of unknown significance, the sparse amounts of archival tumor tissue, the difficulty of establishing the biologic significance (or “actionability”) of somatic events, and the translation of a cohesive and accurate depiction of the genomic portrait of a tumor to treating physicians and their patients. As we strive to integrate next-generation (next-gen) sequencing technology into clinical medicine, forethought about desired outcomes, technical and ethical considerations, and a structured multidisciplinary plan will be needed to extract the most beneficial information from the large amounts of data and genomic results produced.
Actionable events
High-throughput sequencing techniques have now evolved to the point where cancer genomes can be sequenced quickly, sensitively, and accurately. Data from ongoing research initiatives such as the ICGC and the TCGA have demonstrated that the mutation rates vary greatly between tumor types (from 0.28 to 8.15 mutations per megabase in acute myeloid leukemia [AML] and lung squamous carcinoma, respectively), as does the mutational spectrum (8). Some cancers also exhibit significant intratumoral heterogeneity (9). Viewed broadly, these findings show that cancer genomes are convoluted and harbor both driver and passenger mutations.
At this juncture, the cost of high-throughput sequencing is easily within reach of academic medical centers. Furthermore, standard operating protocols (SOPs) have been developed to ensure reproducibility of results with validation in clinical laboratory improvement amendment–certified (CLIA-certified) settings. Therefore, the current bottleneck in delivering genomics-based cancer medicine is rarely data generation, but rather results interpretation, validation, and determination of actionability. If the overarching hypothesis of genomics-driven oncology is that the genomic landscape of tumors can be converted into actionability maps or an “actionability atlas,” as we have chosen to call it, then the careful cataloging of genomic variants and their resultant ability to predict prognosis or treatment response is imperative. Much like the Catalogue of Somatic Mutations in Cancer (COSMIC) contains a comprehensive catalog of over 136,000 somatic coding mutations in over 500,000 tumor samples, we predict that the effectiveness of genomics-based oncology will rely on an operative database linking somatic mutations to therapeutic successes or failures (10).
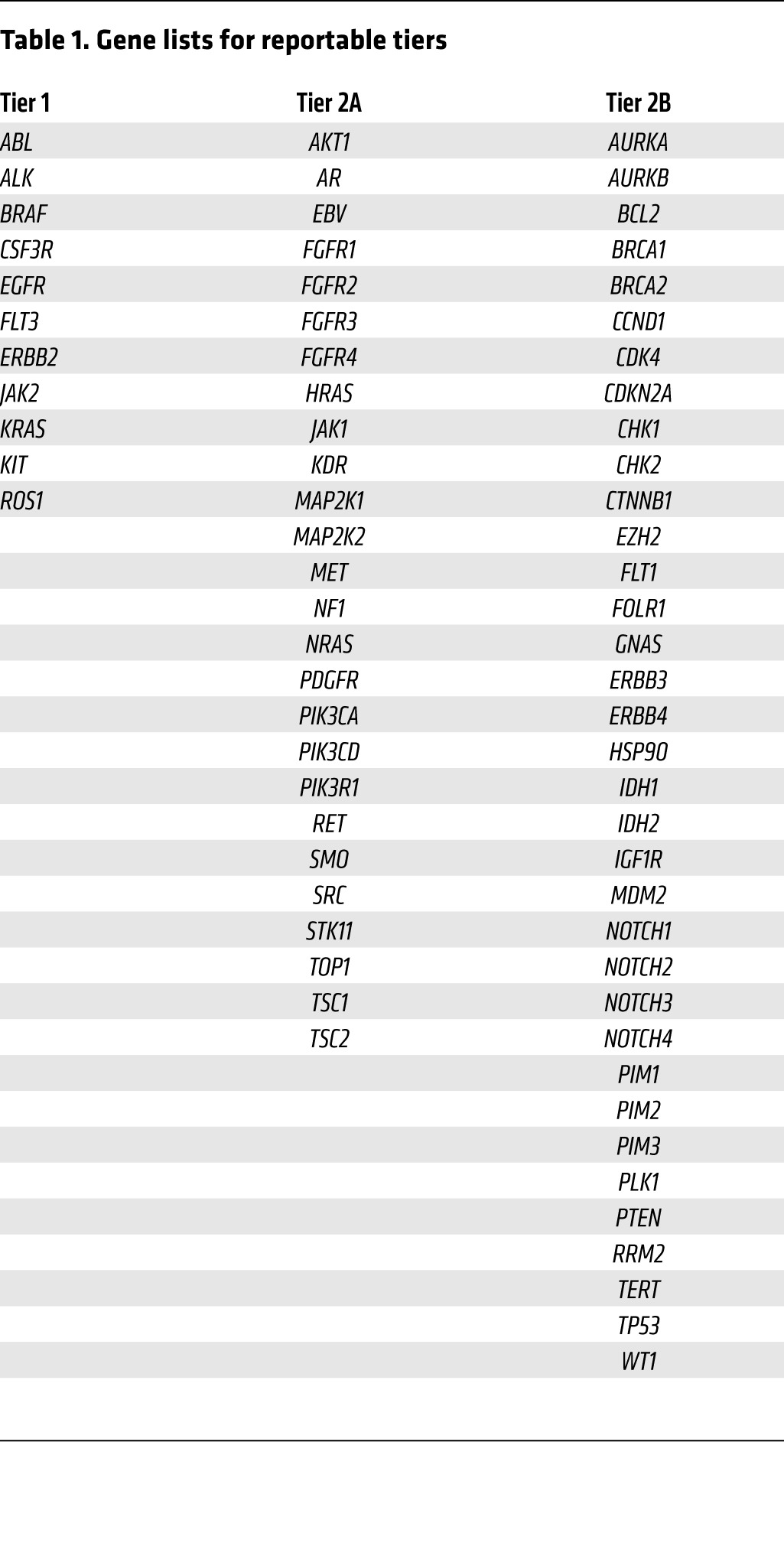
So what attributes make a genomic event clinically actionable? While clearly investigator dependent, we propose some common basic tenets. First, the alteration in a gene or pathway should be clinically informative, either at the level of diagnosis (e.g., mutation of the von Hippel–Lindau tumor-suppressor gene is specific for the clear-cell histologic subtype of renal cell carcinoma; ref. 11), prognosis, or for the prediction of treatment response (e.g., EGFR inhibition in EGFR-mutant NSCLC; refs. 3, 4, 12) or resistance (e.g., KRAS mutations confer resistance to EGFR inhibition in colorectal cancer [CRC]; refs. 13–16). Second, there should be precedent (preferably in the specific tumor type) that the somatic event is a driver mutation. Finally, the genetic aberration should optimally be druggable in a specific manner. Based on these strict criteria, the number of truly actionable mutations is surprisingly low (tier 1 lists those currently defined as actionable at the University of North Carolina at Chapel Hill [UNC] using our next-gen sequencing platform UNCseq) (Tables 1 and 2). Nonetheless, a careful, prospective cataloging of genomic results and a more pathway-driven (as opposed to organ site–driven) approach to therapy will likely lead to a more expansive, yet refined, list of clinically actionable events.
Table 1. Gene lists for reportable tiers.
To date, the vast majority of targeted therapy successes are small-molecule kinase inhibitors. While these stories tell us that such inhibitors can be used successfully to treat cancers driven by specific molecular events, whether such precision targeting can be achieved outside the realm of kinases remains to be seen. Exciting examples of inhibitors of other enzymatic reactions, such as inhibition of the H3K79 methyltransferase DOT1L in mixed-lineage leukemia (MLL), rearranged acute leukemias, or the isocitrate dehydrogenase 1 and 2 (IDH1/2) enzymes in IDH-mutant malignancies (i.e., glioblastoma and AML), have entered clinical trials and shown initial indications of effectiveness in both early-stage clinical trials and preclinical models (17–20). In addition, the enhanced sensitivity of BRCA1/2-deficient cancers to poly(ADP-ribose) polymerase (PARP) inhibition suggests that targeted therapies might be used to target tumor-suppressor gene loss as well (21).
Communication of results to clinicians: molecular tumor boards
A key step in the infrastructure of genomics-based oncology is the communication of genomic alterations to treating physicians and their patients.
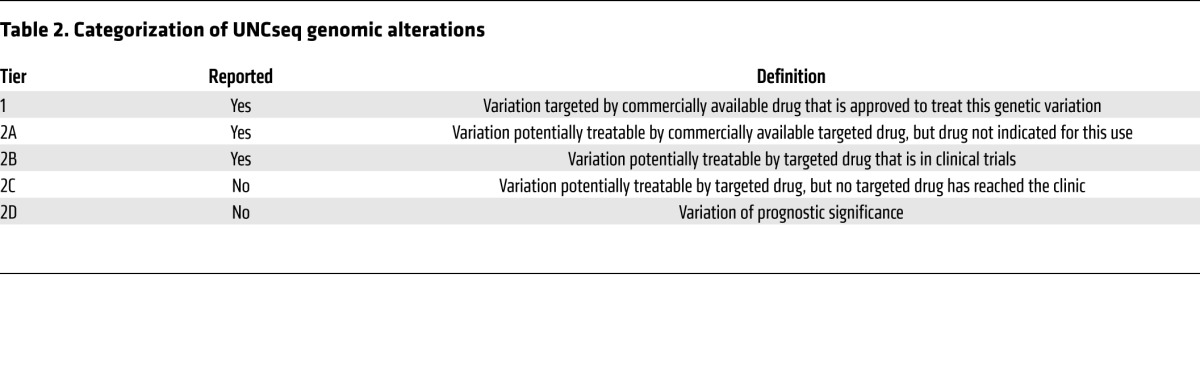
At UNC, we have chosen to stratify genomic alterations based on their likelihood of clinical actionability. Molecular alterations that are considered standard of care (SOC), for example, EGFR or KRAS testing in NSCLC and CRC, respectively (22, 23), are readily determined to be both actionable and reportable. The majority of oncologists also have a good understanding of the clinical implications of the results. These alterations are considered tier 1 (Tables 1 and 2). A second, more challenging group of genomic alterations, tier 2, involves genetic alterations of potential clinical significance; for example, a previously reported mutation that is detected in a class of tumors in which it has not been reported (e.g., EGFR mutations in bladder cancer). This list can be further stratified by whether the targeted drug(s) is commercially available (tier 2A), in clinical trials (tier 2B), or not yet in clinical trials but the target is still considered druggable (tier 2C). Finally, genetic alterations that are considered to have prognostic value are reported as well (tier 2D).
The assignment of genetic alterations to the above categories is relatively straightforward for previously reported variants or copy number alterations that have been compiled into online databases such as COSMIC. More challenging is the attribution of clinical actionability to variants of unknown significance (i.e., those that have not been previously been reported) in known oncogenes or tumor-suppressor genes or to variants clearly altering the function of genes that have not been implicated previously in cancer. We have found it useful to discuss these cases and generate a framework for decision making at a so-called “molecular tumor board” (MTB) composed of clinical and translational oncologists, molecular pathologists, and bioethicists. Within these tumor boards, decisions about the actionability of genomic events and the necessity of CLIA confirmation are made by consensus. Nonetheless, in a reasonable fraction of detected variants of unknown significance, the decision to validate and report remains somewhat arbitrary (Figure 1).
Figure 1. Genomics-based oncology workflow and integration of an actionability atlas.

The sequencing of cancer genomes requires coordinated efforts and constant modification. Patients are consented to access archival tumor tissue or fresh tumor tissue obtained from a new biopsy. Genomic profiles are generated by next-gen sequencing of tumor and normal (germline) tissues and are discussed at MTBs. The decision to report variants back to the patient and physician is made by consensus at the MTB in part based on a categorization of genes predetermined by an independent panel of clinical experts (the Clinical Committee for Genomic Research [CCGR]). Potential outcomes of the reporting of an actionable variant include: enrollment in a clinical trial, off-label use of a current FDA-approved agent, or continued SOC treatment. Systematic cataloging of outcomes into a proposed actionability atlas should aid future decision making.
Integration of next-gen sequencing into clinical trials
If discrete genetic alterations can truly predict therapeutic vulnerabilities, then next-gen sequencing of tumors should allow either the prospective identification and enrichment for potential responders or the retrospective evaluation to identify genetic features associated with response or resistance to therapy. The ability to perform continual mining of data after their initial use for treatment choice is of particular import, as novel therapies are approved for use and as we gain additional knowledge about the utility of genomic biomarkers. Additionally, an unintended consequence of next-gen sequencing of cancer genomes is likely to be a shift from clinical trials based on organ site to those that are based on alterations in oncogenic pathways (Figure 2). An example of this is the treatment of BRAF-mutant cancers with MEK or BRAF inhibitors in multiple tumor types such as melanoma, CRC, thyroid, and urothelial carcinoma (24–27). Nonetheless, it is important to proceed with caution, as genomic alterations may be exquisitely context specific. For example, recent clinical trials have shown that BRAF-mutant melanomas are highly sensitive to BRAF kinase inhibitors (vemurafenib) (28), as are BRAF-mutant papillary thyroid cancers. Thus, many oncologists were surprised when BRAF-mutant CRCs were found to be less responsive to vemurafenib (29). Careful laboratory studies have since revealed that this relative resistance to BRAF inhibition in CRC is secondary to compensatory upregulation of EGFR signaling, which in preclinical models can be cotargeted with PI3K/AKT pathway inhibitors (30, 31). These findings underscore the danger in generalizing therapeutic vulnerabilities across all tumors and demonstrate the importance of validating targets in the context of individual tumor types (Figure 2). Therefore, while it is tempting to imagine a brave new oncologic world in which tumors are categorized by driver pathway alterations rather than organ site, cancer is complex, and this simplistic view will need refinement.
Figure 2. Integration of pathway and organ site clinical trials.

(A) Current oncology clinical trials are generally structured based on organ site, in which novel compounds (A, B, and C) are tested in specific cancers. (B) There are, however, commonalities in oncogenic pathways among cancers such as activation of the EGFR, FGFR3, and BRAF pathways. These pathways can be targeted with relatively specific inhibitors (X, Y, and Z). Some clinical trials are beginning to enroll patients on the basis of pathway activation (i.e., FGFR or BRAF mutations). (C) This approach, while reasonable, will likely require attention to both pathway activation and organ site. For example, BRAF mutations are found in a number of cancers including melanoma, thyroid, colon, and bladder. While BRAF inhibition has shown efficacy in BRAF-mutant melanoma and thyroid cancer, it does not appear to benefit patients with BRAF-mutant CRC. Whether BRAF inhibition in bladder cancer is of use remains unknown.
While not based on a next-gen sequencing platform, several large, adaptively randomized trials using molecular biomarkers, such as BATTLE (biomarker-integrated approaches of targeted therapy for lung cancer elimination) and I-SPY2 TRIAL (investigation of serial studies to predict your therapeutic response with imaging and molecular analysis 2) for NSCLC and breast cancer, respectively, serve as proof of principal that biomarker profiling can occur in real time and that adaptive trial design can validate prespecified hypotheses (32, 33). Nonetheless, additional randomized, controlled studies are needed to further confirm the true predictive value of the biomarkers studied in those trials. Finally, it will likely take some work to define the optimal next-gen biomarker cutoff points for results other than canonical mutations. In the BATTLE and I-SPY2 trials, many of the prespecified biomarker cutoffs were based on a wealth of past clinical experience (e.g., ER and HER2 immunohistochemistry) accumulated over decades (32, 33). Nevertheless, the integration of next-gen sequencing into prospective clinical trials is a reality and, when performed rigorously, will likely yield immense value.
Unresolved issues
Tumor heterogeneity and the development of resistance.
High-resolution tumor-sequencing studies have now defined the startlingly broad scope of intratumoral genomic heterogeneity, which likely contributes to treatment failure as well as the development of drug resistance (34). In retrospect, intratumoral heterogeneity should not have been unexpected, since pathologists have for decades remarked on the variant histologic differentiation that can coexist within what appears to be a solitary tumor. Genomic variability has clear implications for genomics-driven oncology, as genomic characterization of tumors (at least for solid tumors) is typically made from a limited tumor sampling and therefore likely represents only a small percentage of the genomic diversity within a tumor. Furthermore, genomic heterogeneity is not limited to primary tumors, but is clearly an attribute of metastases (35). Therefore, it seems likely that genomics-based oncology will contend with intratumoral heterogeneity and the limited sampling of tumor tissue as intrinsic considerations. While shared mutations should be easily annotated, the detection of private mutations will be dependent on the lesion sampled.
Clinical responses to targeted therapy can often be dramatic (e.g., EGFR-mutant lung cancers and response to erlotinib; refs. 3, 4), yet treatment with targeted therapy invariably results in the development of drug resistance, presumably through positive selection for rare subclonal cell populations within the primary tumor harboring secondary mutations, activation of downstream effectors, or activation of a bypass oncoprotein (36). In the current paradigm of genomics-based oncology, a single genomic snapshot of a patient’s tumor is taken, with all future actionability decisions made on that single sample; however, recent studies demonstrate the value of performing rebiopsies of tumors at the time of clinical progression on targeted therapy (37). Such studies have defined novel mutations that reduce the efficacy of kinase inhibitors (i.e., T790M mutations in NSCLC treated with EGFR kinase inhibitors) but have also defined dramatic changes in histologic subtypes (i.e., a therapy-driven switch between NSCLC and small-cell lung cancer) (37). Therefore, for genomics-based oncology to fully achieve its promise, a genomic snapshot of a patient’s tumor requires repeated updates.
Circulating tumor DNA.
While the next-gen sequencing of cancer genomes has traditionally been performed on banked tissue (either fresh-frozen or formalin-fixed, paraffin-embedded [FFPE]), it has recently been applied to detect circulating tumor DNA (ctDNA) in the peripheral blood of cancer patients. Beyond the obvious ease of sample acquisition (phlebotomy rather than obtaining archival tumor tissue or a new biopsy), the next-gen sequencing of ctDNA is likely to serve a number of broader applications. For example, recent work suggests that it can be used to noninvasively monitor tumor burden (38–40) and, perhaps most excitingly, that it can be used to detect mechanisms of secondary resistance to targeted therapy (e.g., activation of the RAS/MEK/ERK pathway in EGFR inhibitor–treated metastatic CRC patients) (40, 41). While not prohibitive, a current limitation to most of the reported ctDNA techniques is the need for relatively deep and broad sequencing of putative ctDNA.
Tumor microenvironment.
Harnessing the immune system for cancer therapy has undergone a recent renaissance. Impressive therapeutic outcomes have been observed across several immunotherapy platforms, including adoptive cellular therapy (e.g., chimeric antigen receptors [CARs] in hematologic malignancies) (42–46), immune checkpoint blockade (e.g., anti–PD1/PD-L1 antibodies in numerous solid tumors) (47, 48), and various tumor vaccination strategies (49–51). This has led to an intense search for predictive biomarkers of immune responsiveness. While intratumoral expression of PD-L1 has been proposed to have a high negative predictive value for the efficacy of anti–PD-L1 therapy (48), additional biomarkers are under active investigation.
Unfortunately, next-gen sequencing of bulk cancers does not capture important information on the immune infiltrate, such as the level of immune cell infiltration or the cellular milieu of immune cells present, which may be important biomarkers of not only response to immunotherapy but also to cytotoxic chemotherapy (52, 53). Nonetheless, recent studies confirm the utility of exome sequencing for discovery of tumor neoantigens, although it should be noted that the majority of neoantigens identified are in genes that are absent in current targeted exon–sequencing panels (54, 55). Overall, it seems likely that the integration of other genomic platforms such as RNA sequencing and platforms such as proteomics and polychromatic flow cytometry will allow for a more comprehensive “immune portrait” of tumors, including the molecular assessment of lymphocyte clonality and diversity (56) as well as the assessment of other key microenvironmental components such as tumor-associated fibroblasts and endothelial cells, which may be key predictors of response to both immune and antiangiogenic therapies (refs. 57, 58, and Figure 3).
Figure 3. Next-gen sequencing and the complexity of human tumors.

Next-gen sequencing of tumors delivers a high-resolution snapshot of the genomics of tumor cells. With the exception of whole-exome sequencing, which may help identify tumor neoantigens, it does not generally inform us about the tumor microenvironment. Other “omic” platforms, such as RNA sequencing, proteomics and IHC, and polychromatic flow cytometry, would complement the current next-gen sequencing platforms, allowing further characterization of tumor cell–autonomous information and also facilitating the generation of immune, vascular, and stromal portraits that may have prognostic and predictive value. IF, immunofluorescence.
Issues of reimbursement.
Despite the increasing enthusiasm for integrating genomics into cancer care and its promise to advance personalized therapy, there are obvious economic consequences. In general, economic analyses consider the health gained relative to the resources expended, which can be measured by various metrics such as incremental cost–effectiveness ratios (ICERs) or quality-adjusted life years (QALYs) (59). In one analysis, EGFR mutation testing to predict response to EGFR inhibitor treatment in the first-line treatment of metastatic NSCLC showed that it is cost effective, with an ICER of $81,071 per QALY or $46,021 per life year (60). Studies such as these suggest that the integration of next-gen sequencing into cancer care may have a positive impact on health-care economics. Nevertheless, future studies will be required to fully define the economic consequences of technology with both well-defined costs (i.e., the price of actual sequencing) and the less-well-defined expenditures on personnel for data processing, secure data storage, variant interpretation, and determination of actionability at MTBs.
Oncologists’ perception.
It is undeniable that the next-gen sequencing of cancer genomes has become widespread as well as commercialized (e.g., FoundationOne), suggesting that physicians have confidence in the technology and process as well as comfort in discussing genomic results with their patients. A survey of physicians at a tertiary care center prior to the rollout of their next-gen tumor-sequencing platform, however, revealed that there was a wide variability in how physicians planned to incorporate potential results into clinical practice and in their attitudes toward the disclosure of variants of undetermined significance (61). As might be expected, physician opinion on these topics correlated with their perceived level of genomic confidence, with approximately 22% of surveyed physicians reporting that they were “not very confident” or “not confident at all” in their knowledge of genomics. These findings reiterate that vehicles for consensus such as multidisciplinary MTBs and evidence-based guidelines are key components to a successful genomics-driven oncology program. It will be of interest to assess similar perceptions in community-based oncology practices where, at least to our knowledge, genomics tumor boards are not part of routine practice.
The next steps
At many academic medical centers, the building blocks for genomics-driven oncology are now in place. In reality, this has been relatively easy, as capital expenditure to purchase next-gen sequencing machines and reagents is merely a line item on a cancer center’s annual budget. It seems that the coming years will be the most difficult, for to truly benefit, patients will depend on the use of human capital for data processing, the timely interpretation and reporting of actionable results, and, perhaps most importantly, the effective communication about the implications of these results to patients. Indeed, we can sequence as many cancer genomes as we like, but without talented bioinformaticians to decipher the data and so-called “genomic navigators” to guide us, the clinical actionability of unannotated variants remains questionable.
Genomics- or “omics”-driven oncology?
The value of bioinformaticians is likely to increase exponentially, as genomics-driven oncology expands to encompass platforms outside of DNA-based mutations and gene copy number alterations. The future of genomics, or perhaps more appropriately, “omics”-driven oncology, likely holds a multiplatform approach that will allow comprehensive characterization of a tumor on multiple levels; the resulting genomic profile would then be integrated with other “omic” data to predict the functional consequences of genomic alterations on oncogenic pathways. Thus, our current one- or, at best, two-dimensional map of a cancer genome will soon transform into a true genomic landscape replete with multidimensional annotation.
Genomic confidence and genomic navigators.
The survey by Gray and colleagues suggests that even in tertiary care centers, many physicians do not feel confident about their knowledge of genomics (61). While not directly surveyed, this low genomic confidence would suggest a relative reticence toward both ordering next-gen sequencing of cancer genomes as well as discussing their implications directly with patients. Much like medical geneticists, who have specialized in the evaluation, testing, and counseling of patients with a suspected genetic disorder, it would seem that the field of genomics-driven oncology might also usher in a new cadre of genomic navigators. The role of these genomic navigators might be to shepherd patients through the process of testing as well as the potential complications of testing (e.g., detection of a suspected germline variant). Ultimately, however, discussion of the relative actionability of a genomic variant and whether to forgo SOC treatment for a personalized therapy will require significant input from the treating oncologist.
Development of an actionability atlas.
While the future of genomics-driven oncology holds much promise, the human genome is vast, and the majority of cancer genomes are likely to harbor somatic variants of unknown significance. To continue to move the field forward rationally, it is imperative that we develop a central repository such as the above-proposed actionability atlas to catalog novel variants, their purported actionability, and, if attempted, their response to therapy. This should be easy in the context of clinical trials, but will take a concerted effort as more case reports or so-called “N-of-1” studies are undertaken. Despite the catchy name, these N-of-1 studies are to some extent case reports and remain anecdotal. Moreover, while many N-of-1 studies are supported by elegant in vitro work backing the genomic hypotheses, most clearly require prospective validation or validation in large, retrospective datasets before widespread adoption in clinical practice. Nonetheless, only through the careful cartography of cancer genomic landscapes and their responses to therapy can we develop an actionability atlas and genetic taxonomy of cancer based on both tumor type and genetic makeup.
Table 2. Categorization of UNCseq genomic alterations.
Acknowledgments
We acknowledge Jon Serody and Ned Sharpless for useful discussions and the entire UNCseq team (in particular, Michele Hayward and Ashley Salazar) for their dedication and work. This work was supported by NIH grants R01 CA142794 (to W.Y. Kim), U24 CA143848 (to D.N. Hayes), U24 CA143848-02S1 (to D.N. Hayes), and U10 CA181009-01 (to D.N. Hayes); the University Cancer Research Fund (to W.Y. Kim and D.N. Hayes); AACR Kure It (to W.Y. Kim); and the Bladder Cancer Advocacy Network (BCAN) Innovation Award (to W.Y. Kim). W.Y. Kim is a Damon Runyon Merck Clinical Investigator.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(2):462–468. doi:10.1172/JCI68339.
References
- 1.Druker BJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344(14):1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Lynch TJ, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 5.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10(2):130–137. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 8.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swanton C, Caldas C. From genomic landscapes to personalized cancer management-is there a roadmap? Ann N Y Acad Sci. 2010;1210(1):34–44. doi: 10.1111/j.1749-6632.2010.05776.x. [DOI] [PubMed] [Google Scholar]
- 10.Forbes SA, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39(Database issue):D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22(24):4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 12.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 13.Bokemeyer C, et al. Fluorouracil, Leucovorin, and Oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27(5):663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 14.Hecht JR, et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J Clin Oncol. 2009;27(5):672–680. doi: 10.1200/JCO.2008.19.8135. [DOI] [PubMed] [Google Scholar]
- 15.Tol J, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360(6):563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 16.Normanno N, et al. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol. 2009;6(9):519–527. doi: 10.1038/nrclinonc.2009.111. [DOI] [PubMed] [Google Scholar]
- 17.Bernt KM, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20(1):66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daigle SR, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20(1):53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohle D, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang F, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 21.Fong PC, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 22.Ettinger DS, et al. Non-small cell lung cancer. J Natl Compr Canc Netw. 2010;8(7):740–801. doi: 10.6004/jnccn.2010.0056. [DOI] [PubMed] [Google Scholar]
- 23.Benson AB, et al. Metastatic colon cancer, version 3.2013: featured updates to the NCCN Guidelines. J Natl Compr Canc Netw. 2013;11(2):141–152. doi: 10.6004/jnccn.2013.0022. [DOI] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flaherty KT, Hodi FS, Fisher DE. From genes to drugs: targeted strategies for melanoma. Nat Rev Cancer. 2012;12(5):349–361. doi: 10.1038/nrc3218. [DOI] [PubMed] [Google Scholar]
- 27.Flaherty KT, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 28.Sosman JA, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kopetz S, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol. 2010;28(suppl 15s):3534. [Google Scholar]
- 30.Prahallad A, et al. Unresponsiveness of colon cancer to BRAF(V600E)inhibition through feedback activation of EGFR. Nature. 2013;482(7387):100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 31.Mao M, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19(3):657–667. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barker AD, et al. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Nature. 2009;86(1):97–100. doi: 10.1038/clpt.2009.68. [DOI] [PubMed] [Google Scholar]
- 33.Kim ES, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1(1):44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 35.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2(3):214–226. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- 37.Sequist LV, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newman AM, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forshew T, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136):136ra68. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 40.Murtaza M, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2014;497(7447):108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 41.Bettegowda C, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramos CA, Savoldo B, Dotti G. CD19-CAR trials. Cancer J. 2014;20(2):112–118. doi: 10.1097/PPO.0000000000000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brentjens RJ, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brentjens RJ, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123(17):2625–2635. doi: 10.1182/blood-2013-11-492231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maus MV, et al. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol. 2014;32(1):189–225. doi: 10.1146/annurev-immunol-032713-120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brahmer JR, et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Topalian SL, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melero I, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11(9):509–524. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 50.Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 51.Vonderheide RH, Nathanson KL. Immunotherapy at large: the road to personalized cancer vaccines. Nat Med. 2013;19(9):1098–1100. doi: 10.1038/nm.3317. [DOI] [PubMed] [Google Scholar]
- 52.Choi W, et al. Identification of distinct Basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25(2):152–165. doi: 10.1016/j.ccr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Denkert C. Diagnostic and therapeutic implications of tumor-infiltrating lymphocytes in breast cancer. J Clin Oncol. 2013;31(7):836–837. doi: 10.1200/JCO.2012.47.1698. [DOI] [PubMed] [Google Scholar]
- 54.Rajasagi M, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;124(3):453–462. doi: 10.1182/blood-2014-04-567933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Rooij N, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–e442. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iglesia MD, et al. Prognostic B-cell signatures using mRNA-seq in patients with subtype-specific breast and ovarian cancer. Clin Cancer Res. 2013;20(14):3818–3829. doi: 10.1158/1078-0432.CCR-13-3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 58.Özdemir BC, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palmer S, Byford S, Raftery J. Economics notes: types of economic evaluation. BMJ. 1999;318(7194):1349. doi: 10.1136/bmj.318.7194.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Health Quality Ontario Epidermal Growth Factor Receptor Mutation (EGFR) Testing for Prediction of Response to EGFR-Targeting Tyrosine Kinase Inhibitor (TKI) Drugs in Patients with Advanced Non-Small-Cell Lung Cancer: An Evidence-Based Analysis. Ont Health Technol Assess Ser. 2010;10(24):1–48. [PMC free article] [PubMed] [Google Scholar]
- 61.Gray SW, Hicks-Courant K, Cronin A, Rollins BJ, Weeks JC. Physicians’ attitudes about multiplex tumor genomic testing. J Clin Oncol. 2014;32(13):1317–1323. doi: 10.1200/JCO.2013.52.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]