

Abstract
While a host of methods exist to deliver genetic materials or small molecules to cells, very few are available for protein delivery to the cytosol. We describe a modular, light-activated nanocarrier that transports proteins into cells by receptor-mediated endocytosis and delivers the cargo to the cytosol by light triggered endosomal escape. The platform is based on hollow gold nanoshells (HGN) with polyhistidine tagged proteins attached through an avidity-enhanced, nickel chelation linking layer; here, we used green fluorescent protein (GFP) as a model deliverable cargo. Endosomal uptake of the GFP loaded nanocarrier was mediated by a C-end Rule (CendR) internalizing peptide fused to the GFP. Focused femtosecond pulsed-laser excitation triggered protein release from the nanocarrier and endosome disruption, and the released protein was capable of targeting the nucleoli, a model intracellular organelle. We further demonstrate the generality of the approach by loading and releasing Sox2 and p53. This method for targeting of individual cells, with resolution similar to microinjection, provides spatial and temporal control over protein delivery.
Keywords: protein delivery, nanoshell, gold, near-infrared laser, targeted delivery, endosome escape
1. Introduction
Developing novel biochemical approaches to characterize and control intracellular pathways and modify the action of cells and organisms forms a core goal of biology and medicine.1 Ultimately, the synthesis and intracellular delivery of molecules with spatial and temporal control provides a powerful combination. Light-activation offers a means to maintain external spatial and temporal control, demonstrated originally in 1978 by Kaplan and co-workers with light-activated “caged” ATP.2 However, many of the limitations of the original approach that require the specific chemical modification of each molecule to be delivered and the need for high energy UV light to trigger activation still remain.3
Plasmonic gold nanoparticles have attractive applications in biomolecule delivery owing to their ease of surface functionalization and strong absorption of physiologically friendly, near-infrared light.4 Spatially and temporally controlled cargo release from gold nanoparticles has been shown with oligonucleotides for gene regulation and expression5−8 and small molecules for anticancer therapy,9,10 using illumination wavelengths corresponding to the plasmon resonance. Proteins carry out and regulate the majority of the known cellular functions, and changes in their primary sequence or expression levels lead to diverse diseases.11,12 Although extracellular protein therapeutics have been remarkably successful (e.g., insulin, recombinant factor IX, erythropoietin, and interferon13−16), replacing or modulating the function of intracellular proteins remains an unmet challenge.17 The next frontier of protein therapeutics aims to modulate a wide range of intracellular metabolic, signaling, and regulatory activities, as well as guiding stem cell differentiation by transcriptional network perturbation.18−20 For a therapeutic protein to reach the site of action inside the cell it must cross the plasma membrane and retain its biological function.17,21 Transmembrane and endocytic delivery are each confounded by the large macromolecular size, variable net charge, and complex structure of proteins.22 The tendency for proteins to become trapped and degraded inside endosomes and lysosomes,22 the last mile problem, has been approached by using protein transduction domains,23,24 endosomal disrupting agents,25 degradable polymeric carriers,26,27 and membrane interacting supramolecular particles.21,28 Despite these, endosomal escape remains inefficient,24 and furthermore, many of these methods cannot be confined to a specific cell type or area, or the timing of release is uncontrolled.21 For example, electroporation requires high concentrations of protein22,29 and is more commonly used to deliver nucleic acids. Techniques for protein delivery to individual cells in a two-dimensional culture format are limited to (1) microinjection,30 which has low throughput and is not compatible with three-dimensional tissues,31 and (2) light activated polymer degradable methods, which respond to UV rather than NIR irradiation.32,33
Here we report the design of a modular plasmonic nanocarrier platform that delivers His-tagged proteins into the cytosol of cells, based on a novel photocleavable oligonucleotide linker system (Scheme 1). Our molecular linker design uses nickel-chelating groups to overcome limitations posed by covalent immobilization of proteins, thus allowing on-demand temporal control over release of functional proteins from hollow gold nanoshells (HGN) tuned to absorb in the biological “optical” window at 800 nm. By incorporating peptides to induce receptor-mediated endocytosis, we observed efficient and specific cellular targeting that is immediately applicable to ongoing efforts in drug delivery and tissue engineering. Endosomal escape and cargo release were activated through an integration of targeted nanocarriers and two-photon (pulsed laser) patterning microscopy, taking advantage of the exquisite spatial control offered by the laser scanning system to achieve subcellular resolution and real-time monitoring of the protein transduction process.
Scheme 1. Hollow Gold Nanoshell (HGN) Platform Designed for Intracellular Protein Delivery.
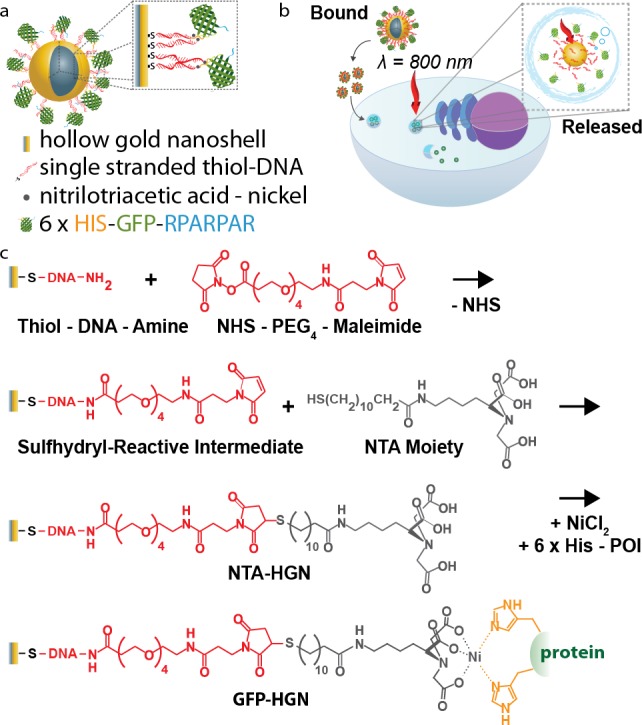
(a) Diagram of the modular plasmonic nanocarrier: a cell-targeting peptide motif (RPARPAR) is fused to His-tag modified green fluorescent protein (R-GFP), which is assembled on DNA-coated HGN. (b) Schematic representation of R-GFP-HGN cellular uptake and protein release. The cell targeting peptide mediates internalization by endocytosis. Laser excitation matching the nanocarrier plasmon resonance releases R-GFP into the cytosol. (c) Nanocarrier synthetic steps: HGN are coated with single stranded thiol-DNA-amine and reacted with NHS-PEG4-maleimide. Thiol NTA moiety addition generates HGN-NTA. NiCl2 facilitates the binding of His tagged proteins by forming NTA-Ni2+.
2. Materials and Methods
2.1. Hollow Gold Nanoshell Synthesis
HGN were synthesized by galvanic replacement of silver seeds, as described previously.34 Silver seed particles were prepared by reducing a stirred solution of 500 mL of 0.2 mM AgNO3 (Sigma) and 0.5 mM sodium citrate (Sigma) in deionized water with 0.5 mL of 1.0 M NaBH4 (EMD) at 60 °C. The solution was stirred for 2 h and then cooled to room temperature before growing the seed particles to a final target size for use as a sacrificial template for the gold nanoshells growth with the addition of 0.75 mL of 2 M NH2OH·HCl (Sigma) and 1.75 mL of 0.1 M AgNO3 and stirred overnight at room temperature. The galvanic replacement of the silver template particles with gold was optimized to have an absorbance peak at around 800 nm by rapid addition of 3.2 mL of 25 mM HAuCl4 (Sigma) at 60 °C.
2.2. Preparation of NTA-HGN
Single stranded thiol modified 25mer DNA of sequence 3′-NH2-(CH2)7-GCCACCACGTCTACTTGAAGTCCCA-PEG18-(CH2)6-SS-(CH2)6-OH-5′ was purchased from Biosearch Technologies and adsorbed onto HGN by the method described by Zhang et al.35 One hundred micromolar DNA was incubated with 10 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (Sigma) for 30 min and then dialyzed against TE buffer pH 8.0 (IDT DNA) overnight at 4 °C. A final concentration of 3 μM TCEP-treated DNA was added to 32 pM HGN at which point low pH adsorption was induced with the addition of 10 mM sodium citrate to a final pH of 3.1. After 10 min the pH was neutralized to 7.4 with the addition of 1 M HEPES (Sigma), and the NaCl concentration was brought to 500 mM. The HGN were then pelleted by centrifugation three times at 9000 × g for 10 min, decanted, and the pellet resuspended in 500 mM NaCl in 10 mM phosphate buffer at pH 7.4 to remove excess DNA. N-[Nα,Nα-Bis(carboxymethyl)-l-lysine]-12-mercaptododecanamide (NTA) (Sigma) was then conjugated to the 3′ amine of the DNA scaffold with the addition of 1 mg/mL NHS-PEG4-maleimide linker (Quanta Biodesign) and incubated for 20 min at room temperature. The maleimide terminated HGN were pelleted 3 times at 9000 × g for 10 min at 4 °C, and 120 μM NTA was added and then incubated for 3 h at room temperature. After pelleting 3 times, the solution was stored at 4 °C until use.
2.3. Protein Expression, Purification, and Loading on HGN
Oligonucleotides encoding the RPARPAR peptides were synthesized and ligated downstream of GFP with a glycine-serine linker placed in between in the backbone pRSET-EmGFP (Invitrogen). pRSET-Sox2 was subcloned by replacing GFP with Sox2 in the RPARPAR-modified GFP construct (R-GFP). Both constructs were confirmed by DNA sequencing. pET15b-Human p53 was purchased from Addgene (plasmid 24859).36 All recombinant proteins were expressed in Escherichia coli BL21(DE3) (Novagen) and purified using nickel-nitrilotriacetic acid affinity chromatography. GFP protein was purified under native conditions, whereas Sox2 and p53 under denaturing conditions and refolded.
The protein of interest (POI) was loaded onto the HGN at a 100,000 to one POI/HGN molar ratio in the presence of 400 μM NiCl2 and incubated for 30 min on ice. The HGN were pelleted by centrifugation at 9000 × g for 10 min at 4 °C for a minimum of 5 times to remove excess protein and resuspended at a final concentration of 320 pM of POI-HGN construct.
2.4. Femtosecond Laser for Protein Release Quantification
Samples were irradiated with pulses generated from a femtosecond Ti:sapphire regenerative amplifier (Spectraphysics Spitfire) running at 1 kHz repetition rate. The laser beam was collimated by a Galilean telescope to achieve a Gaussian diameter of 2.3 mm. In experiments without collimation, the full beam diameter was 5 mm. Pulse duration was monitored by a home-built single-shot optical autocorrelator and was kept at about 130 fs. The spectral full width at half-maximum of the laser radiation was 12 nm centered around 800 nm. The laser beam was directed onto the sample by a series of mirrors, and no focusing optics were used. The energy of the optical pulse was controlled by Schott neutral density glass filters. A thermopile power meter (Newport Inc., Irvine, CA) was used to measure the incident optical power.
2.5. Cell Culture and GFP-HGN Internalization
PPC-1 was maintained in high glucose Dulbecco’s Modified Eagle Medium (DMEM) with phenol red (HyClone) supplemented with 10% fetal bovine serum (HyClone) at 37 °C in 5% CO2. For spatial and temporal controlled release experiments, PPC-1 cells were grown on an 8-well chambered glass slide (Thermo LabTek II) at an initial seeding density of 40,000 cells per well for 24 h 37 °C in 5% CO2 in complete media.
One to 10 μL of GFP-HGN at 320 pM was added per 100 μL of medium. After 2 h of incubation at 37 °C in 5% CO2 atmosphere, the cells were rinsed with Hank’s Balanced Salt Solution (HBSS) (Thermo) prior to imaging.
2.6. Microplate Fluorescence Measurements
Fluorescence measurements of GFP were carried out using a Tecan Infinite 200 Pro microplate reader, exciting at 450 nm (9 nm bandwidth) and reading emission spectra at 490 to 600 nm (20 nm bandwidth) or at a single point at 510 nm. The amount of GFP loaded onto NTA-HGN was determined by chemically competing for the nickel-NTA sites of 3.2 pM nanoparticles with 100 mM imidazole in PBS and incubation for 30 min. The particles were spun down at 12,000 × g for 10 min, and the supernatant was loaded into a 96-well flat clear bottom plastic microtiter plate for fluorescence readout.
Efficiency of GFP release by laser was examined by irradiating a number of samples with particle concentration of 3.2 pM R-GFP-HGN with variable laser powers and exposure times. HGN were then centrifuged at 12,000 × g for 10 min and the supernatants transferred to a 96-well flat clear bottom plastic plate for fluorescence readout. The pellets were then treated with 100 mM imidazole in PBS and then spun down to extract the supernatant containing the GFP retained on the HGN after laser excitation. The supernatant was loaded into a 96-well flat clear bottom plastic microtiter plate for fluorescence readout.
Cellular GFP concentration was determined by incubating 32 pM R-GFP-HGN to a suspension of 500,000 cells for 2 h at 37 °C and 5% CO2. The cells were lysed with a solution of Triton X-100 in PBS and centrifuged to a pellet containing the HGN. The pellet was treated with 100 mM imidazole in PBS and centrifuged to transfer the supernatant containing liberated GFP to a 96-well plate to measure the fluorescence.
Standard curves for GFP concentration quantification in test tubes and in cells were generated by measuring the fluorescence intensity of known concentrations of GFP (determined by absorption spectroscopy with a NanoDrop 1000 (Thermo) using the extinction coefficient of emerald GFP of 57,500 M–1 cm–1 at 487 nm37) serially diluted in the proper experimental buffer.
CBQCA protein staining of Sox2 was performed as directed in the manufacturer’s protocol (Invitrogen). Fluorescence analysis was performed on a black 96-well plate with the Tecan Infinite 200 Pro plate reader exciting at 465 nm (9 nm bandwidth) and reading emission at 550 nm (20 nm bandwidth).
2.7. Western Blot Analysis
Following laser excitation, HGN loaded with Sox2 or p53 was pelleted by centrifugation at 14,000 × g for 10 min. The pellet was etched with KCN to release the remaining protein into solution. A 1× solution of Laemmli sample buffer (Bio-Rad) was added to the samples and then boiled for 10 min. The samples were run on a 12% SDS-PAGE gel and then transferred to a polyvinyl difluoride (PVDF) membrane (Bio-Rad). The membrane was stained with a mouse anti-HIS tag monoclonal antibody (Abgent) at a 1:500 dilution at 4 °C overnight. After stringent wash, the membrane was then stained with Alexa Fluor 488 goat antimouse IgG secondary antibody (Invitrogen) for 3 h at room temperature. The membrane was imaged with a Typhoon Trio (GE) scanner.
2.8. Cell Viability Assay
Five thousand cells in 100 μL of complete medium were seeded in a 96-well plate and grown overnight at 37 °C and 5% CO2. The cells were treated with R-GFP-HGN using the methods described above. The cells were irradiated using an 800 nm pulse-pumped femtosecond Ti:sapphire regenerative amplifier running at 1 kHz repetition rate using a 5 mm full beam diameter with variable exposure times and powers. After 72 h the cell viability was determined by PrestoBlue (Life Technologies) according to the manufacturer’s protocol. The viability was analyzed using a Tecan Infinite 200 Pro microplate reader, exciting at 560 nm (9 nm bandwidth) and reading emission at 590 nm (20 nm bandwidth).
2.9. Two-Photon and Confocal Microscopy
Imaging was performed using an Olympus Fluoview 1000 MPE Microscope. Living cells were excited with a mode-locked Ti:sapphire tunable (690–1020 nm) femtosecond pulsed laser (100 fs pulse duration, 80 MHz repetition rate, Mai Tai HP, Newport-Spectra Physics) regulated with a modulator linked to the Fluoview software. A 25× water immersion objective with a numerical aperture of 1.05 was used. Images were collected in a 12 bit file with 512 × 512 pixels. GFP fluorescence in cells plated on an 8-well glass slide was imaged with a 15 mW blue laser diode exciting at 473 nm raster scanning at a speed of 80,000 Hz. NIR excitation of R-GFP-HGN was performed using the femtosecond pulsed Mai-Tai laser tuned to 800 nm at a raster scan speed of 125,000 Hz up to 35 full-frame cycles. Cells were imaged again with the 473 nm laser diode. Spatially controlled release experiments were performed by selecting a region of interest using the Fluoview software to scan only the selected area with the femtosecond pulsed laser.
2.10. Image Analysis for Quantification of GFP Release by Microscopy
Ratio quantification for Figure 3 was performed using the Ratio Plus plugin in ImageJ to calculate the pixel ratio between images corresponding to after and before NIR excitation. Background subtraction on the images was performed after determining the average background intensity. Line intensity profile was performed on ImageJ.
Figure 3.

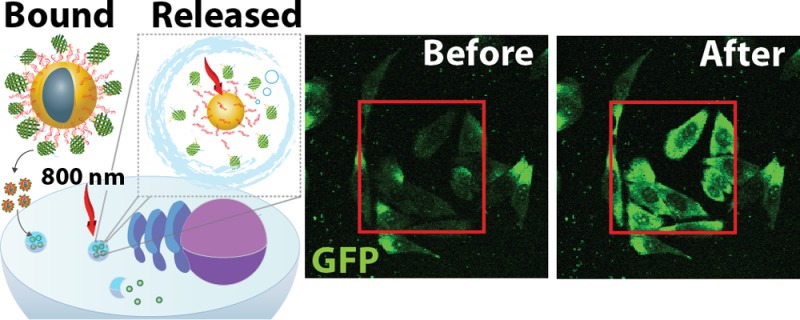
Quantification of R-GFP release in PPC-1 cells after exposure to a focused NIR pulsed laser. (a) Single photon confocal microscopy fluorescence image of PPC-1 cells incubated with R-GFP-HGN for 2 h, prior to NIR excitation (Initial), and after two rounds of 800 nm fs pulsed laser exposure (first pass and second pass), across the full field of view with a total exposure of 280 μs per irradiated area for each pass. R-GFP release and dequenching is indicated by the overall increase in pixel brightness in almost all cells in the first and second passes. Scale bar is 50 μm. (b) The increase of pixel intensities before and after each pass of the laser was quantified by the ratio of after/before and is represented by white pixels (first and second pass ratios). Most of the cells show R-GFP release, but the ratio of increase was higher for the first pass than second pass, indicating first pass rapid release under these laser conditions. (c) Close up of the blue box region in panel a for three frames. Histogram of ratios in the white box region, plotted as percent of pixels from panel b. This plot shows the fluorescence intensity increases most during the first pass of the laser.
Corrected total cell fluorescence (Figure 4) was calculated from the integrated density within an ROI enclosing a specific cell (determined by ImageJ) according to the formula: CTCF = IntDen – (area of cell × mean fluorescence of background).
Figure 4.

Single cell resolution and spatial control of intracellular R-GFP delivery. (a) Red polygons define regions irradiated by the NIR laser. The blue oval (ROI 2) was not exposed to NIR. Fluorescence intensity increased for the irradiated ROIs after laser. The cell in ROI 1 was separated from the control cell ROI 2 by ∼5 μm. Scale bar is 50 μm. (b) Line profiles taken along the cell in ROI 1 shows R-GFP fluorescence intensity increased up to 8-fold after laser, whereas the neighboring control cell in ROI 2 was unchanged. (c) Single photon confocal microscopy of R-GFP-HGN fluorescence before and after NIR laser exposure in the region denoted by the red square. Scale bar is 25 μm. (d) Intensity profile along lines 3 and 4 in panel c showing the GFP fluorescence increase after laser. Nucleoli staining observed after laser irradiation (‡). Subcellular release of R-GFP was elicited mostly (▲), while the cellular area (•), not exposed, showed only a slight increase. Laser power density was ∼8 × 103 W/cm2 (0.1 mJ/cm2), and ∼300 μs exposure time in the region is denoted by the red square. (e) Quantification of corrected total cell fluorescence percent increase in selected cells (outlines shown in panel f) above the initial intensity. N = 6 for cells completely outside the ROI, n = 5 for all other cases. Mean intensity for cells completely inside the ROI was significantly higher than cells completely outside (Kolmogorov–Smirnov test; p < 0.005; ***). Error bars represent standard error of the mean (SEM).
2.11. Statistics
Differences between the viability of the two groups with different laser power intensity and exposure time were analyzed by two-way ANOVA with Bonferroni’s post-tests using GraphPad Prism 5 software (GraphPad Software, La Jolla, USA). The means ± standard deviations of triplicate samples are reported, and a p ≤ 0.05 was considered statistically significant.
Differences between the mean fluorescence for cells irradiated by the laser versus nonirradiated (Figure 4f) were analyzed by 2-sample Kolmogorov–Smirnov test using OriginPro. A p < 0.005 was considered statistically highly significant.
3. Results and Discussion
Exposure of HGN to femtosecond pulsed near-infrared (NIR) laser light generates hot electrons that can cleave a thiol-gold surface bond, without damaging an attached nucleic acid cargo.4−6,38 Also, the laser-supplied energy induces plasmonic heating of a thin (nanometer-scale) shell of water surrounding the metal surface, occurring on a time scale of picoseconds that is too short for heat dissipation to the bulk fluid,4−6,38,39 thus inducing generation of nanobubbles. When HGN are localized intracellularly in endosomes, the nanobubbles can breach the endosomal barrier. This local heating effect has been harnessed to deliver nucleic acids in previous reports, but protein delivery has not been described using such plasmonic nanostructures. Herein, we introduce a protein delivery platform based on HGN coated by a densely packed DNA layer that serves both as anchor for proteins and as a release switch when irradiated by pulsed NIR light matching the HGN plasmon resonance wavelength (Scheme 1).
3.1. Assembly of Nitrilotriacetic Acid Handle on Hollow Gold Nanoshells
HGN of 40 ± 15 nm diameter and 5 ± 2 nm shell thickness were synthesized via the galvanic replacement of gold from sacrificial silver templates as described previously (Figure S1, Supporting Information).6,40 The HGN diameter and shell thickness were optimized to provide a plasmon absorption peak at ∼800 nm since NIR (750–2500 nm) light is physiologically friendly as it penetrates cells, blood, and soft tissues at depths up to several centimeters without damage.6,41 Proteins were attached to the HGN using the histidine (His) tag, which typically consists of five or six His residues added to the N- or C-terminus of the protein by recombinant engineering.42−44 Protein tag sequences have been instrumental for the efficient purification, detection, and surface immobilization of a variety of proteins, and the His-tag is one of the most commonly used methods for immobilization of functional proteins.45 The imidazole moieties of the His residues chelate the free coordination sites of divalent metal ions (e.g., Ni2+ and Co2+), which in turn can be immobilized as chelate complexes of nitrilotriacetic acid (NTA) bound to a solid support via various linkers. NTA chelates Ni2+ strongly, while additional sites on the Ni metal center remain open for chelating a His-tag.46
To secure the NTA-Ni2+ handles on HGN, we first assembled a densely packed monolayer of single stranded thiol-DNA-amine onto the HGN surface by low-pH adsorption.35 The number of linker DNA-amines attached to the HGN was ∼3000, determined by fluorescence labeling of the amine with NHS-fluorescein following a previously described method.5 The DNA layer stabilized the HGN against aggregation, while providing a high density of terminal amine groups47 used to attach the Ni2+ and protein chelator, the NTA-containing moiety N-[Nα,Nα-bis(carboxymethyl)-l-lysine]-12-mercaptododecanamide, via a hydrophilic and flexible NHS-PEG-maleimide linker (Scheme 1). These steps resulted in a novel, generalized carriage system for His-tagged proteins based on a photoreleasing HGN core (NTA-HGN).
The affinity of individual NTA-Ni2+ to His-tags is relatively low, with KD of 10–6 M at neutral pH.42 However, when a support surface has a high density of immobilized NTA-Ni2+, multiple linkages and “rebinding” effects by a His tagged protein provide a greater affinity and long-lasting association.42,43,48 It is with this valency effect in mind that we designed our photoactivated HGN platform. A densely packed NTA-Ni linker layer strongly binds the cargo protein to the HGN. Upon laser treatment the linkers release from the HGN surface and each one, acting on its own, reverts to the much reduced affinity (KD) of the individual NTA-Ni2+-His bond.42 Hence, the linker moieties and His-tag are expected to dissociate rapidly and diffuse away, leaving the protein free and in its active state.
As proof of concept for protein delivery to cells, we designed a model protein with an N-terminal 6-His-tag and a C-terminal cell-targeting peptide motif (RPARPAR), on a green fluorescent protein (GFP) backbone, designated R-GFP. This cell-penetrating GFP variant was generated using recombinant protein engineering, as is possible for a wide variety of potential His-tagged protein cargo. The peptide motif RPARPAR at the C-terminus, which follows the RXXR sequence found in the C-end Rule (CendR), facilitates specific endosome internalization into Neuropilin-1 (NRP-1) expressing cancer cells.23,49 NRP-1 is often overexpressed in tumors and is associated with poor prognosis,23,50,51 generating significant interest as a receptor for targeted drug delivery. Importantly, R-GFP serves as both fluorescent tracer and as an indicator of the mildness of the photorelease event since it fluoresces only if its tertiary structure is not denatured.52−54
3.2. Characterization of Release from Nanocarrier Platform
To assemble the protein onto the nanoparticles, R-GFP was incubated with the HGN-NTA at 100,000:1 R-GFP/HGN molar ratio in the presence of NiCl2. The large excess of protein was chosen to ensure maximal loading onto the particles and resulted in colloidally stable complexes (R-GFP-HGN). After washing, the amount of R-GFP successfully loaded onto the HGN was determined spectroscopically by His-tag elution with imidazole (Figure 1a). Imidazole is known to compete with the NTA:Ni2+ for the His-tag, and we found that this treatment completely released R-GFP from HGN (Figure 1a). The loading of R-GFP on HGN caused a reversible ∼80% quenching of the protein fluorescence, relative to free R-GFP (Figure 1a), attributed to the well-known fluorescence quenching properties of gold. The dequenching of R-GFP upon release from HGN was used to characterize the treatment efficiency under various laser conditions (Figure 1b).55
Figure 1.
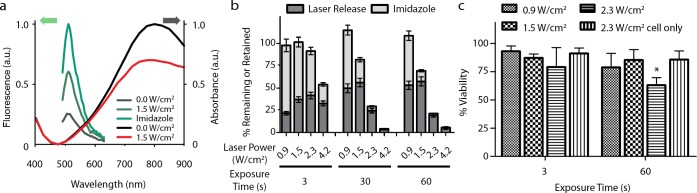
Characterization of the nanocarrier platform. (a) R-GFP fluorescence was partially quenched when bound to HGN; however, fluorescence increased upon exposure to laser excitation power of 1.5 W/cm2 for 30 s. Imidazole competes with histidine for the linker chelator groups on the HGN, chemically releasing the protein from the nanocarrier, resulting in maximum dequenching. R-GFP-HGN UV–vis absorbance decreased after laser exposure, indicative of changes to the core structure. (b) Quantification of R-GFP release from HGN after laser treatment at various laser powers and exposure times. R-GFP remaining on the HGN after laser exposure is released by imidazole competition and presented as the “retained” value, which is greater for lower laser powers and exposure times. Error bars represent the standard deviation. The expected sum of laser followed by imidazole is ∼100%. Reabsorption of R-GFP onto freshly exposed gold surfaces and partial protein denaturation may explain the incomplete release under certain conditions, as nonspecifically bound proteins would not be susceptible to imidazole competition. (c) R-GFP-HGN was internalized into PPC-1 cells, which were then treated with various combinations of laser power and exposure time. Decreased viability was observed in cells exposed to 2.3 W/cm2 fluence for 60 s (p < 0.05, *), well above the optimal release conditions. No loss in cell viability was observed with laser in the absence of HGN.
Our optimized nanoplatform carried ∼800 R-GFP per HGN (Figure S2, Supporting Information), which, if we consider binding to occur only to the exterior of the HGN, indicates a footprint of ∼7 nm2 per protein. This is a reasonable estimate since GFP is a barrel-shaped protein 4.2 nm long and 2.4 nm in diameter and has a cross-sectional area of ∼4.5 nm2. Assuming that each amine (∼3000 per HGN) was converted to an NTA-Ni2+ during synthesis, there were ∼4 NTA-Ni2+ available per GFP. We note that this value is above the expected minimum of two NTA-Ni groups per His tag required for stable, avidity-enhanced association.42 Furthermore, we observed minimal nonspecific binding in the absence of Ni2+ ions (Figure S3, Supporting Information), substantiating the specificity of the NTA-Ni2+ linkage.56
To characterize release of the protein, a wide-beam pulsed laser (diameter ∼5 mm) was directed at R-GFP-HGN in solution. We found that the R-GFP fluorescence increased in a laser power and exposure time-dependent manner, indicating a controlled detachment of the R-GFP from the HGN.6 The laser exposure was expected to break, or thermalize, the gold–thiol bonds holding the DNA5,6,57 and, by extension, cause the NTA-Ni2+ handles to release from the HGN. The linker length was chosen to position the protein several nanometers from the surface (Scheme 1) so that protein temperature remains near ambient during laser release,58 and thermal denaturation should be minimal.59 A release of ∼55% R-GFP, as compared to imidazole release, was possible for the optimal conditions (Figure 1b): irradiation at 800 nm at 0.9–1.5 W/cm2 (corresponding to a pulse fluence of 0.9–1.5 mJ/cm2, using 130 fs pulses at 1 kHz pulse repetition rate), for 30–60 s. For 3 s exposure at 1.5 W/cm2 a moderate fraction of ∼35% R-GFP was released. Since GFP fluorescence is known to directly correlate with having a natively folded structure,52 the fluorescence we observe after laser release supports a conclusion that the protein was released intact. We found that the remaining bound fraction of R-GFP was releasable by subsequent imidazole treatment of the particles (Figure 1b), and the sum of laser and imidazole released R-GFP approximately matched that from using imidazole alone. This result attests to the mildness of the release. We conclude that at least 50% of the R-GFP cargo could be released from HGN, with optical control, and without significant protein denaturation.
We demonstrate that our nanoparticle assembly can also support the loading and release of transcription factors Sox2 and p53 (Figure 2), establishing the versatility of the nanocarrier platform. Sox2-carrying HGN (Sox2-HGN) was subjected to laser irradiation and release assessed to be up to 60% of total protein, using the protein-reactive fluorescent dye 3-(4-carboxybenzoyl) quinoline-2-carboxaldehyde (CBQCA), as shown in Figure 2a. Following laser treatment and HGN centrifugation of Sox2-HGN and p53-HGN, the pellet was etched using KCN, to quantify the amount of protein retained on the nanocarrier by Western Blot and densitometry analysis (Figure 2b,c). A sharp extra Sox2 band is observed indicating site specific degradation of the protein as a result of the extensive processing during the sample analysis and lack of protease inhibitors. Similar degradation was observed in previous reports and suggests that degradation is not dependent on the laser.60 Sox2 shows a laser power-dependent release with approximately 40% of the protein retained on the HGN after irradiation with 2.3 W/cm2 for 30 s (Figure 2a), confirming the quantification done by CBQCA. Finally, we demonstrate that p53 shows a similar release pattern upon laser irradiation (Figure 2c).
Figure 2.
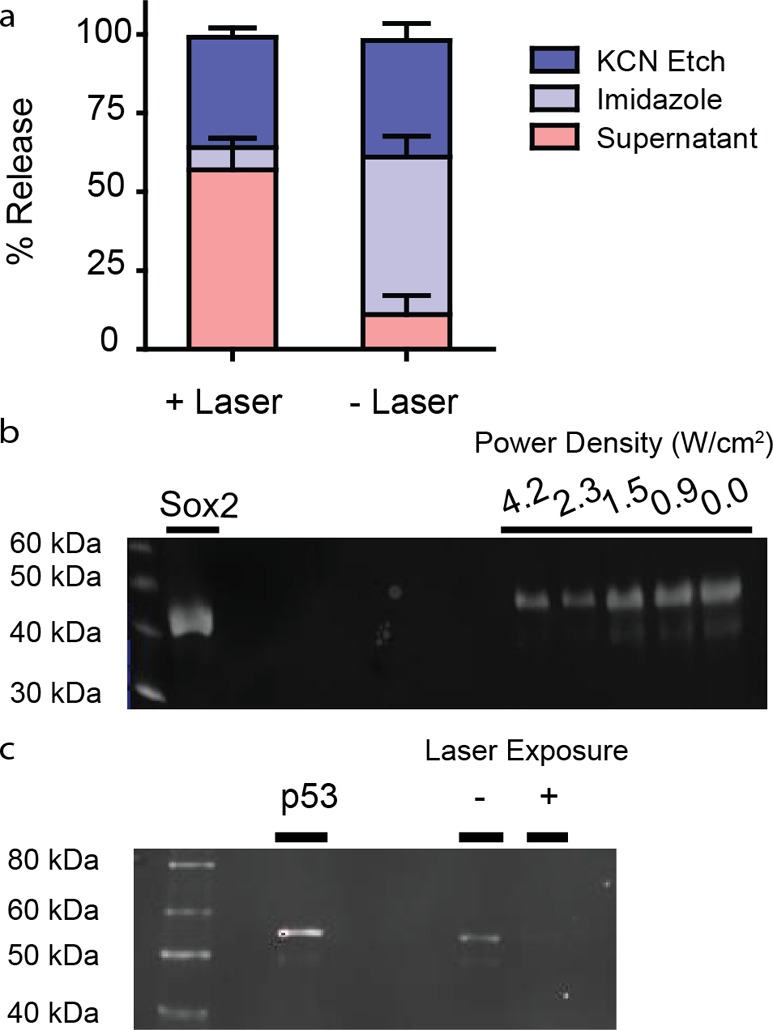
Characterization of Sox2 and p53 loading and release. (a) Bar graph showing the fraction of Sox2 that could be released from Sox2-HGN by each of the consecutive steps: ±laser, imidazole, and KCN etching of the HGN. For −Laser the fraction of Sox2 released by KCN was similar to the +Laser step. Sox2 release from Sox2-HGN was determined by labeling with an amine reactive dye CBQCA after laser exposure at 2.3 W/cm2 for 30 s. The amount of Sox2 that remained on the HGN was evaluated by pelleting and treating with imidazole and KCN, respectively. (b) Anti-HIS Western Blot of Sox2 retained on HGN after laser irradiation. Upon laser irradiation, samples were pelleted and washed, then HGN was etched with KCN, and solutions were run on a 12% SDS PAGE gel. Etching the HGN with KCN after laser excitation showed decreasing band intensity with increasing laser power density revealing release of Sox2 from the nanoparticle surface. An additional Sox2 band is observed from nonlaser-dependent degradation due to sample processing. Laser power densities were varied at a constant exposure time of 30 s. (c) p53 release from p53-HGN was evaluated by Western blot staining as in panel b after laser exposure at 2.3 W/cm2 for 30 s.
3.3. Cytotoxicity
NRP-1 expressing PPC-1 cells were incubated with R-GFP-HGN optimized to allow maximal internalization while minimizing the incubation time to 2 h (yielding an estimated count of 120 R-GFP-HGN per cell, see Supporting Information for details). The cells were exposed to the same range of NIR laser conditions used above for characterizing R-GFP, then cell viability was assessed 72 h post-treatment (Figure 1c). There was no significant reduction of cell viability by GFP-HGN for exposure time of 60 s and 1.5 W/cm2, a condition giving maximum release in Figure 1b. R-GFP-HGN in combination with laser treatment showed significant viability loss (30% or more) only for the highest laser power (2.3 W/cm2) and longest exposure times (60 s). As a control experiment the NIR treatment of cells without HGN showed negligible toxicity even at the highest light intensity.61,62 The NRP-1 deficient melanoma cell line M21 served as a negative control for uptake (Figure S4, Supporting Information).63 We quantified the GFP-HGN associated with PPC-1 by cell lysate fluorescence to be ∼105 GFP molecules per cell or ∼100 nM GFP assuming 2 pL cell volume (Figure S6, Supporting Information).64 This is competitive to typical intracellular concentrations of transcription factors such as p53, which has copy numbers ranging from 104 to 105 depending on tissue type and within the active concentration range of 10 nM to 10 μM of transcription factors transduced by peptide transduction domains.65−67
3.4. Intracellular Release of GFP by NIR Laser Irradiation of GFP-HGN
Next, we tested the ability of R-GFP-HGN to release protein cargo from endosomes into the cytosol in response to NIR laser pulses. In Figure 3, we explored protein release in PPC-1 cells using a laser scanning confocal two-photon microscope (Olympus Fluoview 1000 MPE) equipped with a tunable (690–1020 nm) fs pulsed laser, as well as a 473 nm continuous wave laser. The instrument allows switching between these two modes of excitation, with fine control over imaging and submicron resolution in laser patterning. PPC-1 cells were cultured on chambered glass slides and incubated with R-GFP-HGN for 2 h to allow internalization to occur. Internalization of RPARPAR labeled nanoparticles is about 60% complete within an hour.68 RPARPAR is well-known to promote endocytosis.49 The field of view was irradiated with 800 nm fs pulsed laser light (80 MHz), then the imaging mode was switched back to single photon to compare with the initial image. By increasing the power of the pulsed-NIR laser we defined a working range for R-GFP release from the HGN. A laser power of 8 × 103 W/cm2 was chosen (0.1 mJ/cm2 pulse fluence, using 100 fs pulses at 80 MHz pulse repetition rate, with beam diameter of ∼6 μm), whereby an average exposure time of 280 μs caused a significant increase of R-GFP fluorescence intensity in cells, indicating dequenching and release from HGN (Figure 3). Furthermore, an additional irradiation cycle released more R-GFP from the HGN but with diminishing returns (Figure 3, second pass). Autofluorescence intensity from PPC-1 without R-GFP-HGN did not change upon laser exposure (Figure S7, Supporting Information).
3.5. Spatial Release by NIR Pulsed Laser Irradiation
To understand the spatial release capability of the R-GFP in greater detail, we next defined regions of interest (ROIs) for the focused scanning laser to specifically illuminate (Figure 4). The laser was rastered within the user-defined shapes, modulated by a beam deflector that enables precise control over the exposure time applied at the pixel (sample volume) level. We incubated R-GFP-HGN with PPC-1 cells as in Figure 3, and for the laser operation, we defined ROIs that contained single cells, or parts of cells, that were interspersed between negative control cells not to be exposed. In the first example (Figure 4a,b) individual cells were targeted by ROI to test the cell level accuracy of the release process, whereas in Figure 4c–f, we targeted cells and parts of cells within a high-density cell cluster. For each experiment the pulsed laser was rastered exclusively across the areas within each red ROI (Figure 4a,c). Single-photon confocal imaging (473 nm laser) of R-GFP in the full field of view, before and ∼1 min after pulsed laser, revealed up to a 5-fold increase in intensity for the cells outlined by the ROIs (Figure 4b,d). Figure 4b shows that for cells external to the laser-exposed ROIs, one example being the cell within the blue oval (not targeted by laser), R-GFP was not released. The separation between it and the closest targeted ROI was ∼5 μm, indicating the laser patterning technique indeed has high fidelity.
Subcellular resolution over release was possible by defining a ROI smaller than the cells. In Figure 4c,d, the portion of the cell (line scan 3) within the irradiated ROI areas showed increased intensity while the portion outside the ROI was not much increased. In quantification for several cells, Figure 4e, those cells completely within the red square gave a total integrated cell fluorescence increase of ∼300% greater than the initial intensity, in good agreement with decreased R-GFP quenching in Figure 4a,c. Two photon real-time imaging during NIR laser stimulation further confirmed an inverse relationship between cytosolic (diffuse) and endosomal (punctate) fluorescence intensity (Figure S8, Supporting Information). Cells that were completely outside the area (Figure 4e,f) showed negligible fluorescence increase (∼8%) likely due to changes in the focal plane when switching from single to multiphoton modality. For cells lying across the red lines, those cellular areas within the ROI showed increases of ∼300%, whereas regions of the same cells that were outside the ROI showed a moderate fluorescence increase, ∼60%, attributable to diffusion of intracellularly released R-GFP, occurring on a time scale of ∼1 min between stimulation and imaging. The reported intracellular diffusion coefficient of GFP in mammalian cells is ∼25 μm2/s,69 although R-GFP may diffuse at a different rate if the RPARPAR peptide motif interacts with cellular organelles, as GFP fused with arginine rich HIV-derived TAT peptide (YGRKKRRQRRR) was reported to have an impaired diffusion coefficient of 5.9 μm2/s.70 Interestingly, the RPARPAR peptide on the GFP seems to impart nuclear targeting properties since we observed that when the protein was delivered into the cytosol the nucleoli showed increased brightness relative to the nucleus as a whole (Figure 4c). This effect is not surprising since RPARPAR resembles known nuclear localization sequences having cationic residues,71 as well as the putative histone binding peptide CQRPPR.72 Further studies may examine intracellular targeting of laser-released cargo in more detail.
4. Conclusions
In summary, we utilized a commercial two-photon microscope to provide unprecedented control and real-time monitoring of payload release by “painting” individual cells with NIR light, providing protein delivery with cell-level resolution with no additional equipment or effort. We demonstrate cellular delivery of a His-tagged protein by a nanocarrier in response to a focused fs pulsed laser and show that the protein cargo retains a functional conformation. Spatial (of the order of 1 μm) and temporal (within seconds) control over delivery resulted from light absorption at the plasmon resonance of an optimized hollow gold nanoshell. We used a fusion protein containing a cell targeting peptide to drive uptake into endosomes, here demonstrated for the PPC-1 prostate cancer cells that express the NRP-1 receptor. The thiol-DNA-NTA anchoring system confers control over laser release; the gold–thiol bonds are thermalized and released by the NIR laser pulses, presumably reducing the valency of the NTA toward the protein and ultimately separating the protein from the HGN carrier. The NIR laser pulses also induce vapor bubble formation that leads to rupture of the endosomes and release of the GFP throughout the cell. We observed nuclear accumulation of released R-GFP, as well as increased fluorescence over distant cytosolic regions of the cell, which together indicate successful R-GFP delivery throughout the cell. There were minimal adverse effects on cell viability due to either the nanoparticles or irradiation with NIR light. This technique opens the possibility of delivering various His-tagged proteins to the cytosol of cells, individually or in concert, and could enable optical control for stem cell differentiation, or for selective apoptosis. Although different cells may be affected differently by the treatment, we anticipate that the platform can be tuned to best suit each cell line’s specific requirements.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant R01 EB012637. The authors thank T. Teesalu, E. Ruoslahti, and M. Moskovits for cell lines and helpful discussion, and A. Mikhailovsky for laser use. We thank M. Raven for aid with microscopy and acknowledge the support for the Olympus Fluoview 1000 MPE microscope from the NIH (1S10RR022585-01A1). D.P.M. acknowledges support from the NSF (NSF-OISE-0968399). G.B.B. acknowledges support from the NIH (R01 CA 152327, T32 CA 121949) and the Cancer Center of Santa Barbara.
Supporting Information Available
Experimental methods and additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ Center for Bioengineering, University of California Santa Barbara, CA 93106, United States
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Heim R.; Prasher D. C.; Tsien R. Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. U.S.A. 1994, 912612501–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J. H.; Forbush B. III; Hoffman J. F. Rapid photolytic release of adenosine 5′-triphosphate from a protected analogue: utilization by the Na:K pump of human red blood cell ghosts. Biochemistry 1978, 17101929–35. [DOI] [PubMed] [Google Scholar]
- Lee H. M.; Larson D. R.; Lawrence D. S. Illuminating the chemistry of life: design, synthesis, and applications of ″caged″ and related photoresponsive compounds. ACS Chem. Biol. 2009, 46409–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreaden E. C.; Alkilany A. M.; Huang X.; Murphy C. J.; El-Sayed M. A. The golden age: gold nanoparticles for biomedicine. Chem. Soc. Rev. 2012, 4172740–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Pallaoro A.; Braun G. B.; Morales D. P.; Ogunyankin M. O.; Zasadzinski J.; Reich N. O. Modular plasmonic nanocarriers for efficient and targeted delivery of cancer-therapeutic siRNA. Nano Lett. 2014, 1442046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun G. B.; Pallaoro A.; Wu G.; Missirlis D.; Zasadzinski J. A.; Tirrell M.; Reich N. O. Laser-activated gene silencing via gold nanoshell–siRNA conjugates. ACS Nano 2009, 372007–2015. [DOI] [PubMed] [Google Scholar]
- Chen C.-C.; Lin Y.-P.; Wang C.-W.; Tzeng H.-C.; Wu C.-H.; Chen Y.-C.; Chen C.-P.; Chen L.-C.; Wu Y.-C. DNA–gold nanorod conjugates for remote control of localized gene expression by near infrared irradiation. J. Am. Chem. Soc. 2006, 128113709–3715. [DOI] [PubMed] [Google Scholar]
- Huschka R.; Barhoumi A.; Liu Q.; Roth J. A.; Ji L.; Halas N. J. Gene silencing by gold nanoshell-mediated delivery and laser-triggered release of antisense oligonucleotide and siRNA. ACS Nano 2012, 697681–7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J.; Zhang G.; Li C. Exceptionally high payload of doxorubicin in hollow gold nanospheres for near-infrared light-triggered drug release. ACS Nano 2010, 421033–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Liu X.; Liu Z.; Pu F.; Ren J.; Qu X. Near-infrared light-triggered, targeted drug delivery to cancer cells by aptamer gated nanovehicles. Adv. Mater. 2012, 24212890–2895. [DOI] [PubMed] [Google Scholar]
- Greenblatt M. S.; Bennett W. P.; Hollstein M.; Harris C. C. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54184855–4878. [PubMed] [Google Scholar]
- Ingram V. M. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature 1957, 1804581326–328. [DOI] [PubMed] [Google Scholar]
- Keen H.; Glynne A.; Pickup J. C.; Viberti G. C.; Bilous R. W.; Jarrett R. J.; Marsden R. Human insulin produced by recombinant DNA technology: safety and hypoglycaemic potency in healthy men. Lancet 1980, 28191398–401. [DOI] [PubMed] [Google Scholar]
- Roth D. A.; Kessler C. M.; Pasi K. J.; Rup B.; Courter S. G.; Tubridy K. L. Human recombinant factor IX: safety and efficacy studies in hemophilia B patients previously treated with plasma-derived factor IX concentrates. Blood 2001, 98133600–6. [DOI] [PubMed] [Google Scholar]
- Corwin H. L.; Gettinger A.; Pearl R. G.; Fink M. P.; Levy M. M.; Shapiro M. J.; Corwin M. J.; Colton T. Efficacy of recombinant human erythropoietin in critically ill patients: a randomized controlled trial. JAMA 2002, 288222827–35. [DOI] [PubMed] [Google Scholar]
- Quesada J. R.; Hersh E. M.; Manning J.; Reuben J.; Keating M.; Schnipper E.; Itri L.; Gutterman J. U. Treatment of hairy cell leukemia with recombinant alpha-interferon. Blood 1986, 682493–7. [PubMed] [Google Scholar]
- Fu A.; Tang R.; Hardie J.; Farkas M. E.; Rotello V. M. Promises and Pitfalls of Intracellular Delivery of Proteins. Bioconjugate Chem. 2014, 2591602–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock K.; Nolden L.; Edenhofer F.; Quandel T.; Brustle O. Transcription factor-based modulation of neural stem cell differentiation using direct protein transduction. Cell. Mol. Life Sci. 2010, 67142439–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenobu T.; Tomizawa K.; Matsushita M.; Li S. T.; Moriwaki A.; Lu Y. F.; Matsui H. Development of p53 protein transduction therapy using membrane-permeable peptides and the application to oral cancer cells. Mol. Cancer Ther. 2002, 1121043–9. [PubMed] [Google Scholar]
- Jo J.; Hong S.; Choi W. Y.; Lee D. R. Cell-penetrating peptide (CPP)-conjugated proteins is an efficient tool for manipulation of human mesenchymal stromal cells. Sci. Rep. 2014, 4, 4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang R.; Kim C. S.; Solfiell D. J.; Rana S.; Mout R.; Velazquez-Delgado E. M.; Chompoosor A.; Jeong Y.; Yan B.; Zhu Z. J.; Kim C.; Hardy J. A.; Rotello V. M. Direct delivery of functional proteins and enzymes to the cytosol using nanoparticle-stabilized nanocapsules. ACS Nano 2013, 786667–6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z.; Biswas A.; Zhao M.; Tang Y. Tailoring nanocarriers for intracellular protein delivery. Chem. Soc. Rev. 2011, 4073638–55. [DOI] [PubMed] [Google Scholar]
- Chen R.; Braun G. B.; Luo X.; Sugahara K. N.; Teesalu T.; Ruoslahti E. Application of a proapoptotic peptide to intratumorally spreading cancer therapy. Cancer Res. 2013, 7341352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeneman K.; Delehanty J. B.; Blanco-Canosa J. B.; Susumu K.; Stewart M. H.; Oh E.; Huston A. L.; Dawson G.; Ingale S.; Walters R.; Domowicz M.; Deschamps J. R.; Algar W. R.; DiMaggio S.; Manono J.; Spillmann C. M.; Thompson D.; Jennings T. L.; Dawson P. E.; Medintz I. L. Selecting Improved Peptidyl Motifs for Cytosolic Delivery of Disparate Protein and Nanoparticle Materials. ACS Nano 2013, 753778–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi T.; Nielsen P. E. Enhanced delivery of cell-penetrating peptide-peptide nucleic acid conjugates by endosomal disruption. Nat. Protoc. 2006, 12633–6. [DOI] [PubMed] [Google Scholar]
- Zhao M.; Hu B.; Gu Z.; Joo K.-I.; Wang P.; Tang Y. Degradable polymeric nanocapsule for efficient intracellular delivery of a high molecular weight tumor-selective protein complex. Nano Today 2013, 8111–20. [Google Scholar]
- Zheng Y.; Cai Z.; Song X.; Chen Q.; Bi Y.; Li Y.; Hou S. Preparation and characterization of folate conjugated N-trimethyl chitosan nanoparticles as protein carrier targeting folate receptor: in vitro studies. J. Drug Targeting 2009, 174294–303. [DOI] [PubMed] [Google Scholar]
- Ghosh P.; Yang X.; Arvizo R.; Zhu Z. J.; Agasti S. S.; Mo Z.; Rotello V. M. Intracellular delivery of a membrane-impermeable enzyme in active form using functionalized gold nanoparticles. J. Am. Chem. Soc. 2010, 13282642–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X.; Xu A. M.; Leal-Ortiz S.; Cao Y.; Garner C. C.; Melosh N. A. Nanostraw-electroporation system for highly efficient intracellular delivery and transfection. ACS Nano 2013, 7, 4351–4358. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yu L. C. Single-cell microinjection technology in cell biology. BioEssays 2008, 306606–10. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Yu L. C. Microinjection as a tool of mechanical delivery. Curr. Opin. Biotechnol. 2008, 195506–510. [DOI] [PubMed] [Google Scholar]
- Gu Z.; Yan M.; Hu B.; Joo K.-I.; Biswas A.; Huang Y.; Lu Y.; Wang P.; Tang Y. Protein Nanocapsule Weaved with Enzymatically Degradable Polymeric Network. Nano Lett. 2009, 9124533–4538. [DOI] [PubMed] [Google Scholar]
- Peng K.; Tomatsu I.; Kros A. Light controlled protein release from a supramolecular hydrogel. Chem. Commun. 2010, 46234094–6. [DOI] [PubMed] [Google Scholar]
- Prevo B. G.; Esakoff S. A.; Mikhailovsky A.; Zasadzinski J. A. Scalable routes to gold nanoshells with tunable sizes and response to near-infrared pulsed laser irradiation. Small 2008, 4, 1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Servos M. R.; Liu J. Instantaneous and quantitative functionalization of gold nanoparticles with thiolated DNA using a pH-assisted and surfactant-free route. J. Am. Chem. Soc. 2012, 134177266–9. [DOI] [PubMed] [Google Scholar]
- Ayed A.; Mulder F. A.; Yi G. S.; Lu Y.; Kay L. E.; Arrowsmith C. H. Latent and active p53 are identical in conformation. Nat. Struct. Biol. 2001, 89756–60. [DOI] [PubMed] [Google Scholar]
- Shaner N. C.; Steinbach P. A.; Tsien R. Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 212905–9. [DOI] [PubMed] [Google Scholar]
- Jain P. K.; Qian W.; El-Sayed M. A. Ultrafast cooling of photoexcited electrons in gold nanoparticle-thiolated DNA conjugates involves the dissociation of the gold-thiol bond. J. Am. Chem. Soc. 2006, 12872426–33. [DOI] [PubMed] [Google Scholar]
- Wu G.; Mikhailovsky A.; Khant H. A.; Fu C.; Chiu W.; Zasadzinski J. A. Remotely triggered liposome release by near-infrared light absorption via hollow gold nanoshells. J. Am. Chem. Soc. 2008, 130268175–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevo B. G.; Esakoff S. A.; Mikhailovsky A.; Zasadzinski J. A. Scalable routes to gold nanoshells with tunable sizes and response to near-infrared pulsed-laser irradiation. Small 2008, 481183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissleder R. A clearer vision for in vivo imaging. Nat. Biotechnol. 2001, 194316–7. [DOI] [PubMed] [Google Scholar]
- Nieba L.; Nieba-Axmann S. E.; Persson A.; Hämäläinen M.; Edebratt F.; Hansson A.; Lidholm J.; Magnusson K.; Karlsson Å. F.; Plückthun A. BIACORE analysis of histidine-tagged proteins using a chelating NTA sensor chip. Anal. Biochem. 1997, 2522217–228. [DOI] [PubMed] [Google Scholar]
- De M.; Rana S.; Rotello V. M. Nickel-ion-mediated control of the stoichiometry of his-tagged protein/nanoparticle interactions. Macromol. Biosci. 2009, 92174–8. [DOI] [PubMed] [Google Scholar]
- Sapsford K. E.; Pons T.; Medintz I. L.; Higashiya S.; Brunel F. M.; Dawson P. E.; Mattoussi H. Kinetics of metal-affinity driven self-assembly between proteins or peptides and CdSe–ZnS quantum dots. J. Phys. Chem. C 2007, 1113111528–11538. [Google Scholar]
- Arnau J.; Lauritzen C.; Petersen G. E.; Pedersen J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expression Purif. 2006, 4811–13. [DOI] [PubMed] [Google Scholar]
- Porath J.; Carlsson J.; Olsson I.; Belfrage G. Metal chelate affinity chromatography, a new approach to protein fractionation. Nature 1975, 2585536598–9. [DOI] [PubMed] [Google Scholar]
- Hurst S. J.; Lytton-Jean A. K.; Mirkin C. A. Maximizing DNA loading on a range of gold nanoparticle sizes. Anal. Chem. 2006, 78248313–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapsford K. E.; Algar W. R.; Berti L.; Gemmill K. B.; Casey B. J.; Oh E.; Stewart M. H.; Medintz I. L. Functionalizing nanoparticles with biological molecules: developing chemistries that facilitate nanotechnology. Chem. Rev. 2013, 11331904–2074. [DOI] [PubMed] [Google Scholar]
- Teesalu T.; Sugahara K. N.; Kotamraju V. R.; Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. U.S.A. 2009, 1063816157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H.; Cheng L.; Tickner M.; Bagherzadeh A.; Selwood D.; Zachary I. Neuropilin-1 antagonism in human carcinoma cells inhibits migration and enhances chemosensitivity. Br. J. Cancer 2010, 1023541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallaoro A.; Braun G. B.; Moskovits M. Quantitative ratiometric discrimination between noncancerous and cancerous prostate cells based on neuropilin-1 overexpression. Proc. Natl. Acad. Sci. U.S.A. 2011, 1084016559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar D. M.; Aoudjane S.; Gaudet M.; Aeppli G.; Dalby P. A. Optically induced thermal gradients for protein characterization in nanolitre-scale samples in microfluidic devices. Sci. Rep. 2013, 3, 2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkaabi K. M.; Yafea A.; Ashraf S. S. Effect of pH on thermal- and chemical-induced denaturation of GFP. Appl. Biochem. Biotechnol. 2005, 1262149–156. [DOI] [PubMed] [Google Scholar]
- Postupalenko V.; Sibler A.-P.; Desplancq D.; Nominé Y.; Spehner D.; Schultz P.; Weiss E.; Zuber G. Intracellular delivery of functionally active proteins using self-assembling pyridylthiourea-polyethylenimine. J. Controlled Release 2014, 178, 86–94. [DOI] [PubMed] [Google Scholar]
- Bajaj A.; Rana S.; Miranda O. R.; Yawe J. C.; Jerry D. J.; Bunz U. H. F.; Rotello V. M. Cell surface-based differentiation of cell types and cancer states using a gold nanoparticle-GFP based sensing array. Chem. Sci. 2010, 11134. [Google Scholar]
- Rana S.; Yeh Y. C.; Rotello V. M. Engineering the nanoparticle-protein interface: applications and possibilities. Curr. Opin. Chem. Biol. 2010, 146828–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana N. R.; Gearheart L.; Murphy C. J. Wet chemical synthesis of silver nanorods and nanowires of controllable aspect ratio. Chem. Commun. 2001, 7617–618. [Google Scholar]
- Hill H. D.; Millstone J. E.; Banholzer M. J.; Mirkin C. A. The role radius of curvature plays in thiolated oligonucleotide loading on gold nanoparticles. ACS Nano 2009, 32418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris N.; Ford M. J.; Cortie M. B. Optimization of plasmonic heating by gold nanospheres and nanoshells. J. Phys. Chem. B 2006, 1102210701–10707. [DOI] [PubMed] [Google Scholar]
- Pan C.; Lu B.; Chen H.; Bishop C. E. Reprogramming human fibroblasts using HIV-1 TAT recombinant proteins OCT4, SOX2, KLF4 and c-MYC. Mol. Biol. Rep. 2010, 3742117–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukianova-Hleb E. Y.; Ren X.; Zasadzinski J. A.; Wu X.; O L. D. Plasmonic nanobubbles enhance efficacy and selectivity of chemotherapy against drug-resistant cancer cells. Adv. Mater. 2012, 24, 3831–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Kang B.; Qian W.; Mackey M. A.; Chen P. C.; Oyelere A. K.; El-Sayed I. H.; El-Sayed M. A. Comparative study of photothermolysis of cancer cells with nuclear-targeted or cytoplasm-targeted gold nanospheres: continuous wave or pulsed lasers. J. Biomed. Opt. 2010, 155058002. [DOI] [PubMed] [Google Scholar]
- Sugahara K. N.; Teesalu T.; Karmali P. P.; Kotamraju V. R.; Agemy L.; Girard O. M.; Hanahan D.; Mattrey R. F.; Ruoslahti E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 166510–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R.; Jorgensen P.; Moran U.; Weber G.; Springer M. BioNumbers-the database of key numbers in molecular and cell biology. Nucleic Acids Res. 2009, 38, D750–D753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggin M. D. Animal transcription networks as highly connected, quantitative continua. Dev. Cell 2011, 214611–626. [DOI] [PubMed] [Google Scholar]
- Michiue H.; Tomizawa K.; Wei F. Y.; Matsushita M.; Lu Y. F.; Ichikawa T.; Tamiya T.; Date I.; Matsui H. The NH2 terminus of influenza virus hemagglutinin-2 subunit peptides enhances the antitumor potency of polyarginine-mediated p53 protein transduction. J. Biol. Chem. 2005, 28098285–9. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Ma Y.; Gu J.; Liao B.; Li J.; Wong J.; Jin Y. Reprogramming of somatic cells via TAT-mediated protein transduction of recombinant factors. Biomaterials 2012, 33205047–55. [DOI] [PubMed] [Google Scholar]
- Braun G. B.; Friman T.; Pang H. B.; Pallaoro A.; de Mendoza T. H.; Willmore A. M.; Kotamraju V. R.; Mann A. P.; She Z. G.; Sugahara K. N.; Reich N. O.; Teesalu T.; Ruoslahti E. Etchable plasmonic nanoparticle probes to image and quantify cellular internalization. Nat. Mater. 2014, 13, 904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayel M. J.; Hom E. F.; Verkman A. S. Diffusion of green fluorescent protein in the aqueous-phase lumen of endoplasmic reticulum. Biophys. J. 1999, 7652843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardarelli F.; Serresi M.; Bizzarri R.; Giacca M.; Beltram F. In vivo study of HIV-1 Tat arginine-rich motif unveils its transport properties. Mol. Ther. 2007, 1571313–22. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Melikov K.; Vives E.; Ramos C.; Verbeure B.; Gait M. J.; Chernomordik L. V.; Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 2781585–90. [DOI] [PubMed] [Google Scholar]
- Wang K.; Purushotham S.; Lee J.-Y.; Na M.-H.; Park H.; Oh S.-J.; Park R.-W.; Park J. Y.; Lee E.; Cho B. C.; Song M.-N.; Baek M.-C.; Kwak W.; Yoo J.; Hoffman A. S.; Oh Y.-K.; Kim I.-S.; Lee B.-H. In vivo imaging of tumor apoptosis using histone H1-targeting peptide. J. Controlled Release 2010, 1483283–291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.