

Abstract
3-methylglutaconic aciduria (3-MGA-uria) is a nonspecific finding associated with mitochondrial dysfunction, including defects of oxidative phosphorylation. 3-MGA-uria is classified into five groups, of which one, type IV, is genetically heterogeneous. Here we report five children with a form of type IV 3-MGA-uria characterized by cataracts, severe psychomotor regression during febrile episodes, epilepsy, neutropenia with frequent infections, and death in early childhood. Four of the individuals were of Greenlandic descent, and one was North American, of Northern European and Asian descent. Through a combination of homozygosity mapping in the Greenlandic individuals and exome sequencing in the North American, we identified biallelic variants in the caseinolytic peptidase B homolog (CLPB). The causative variants included one missense variant, c.803C>T (p.Thr268Met), and two nonsense variants, c.961A>T (p.Lys321∗) and c.1249C>T (p.Arg417∗). The level of CLPB protein was markedly decreased in fibroblasts and liver of affected individuals. CLPB is proposed to function as a mitochondrial chaperone involved in disaggregation of misfolded proteins, resulting from stress such as heat denaturation.
Main Text
3-methylglutaconic aciduria (3-MGA-uria) is a nonspecific biochemical finding associated with a group of inborn errors of metabolism, particularly mitochondrial disorders. 3-MGA is a branched-chain organic acid and intermediate of leucine degradation and the mevalonate shunt pathway. The latter is essential for the synthesis of cholesterol, coenzyme Q, dolichols, and isoprenoids, which are needed for sterol synthesis and acetyl-CoA metabolism in mitochondria.1–4 The clinical features of the 3-MGA-uria syndromes are diverse and are classified into five types, each with significant heterogeneity.5,6 In all types, with the exception of 3-MGA-uria type I (MIM 250950), the activities of 3-methylglutaconyl-CoA hydratase and other enzymes of leucine degradation are normal, and the 3-MGA-uria is thought to be secondary to defects in phospholipid remodeling or integrity of mitochondrial membranes, leading to electron transport chain dysfunction.7,8 3-MGA-uria type I is an inborn error of leucine metabolism, caused by variants in AUH (MIM 600529). AUH encodes 3-methylglutaconyl-CoA hydratase, which catalyzes the fifth step of leucine catabolism, whereby 3-methylglutaconyl-CoA is converted to 3-hydroxy-3-methylglutaryl-coenzyme A.9 3-MGA-uria type I might present as an acute, life-threatening condition in childhood with nonspecific features such as seizures, intellectual disability, or abnormal liver function. It might also present as leukoencephalopathy in adults.5
3-MGA-uria type II (MIM 302060), or Barth syndrome, is an X-linked recessive disorder caused by variants in TAZ (MIM 300394), which encodes taffazin, a cardiolipin transacylase in the inner mitochondrial membrane.10 Clinical findings include cardiomyopathy, myopathy, short stature, neutropenia, hypocholesterolemia, dysmorphism, and cognitive difficulties. 3-MGA-uria type III (MIM 258501), or Costeff syndrome, is caused by variants in OPA3 (MIM 165300), characterized by early-onset optic atrophy, extrapyramidal signs, spasticity, ataxia, dysarthria, and cognitive deficiency.11 OPA3 is a mitochondrial outer membrane protein involved in maintenance of the respiratory chain. 3-MGA-uria type V (MIM 610198) features early-onset dilated cardiomyopathy, nonprogressive cerebellar ataxia, testicular dysgenesis, and growth failure. Type V is caused by variants in DNAJC19 (MIM 608977), which encodes a mitochondrial cochaperone.7,12 DNAJC19 forms a complex with prohibitins (PHB) and lipid scaffolds in the inner membrane of mitochondria that is necessary for mitochondrial morphogenesis, neuronal survival, and phospholipid remodeling. In particular, this complex regulates cardiolipin remodeling in cardiac mitochondrial membranes through tafazzin.7,8 Thus, 3-MGA-uria types V and II affect a common pathway related to mitochondrial membrane metabolism, defects in which lead to loss of electron transport chain function and energy production.
3-MGA-uria type IV (MIM 250951), the “unclassified type,” includes all other forms of 3-MGA-uria with normal 3-methylglutaconyl-CoA hydratase enzyme activity. A diagnosis of 3-MGA-uria type IV is complicated by the large number of implicated genes, including those involved in mitochondrial DNA depletion syndromes, mitochondrial DNA deletion syndromes, MELAS (MIM 540000), Smith-Lemli-Opitz syndrome (MIM 270400), and glycogen storage disease type 1b (MIM 232220).5,6,13,14 Many individuals with type IV 3-MGA-uria have pathogenic variants in nuclear-encoded mitochondrial genes involved in respiratory chain function.5,15,16 In addition, two genes have been associated with specific type IV 3-MGA-uria disorders. First, variants in SERAC1 (MIM 614725), which encodes an enzyme involved in phosphatidylglycerol remodeling, are associated with MEGDEL syndrome (MIM 614739), characterized by progressive spasticity, dystonia, deafness, psychomotor retardation, hypocholesterolemia, and Leigh-like lesions on MRI.17 Second, variants in TMEM70 (MIM 612418) are associated with an isolated mitochondrial complex V deficiency and neonatal hypotonia, hypertrophic cardiomyopathy, psychomotor retardation, cataract, and hyperammonemia.6
Molecular diagnosis of 3-MGA-uria is complicated by marked clinical and locus heterogeneity, and many affected individuals lack a specific genetic etiology because of the number of diverse targets, some of which have yet to be identified. Individuals with 3-MGA-uria present with variable symptoms and organ involvement, with predominantly progressive neurological impairment in combination with 3-MGA-uria and other biochemical findings suggestive of mitochondrial dysfunction.
Here, we describe the identification of variants in CLPB, associated with a distinct 3-MGA-uria syndrome characterized by cataracts, severe psychomotor regression during febrile episodes, epilepsy, neutropenia with frequent infections, and death in early childhood. Four individuals, including one pair of siblings, were from Greenland; the fifth was a North American, of Northern European and Asian descent. None of the families from Greenland were known to be related.
Written informed consent was obtained from all individuals investigated or their guardians, and the research protocols were performed in accordance with the ethical standards of the respective national and international committees on human subject research.
Individual 1 was female, the first child born to healthy parents after an uncomplicated pregnancy, weighing 2,680 g at term. At the age of 6 months, she was diagnosed with bilateral cataracts. Although psychomotor development was apparently normal, by 9 months of age, hypotonia was identified. Severe dehydration and respiratory distress was precipitated by a febrile illness at 11 months of age, after which developmental regression occurred along with severe hypotonia and development of extrapyramidal symptoms (buccolingual movements, myoclonus, and choreoathetosis) and, subsequently, seizures. No dysmorphic features were noted. Hepatic transaminases were elevated and associated with marked macronodular steatosis on liver biopsy. Her clinical course featured intermittent leukopenia, moderate to severe neutropenia, and frequent infections (Table 1). Bone marrow examination showed a profound maturation arrest. Blood and cerebrospinal fluid (CSF) lactic acid levels were elevated, and urine organic acid screening demonstrated moderate 3-MGA-uria, with marked increase during the first febrile episode. She died at 11 months of age with heart failure, kidney failure, and pneumothorax.
Table 1.
Biochemical Findings in Individuals with CLPB Variants
| Subject | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| White blood count (WBC), × 109/l (reference, 4.0–14.0 × 109/l) | 1.4–9.3 | 2.8–7.1 | 2.3–7.7 | NA | 2.69–8.19 × 103 mcl (reference, 5–21) |
| Absolute neutrophil count (ANC), × 109/l (reference: neutropenia < 1.5 × 109/l, severe neutropenia < 0.5 × 109/l) | 0–0.8 | 0.1–1.9 | 0.3 | NA | 0.00–0.39 × 103 mcl (reference, 1.5–9.0) |
| Platelets | NA | NA | NA | NA | 66–188 × 103 mcl (reference, 150–450) |
| Blood lactate, mmol/l (reference, <1.7 mmol/l) | <7.7 | 3.8 | NA | NA | 2.2–5.5 (0.7–2.1) |
| CSF lactate, mmol/l (reference, <1.7 mmol/l) | 5.89 | 2.11 | NA | NA | |
| Urine methylglutaconic acid, mg/g creatinine (reference, <15 mg/g creatinine) | 70–250 | 250 | 210–260 | 80–220 | 193 mmol/mol creatinine (reference, <15) |
NA, not available.
Individual 2 was the younger brother of individual 1. Pregnancy and birth were uncomplicated. He was born at term with a birth weight of 3,090 g and a head circumference of 34 cm. Like his sister, his early development was normal until a febrile illness at 9 months of age, when he developed a very similar phenotype and laboratory findings. No dysmorphic features were noted. He required tube feeding (Table 1). Analysis of bone marrow showed deficient granulopoiesis. Magnetic resonance imaging (MRI) of the brain showed symmetrical hyperintensities in the globus pallidus and cerebral atrophy. Muscle histology was normal. He died at 3 years of age; cause of death was not reported.
Individual 3 was female, and the first child born to healthy parents, weighing 3,000 g and measuring 51 cm at birth. Her early development was apparently normal, but from age 6 months, she was noted to have psychomotor retardation with regression and hypotonia. No dysmorphic features were noted. At 1 year of age, she was admitted to the hospital because of generalized epilepsy. She was microcephalic (42.7 cm, 2 SD below the normal range), with hypertonicity of the upper extremities and hyperreflexia. Ophthalmologic examination showed slight zonular cataracts bilaterally. A brain MRI was normal. She had leukopenia and severe neutropenia. Urine 3-MGA was moderately elevated. She died at 2.5 years of age from pneumonia.
Individual 4 was female, the third child born to healthy parents after an uncomplicated pregnancy and term birth, weighing 2,700 g and measuring 50 cm. Her two older siblings were healthy. She had pneumonia three times in the first 4 months of life. During the last episode, she was lethargic and had convulsions, featuring opisthotonus. She was nondysmorphic. She subsequently had severe hypotonia, psychomotor retardation, repeated episodes of opisthotonus, hyperreflexia, dystonia, and few spontaneous movements. She had growth retardation and microcephaly (−3 SD). She developed severe generalized epilepsy at the age of 2 years. Liver transaminases were normal. There was chronic neutropenia but not leukopenia. 3-MGA excretion was moderately elevated. Brain CT at age 6 months was normal. She died at 4 years of age; cause of death was not reported.
Individual 5 was a female infant born to a 26-year-old G2P1 011 mother. She was delivered at 39 weeks gestation by elective Caesarean section, because of breech presentation. Her birth weight was 2,466 g. Pregnancy was complicated by polyhydramnios, growth retardation, poor fetal movements, and suspected arthrogryposis. Prenatal testing included normal maternal serum screening and a normal 46, XX karyotype. At birth, she was cyanotic with no observed respiratory effort. She was noted to have a rigid trunk, extremities, neck, and jaw; her extremities were fixed in partial flexion with fisting of hands in upper extremities and with hip flexion and lower leg extension. In addition, she had mild upper and lower extremity rhizomelia and contractures at elbows, wrists, fingers, and hips. Her facial appearance was unusual, including a slanting forehead, downslanting palprebral fissures with periorbital fullness, small appearing globes, prominent nasal bridge, bulbous nose with perinasal creases, and micrognathia. Neurologic exam revealed tremors, 1+/4 deep tendon reflex at both knees, and absent deep tendon reflexes at the elbows. In addition, she had congenital cataracts and mild bilateral pelviectasis. She had at least two events featuring oxygen desaturation and bradycardia that required positive pressure ventilation. Due to poor neurological prognosis, care was redirected and she expired on day 8 of life after respiratory failure.
A brain MRI was consistent with observed microcephaly and demonstrated generalized decreased parenchymal volume without evidence of brain malformations or recent ischemia, although a left choroid plexus hemorrhage without hydrocephalus was incidentally identified. An electroencephalogram (EEG) showed long runs of generalized attenuation without seizure activity. Biochemical testing, including serum amino acid, urine organic acid, and urine amino acid profiles, revealed moderately elevated levels of 3-MGA in addition to nonspecific elevations of urinary amino acids. Other notable laboratory findings included significant neutropenia and thrombocytopenia. In addition, she had a bleeding diathesis with elevated prothrombin time, partial thromboplastin time, and decreased fibrinogen, which required infusion of blood products. Testing for factor V Leiden and prothrombin variants was negative.
The current Greenlandic population of ∼55,000 descended from a presumably small number of Inuit founders from Canada who lived in relative isolation for centuries, accounting for a founder effect and high carrier rate for certain rare conditions. Assuming a common founder in the four Greenlandic individuals, a genome-wide search for homozygosity was performed with the Affymetrix GeneChip 50cK Xba array, v.2.0 (Affymetrix). This revealed a single homozygous region of 4.5 Mb on chromosome 11, between markers rs826056 and rs1938685, encompassing 62 genes. The Maestro database was used to search for genes with a predicted mitochondrial localization (score ≥ 4).22 The highest-scoring genes were UCP2 (8.6), MRPL48 (4.7), CLPB (4.1), and FAM86C (1.6). Sequencing analysis revealed no variants in UCP2, MRPL48, or FAM86C. Analysis of CLPB (RefSeq accession number NM_030813.3) showed a homozygous missense variant, c.803C>T (p.Thr268Met), in all four affected children (Figure 1A). The parents of individuals 1 and 2 were each heterozygous for the variant; the parents of the other two children were not available. To assess the potential pathogenicity of this variant, the relative incidence, conservation, in silico predictions for pathogenicity, and potential effect on splicing was evaluated. To rule out a splicing defect, cDNA derived from fibroblasts was amplified and sequenced, revealing a normally spliced mRNA (data not shown) with band of normal size (Figure 1B). The variant results in the substitution of a highly conserved polar threonine for a nonpolar methionine (Figure 1C), which is located in one of the ankyrin domains (Figure 1D) and predicted to be pathogenic by SIFT, PolyPhen-2, and MutationTaster. A TaqMan assay was developed to assess the carrier frequency of c.803C>T variant in Greenlandic controls: 6 of 184 samples were determined to be heterozygous, corresponding to a carrier frequency of 3.3%, which is comparable to carrier frequencies of other founder variants in the Greenlandic population. It was not found in 2,180 samples sequenced at Children’s Mercy Hospital (CMH) or in 13,000 alleles in the Exome Variant Server but was reported with a carrier frequency of 1 out of 662 in ClinSeq (rs200032855). Sequencing of CLPB was performed in two additional individuals of Danish descent with a similar phenotype, but no variants were identified.
Figure 1.
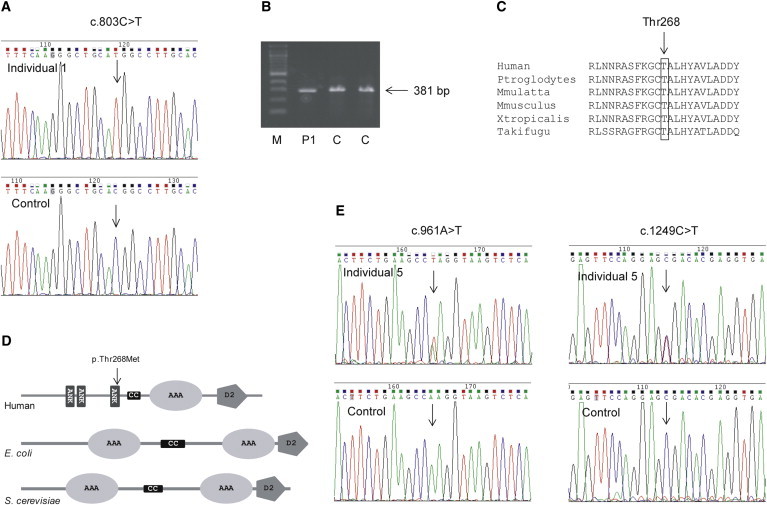
Identification of CLPB Variants in Five Individuals
(A) DNA sequence analysis of CLPB shows the position of the homozygous c.803C>T variant in the subject compared to the control.
(B) Analysis of cDNA encompassing the c.803C>T variant shows a band of normal size in individual 1. To assess the functional effect of the c.803C>T variant on splicing, RNA was extracted from fibroblasts and reverse transcribed to cDNA with the SuperScript II Reverse Transcriptase kit (Invitrogen), and PCR of a 381 bp cDNA fragment encompassing exons 4–8 was performed. To assess the carrier frequency for the c.803C>T variant, a TaqMan assay was developed (Applied Biosystems). The PCR conditions were: 10 μl Universal PCR Master Mix, 0.5 μl 40 × assay mix, and 20–100 ng DNA in a total volume of 20 μl. The PCR program was 95°C for 10 min, and 50 cycles at 92°C for 15 s and 60°C for 1 min. The samples were run on an ABI Prism 7000 and analyzed with ABI SDS software.
(C) The alignment of the amino acid sequences of CLPB homologs in different vertebrate species shows the conservation of the mutated threonine at position 268.
(D) A schematic representation of human CLPB (not to scale) shows the predicted domains and the position of the p.Thr268Met substitution. The following abbreviations are used: ANK, ankyrin repeat; CC, coiled-coil domain; AAA+, AAA+ ATPase; and D2-small, ClpB-D2-small.
(E) DNA sequence analysis of CLPB DNA shows the position of the heterozygous c.961A>T variant in subject 5 compared to the control (left) and of the heterozygous c.1249C>T (right).
In parallel, independent exome sequencing analysis was performed on individual 5 (CMH193) and her two healthy parents. DNA was prepared utilizing the KAPA Biosystems library preparation kit (KAPA Biosystems) followed by Illumina TruSeqExome enrichment (Illumina). Samples were sequenced on an Illumina HiSeq 2000 instrument with TruSeq v.3 reagents, as paired 100 nucleotide reads to a depth of 7.7 gigabases resulting in median target coverage of 135×; the mitochondrial genome was represented at an average depth of 200×. Alignment and variant calling was performed as previously reported,18,19 resulting in the identification of ∼170,000 nucleotide variants. Variants were filtered to 1% minor allele frequency in an internal database of 1,913 samples, then prioritized by the American College of Medical Genetics (ACMG) categorization,20 OMIM identity, and phenotypic assessment. Genomic sequence data are available at dbGAP (accession phs000564). No rare homozygous or compound heterozygous variants comprising a diagnostic genotype were identified in a previously reported disease-associated gene, but one de novo variant was identified in ATP6VOA2, associated with autosomal-recessive cutis laxa (MIM 219200). Because of minimal phenotypic overlap and the absence of a second variant, this finding was not pursued. In addition, this individual was found to be compound heterozygous for two nonsense variants in CLPB, c.961A>T (p.Lys321∗) and c.1249C>T (p.Arg417∗) (Figure 1E). The two variants were confirmed by Sanger sequencing. Segregation analysis confirmed that the variants were inherited from carrier parents, consistent with an autosomal-recessive inheritance pattern. In addition, genotyping of two subsequently born healthy siblings revealed that they were each heterozygous for one of the variants. Both CLPB variants were absent from the NHLBI Exome Sequencing Project (EVS) and an internal variant database of 2,180 samples. In addition, truncating CLPB variants were absent from the internal data set and rare in the EVS; homozygous truncating variants were absent. CLPB c.1249C>T has been reported in dbSNP as rs200203460, but with unknown frequency.
Respiratory chain enzyme assays were performed on frozen muscle and liver from individual 5 as part of a metabolic autopsy at the Center For Inherited Disorders of Energy Metabolism (CIDEM) (Cleveland, OH). Muscle tissue showed generally decreased enzyme activities, probably due to tissue deterioration. In frozen liver, the activity of rotenone-sensitive NADH-cytochrome c reductase (reflecting complexes I and III) was significantly below the control range (14%), as was complex III activity (28%), whereas complexes I and II had normal activity (88% and 73%, respectively). Complex IV activity was not assayed. Citrate synthase activity was 90% of control. In addition, ATP production in digitonin-permeabilized fibroblasts and in-gel enzyme activity in frozen liver was performed as reported.21,23 ATP production in digitonin-permeabilized fibroblasts from individual 1 energized with either 10 mM pyruvate plus 10 mM malate, 10 mM glutamate plus 10 mM malate, or 10 mM succinate plus rotenone was normal (37.9 nmol/min/mg protein, reference 31.8 ± 4.5 [mean ± SD]; 41.0 nmol/min/mg protein, reference 33.0 ± 5.8; and 19.2 nmol/min/mg, reference 14.9 ± 3.3, respectively). Analysis of in-gel enzyme activity showed normal activities of complexes I and IV in the liver of individual 5 (Figure 2A).
Figure 2.
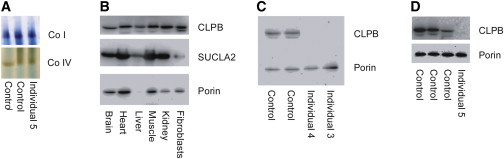
Analysis of CLPB Protein Levels and In-Gel Activity of Complexes I and IV
(A) In-gel enzyme activity in liver from individual 5 shows normal activity of complexes I and IV.
(B) Analysis by SDS-PAGE of CLPB protein in different human tissues showing ubiquitous localization of CLPB.
(C and D) Immunoblot analysis of CLPB in fibroblasts from individuals 3 and 4 and liver from subject 5 (D) shows absence of CLPB protein. In brief, mitochondrial protein was isolated from fibroblasts and liver as previously reported.23 The samples (25–30 cμg protein/lane) were run on a 12% SDS polyacrylamide gel and transferred to a PVDF membrane. The membrane was probed with a polyclonal antibody against CLPB (Atlas Antibodies) at a 1:1,000 dilution and developed with a 1:1,000 dilution of goat anti-rabbit antibody (Dako). An antibody against porin (Proteintech) was used as a loading control at a 1:1,000 dilution, and SUCLA2 was used as a reference at a 1:1,000 dilution. The secondary antibody was goat anti-mouse at a 1:1,000 dilution (Dako). The bands were visualized with the Supersignal West Pico and Femto substrates (Thermo Fisher Scientific) and MicroChemi imaging (DNR Bioimaging Systems).
To investigate normal localization of CLPB protein, immunoblot analysis of several human tissues was performed, demonstrating ubiquitous presence of CLPB, although protein abundance was more variable than other mitochondrial proteins (Figure 2B). Immunoblot analysis performed on fibroblasts from individuals with the p.Thr268Met variant (Figure 2C), as well as in liver from individual 5 (with two nonsense variants) showed absence of CLPB protein (Figure 2D).
Our data indicate that biallelic recessive variants in CLPB result in a specific clinical and biochemical phenotype associated with 3-MGA-uria (summarized in Table 1). The onset of clinical disease in CLPB-associated 3-MGA-uria ranged from prenatal in one individual with polyhydramnios, intrauterine growth retardation, and poor movements to postnatal onset between 4 and 9 months of age. Postnatal onset was characterized by development of cataracts, psychomotor regression or lethargy, and frequent febrile illnesses complicated by development of seizures and psychomotor regression. Neutropenia was a consistent finding in all. Cataracts were identified in four of five individuals and microcephaly in three of five. Life expectancy was short, with death by 8 days in the child with prenatal onset, and from 11 months to 4 years of age in the remaining individuals.
Many of the clinical findings in the individuals with CLPB-associated 3-MGA-uria were similar to those seen in other mitochondrial disorders, such as onset coincident with systemic illness and the presence of epilepsy, psychomotor regression, and microcephaly. Neutropenia, however, is an uncommon finding in those with other mitochondrial disorders, except MGA-uria type II (Barth syndrome), where it is found in the majority of affected individuals.22 As in Barth syndrome, bone marrow histology showed arrested granulopoiesis as the proximal cause of neutropenia.
Cataract is infrequently reported in mitochondrial disorders, although it is present in Sengers syndrome (MIM 212350)24 and in individuals with TMEM70- and GFER-related (MIM 60094) disease.25–27 The main mechanism of cataract development is unknown. However, variants in GFER, AGK (MIM 610345), TMEM70, TAZ, and SERAC1 affect mitochondrial membrane integrity28 or lipid metabolism of the mitochondrial membranes.29 Hence, cataract formation in the individuals reported here can be explained by disrupted protein-lipid interactions affecting oxidative phosphorylation enzymes, increasing oxidative stress, and stress-induced protein damage. In addition, it might be speculated that a contributing factor to CLPB-related disease is the resultant accumulation of aggregated proteins.
Although cerebral MRI was normal in one individual, two others showed cerebral atrophy, of whom one also had hyperintensities in the globus pallidus, similar to findings in Leigh syndrome (MIM 256000). In addition, although not apparent on MRI, it should be noted that all of the individuals with pathogenic CLPB variants had signs and symptoms of basal ganglia degeneration.
No evidence of a severe respiratory chain enzyme deficiency was found in children with CLPB-related disease, although lactic acidosis was present in all. This is similar to other types of 3-MGA-uria, the exception being individuals with TMEM70-related disease, which is usually associated with complex V deficiency.
The levels of 3-MGA-uria in the individuals reported here were similar to those seen in Barth syndrome.30 In type I 3-MGA-uria, 3-MGA is produced through catabolism of leucine, which is not likely the case in the other types, because these individuals do not excrete higher amounts of 3-MGA after a protein-rich meal, as seen in type I.31 Also, 3-MGA level might fluctuate substantially in non-type I 3-MGA-uria and is apparently unrelated to the clinical course.6 All types of 3-MGA-uria are associated with some degree of mitochondrial respiratory chain dysfunction, but the exact mechanism is unknown. However, in vivo experiments indicate that oxidative stress can be induced by accumulation of 3-MGA in tissue and biological fluids in affected individuals, which presumably contributes to the mitochondrial pathophysiology.32
The phenotype of individual 5, who exhibited prenatal onset of symptoms and neonatal death, was more severe than in the children homozygous for the p.Thr268Met missense variant. The compound heterozygous nonsense variants led to absence of CLPB protein in immunoblots of liver. In addition, CLPB protein was not detected in fibroblasts from the individuals homozygous for the p.Thr268Met variant. Such missense variants might be expected to produce residual amounts of partially functional protein, perhaps below the detection limits of this assay in fibroblasts, and might account for the slightly milder phenotype in the p.Thr268Met homozygotes. Whether the earlier diagnosis of such individuals could lead to a change in management should be investigated, such as metabolomic analysis to identify the pathways involved and possible alternative routes for energy provision.
Most knowledge of CLPB function has come from studies in E. coli (ClpB) and yeast (Hsp78) homologs, which belong to the Clp/Hsp104 family (Hsp100), a group of AAA+ proteins containing two consensus ATP-binding sites. Hsp100 family proteins are chaperones that disaggregate proteins misfolded due to stress such as heat shock.33 The current structural model of ClpB is a ring-shaped hexamer, where the aggregated target polypeptides bind near the central cavity and are threaded through the ring complex, thereby becoming disaggregated.34 The ClpB homohexamer is part of a larger oligomeric protein complex. In contrast, Hsp78 occurs mainly as a homotrimer.35 ClpB/Hsp78 act in close collaboration with DnaK/Hsp70 chaperones that bind aggregates and target them to ClpB/Hsp78. Both bacterial Clpb and yeast Hsp78 have been shown to bind newly imported proteins and to prevent aggregation of misfolded proteins in the mitochondrial matrix under conditions of impaired DnaK/Hsp70 function.36 Hsp78 deletion mutants exhibit no abnormal phenotype under normal conditions but show effects on mitochondrial protein biosynthesis37 and mtDNA replication38 at high temperatures. Hsp78 deletion mutants harboring an additional variant in Hsp70 exhibit loss of mitochondrial DNA and aggregation of mutant Hsp70, suggesting that one function of Hsp78 might be to keep mutant forms of Hsp70 in a functional and soluble state.33,39 In E. coli, deficiency of ClpB leads to increased heat sensitivity.40
E. coli ClpB and yeast Hsp78 contain two AAA+ domains (Figure 1D), which function to mediate oligomerization of the protein.41 In contrast, mammalian CLPB has a single AAA+ domain, along with three ankyrin-like repeats, which are not found in the E. coli and yeast homologs. Ankyrin repeats are involved in protein-protein interaction and could aid in oligomerization of CLPB.
A striking finding of the children reported here was onset and/or severe psychomotor regression during febrile episodes, which could be consistent with a function of human CLPB in disaggregation of proteins misfolded due to heat denaturation, similar to that of its proposed homologs, although this is also a common feature in other mitochondrial disorders. Another important mitochondrial chaperone, Hsp60 (GroEL in E. coli), and its partner, HSP10, assume a similar role to DnaK/Hsp70 in mediating the ATP-dependent mitochondrial import of nuclear-encoded proteins, as well as protein folding and refolding both under normal and cellular stress conditions.42,43 Variants in HSPD1 (MIM 118190) encoding Hsp60 have been reported in association with MitCHAP60 disease (MIM 612233), an early-onset neurodegenerative disorder similar to CLPB-related disease. Individuals with MitCHAP60 disease exhibit leukodystrophy, hypotonia, developmental delay, pyramidal signs, seizures, microcephaly, episodes of abnormal breathing, growth failure, and mildly elevated plasma lactate during acute illness.44 Mild reductions in COX activity were observed in only the most severely affected individuals; muscle biopsies revealed giant mitochondria with abnormal cristea.44 Knockout of Hspd1 in mice results in embryonal lethality45 and null variants in Hsp60 are nonviable in yeast, secondary to severe mitochondrial-protein folding defects, whereas conditional mutations result in the accumulation of misfolded proteins incapable of forming active oxidative phosphorylation enzyme complexes.44 These studies suggest a key role of Hsp60 and Clpb in regulating protein folding inside the mitochondrial matrix. Defects in mitochondrial chaperones exert a profound impact on overall mitochondrial biogenesis, membrane integrity, and function.44,46,47
In conclusion, we report a severe, recessive form of type IV 3-MGA-uria associated with mitochondrial dysfunction caused by loss-of-function variants in CLPB. Further studies are needed to explore the exact function of human CLPB.
Acknowledgments
We thank the families for their participation. We thank Dr. Lili Miles for the EM studies. This work was supported by the Clare Giannini Fund and a grant from The Danish Council for Independent Research | Medical Sciences to E.O. (12-127702).
Contributor Information
Carol Saunders, Email: csaunders@cmh.edu.
Elsebet Ostergaard, Email: elsebet.ostergaard@dadlnet.dk.
Web References
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
ClinSeq, http://genome.gov/20519355
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
PolyPhen-2, www.genetics.bwh.harvard.edu/pph2/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Gunay-Aygun M. 3-Methylglutaconic aciduria: a common biochemical marker in various syndromes with diverse clinical features. Mol. Genet. Metab. 2005;84:1–3. doi: 10.1016/j.ymgme.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Marinier E., Lincoln B.C., Garneau M., David F., Brunengraber H. Contribution of the shunt pathway of mevalonate metabolism to the regulation of cholesterol synthesis in rat liver. J. Biol. Chem. 1987;262:16936–16940. [PubMed] [Google Scholar]
- 3.Rauthan M., Pilon M. The mevalonate pathway in C. elegans. Lipids Health Dis. 2011;10:243. doi: 10.1186/1476-511X-10-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weinstock S.B., Kopito R.R., Endemann G., Tomera J.F., Marinier E., Murray D.M., Brunengraber H. The shunt pathway of mevalonate metabolism in the isolated perfused rat liver. J. Biol. Chem. 1984;259:8939–8944. [PubMed] [Google Scholar]
- 5.Wortmann S.B., Duran M., Anikster Y., Barth P.G., Sperl W., Zschocke J., Morava E., Wevers R.A. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J. Inherit. Metab. Dis. 2013;36:923–928. doi: 10.1007/s10545-012-9580-0. [DOI] [PubMed] [Google Scholar]
- 6.Wortmann S.B., Rodenburg R.J., Jonckheere A., de Vries M.C., Huizing M., Heldt K., van den Heuvel L.P., Wendel U., Kluijtmans L.A., Engelke U.F. Biochemical and genetic analysis of 3-methylglutaconic aciduria type IV: a diagnostic strategy. Brain. 2009;132:136–146. doi: 10.1093/brain/awn296. [DOI] [PubMed] [Google Scholar]
- 7.Richter-Dennerlein R., Korwitz A., Haag M., Tatsuta T., Dargazanli S., Baker M., Decker T., Lamkemeyer T., Rugarli E.I., Langer T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014;20:158–171. doi: 10.1016/j.cmet.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 8.Lamari F., Mochel F., Sedel F., Saudubray J.M. Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: toward a new category of inherited metabolic diseases. J. Inherit. Metab. Dis. 2013;36:411–425. doi: 10.1007/s10545-012-9509-7. [DOI] [PubMed] [Google Scholar]
- 9.IJlst L., Loupatty F.J., Ruiter J.P., Duran M., Lehnert W., Wanders R.J. 3-Methylglutaconic aciduria type I is caused by mutations in AUH. Am. J. Hum. Genet. 2002;71:1463–1466. doi: 10.1086/344712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnston J., Kelley R.I., Feigenbaum A., Cox G.F., Iyer G.S., Funanage V.L., Proujansky R. Mutation characterization and genotype-phenotype correlation in Barth syndrome. Am. J. Hum. Genet. 1997;61:1053–1058. doi: 10.1086/301604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anikster Y., Kleta R., Shaag A., Gahl W.A., Elpeleg O. Type III 3-methylglutaconic aciduria (optic atrophy plus syndrome, or Costeff optic atrophy syndrome): identification of the OPA3 gene and its founder mutation in Iraqi Jews. Am. J. Hum. Genet. 2001;69:1218–1224. doi: 10.1086/324651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davey K.M., Parboosingh J.S., McLeod D.R., Chan A., Casey R., Ferreira P., Snyder F.F., Bridge P.J., Bernier F.P. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J. Med. Genet. 2006;43:385–393. doi: 10.1136/jmg.2005.036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelley R.I., Kratz L. 3-methylglutaconic acidemia in Smith-Lemli-Opitz syndrome. Pediatr. Res. 1995;37:671–674. doi: 10.1203/00006450-199505000-00020. [DOI] [PubMed] [Google Scholar]
- 14.Law L.K., Tang N.L., Hui J., Lam C.W., Fok T.F. 3-methyglutaconic aciduria in a Chinese patient with glycogen storage disease Ib. J. Inherit. Metab. Dis. 2003;26:705–709. doi: 10.1023/b:boli.0000005603.04633.21. [DOI] [PubMed] [Google Scholar]
- 15.Wortmann S.B., Kluijtmans L.A., Rodenburg R.J., Sass J.O., Nouws J., van Kaauwen E.P., Kleefstra T., Tranebjaerg L., de Vries M.C., Isohanni P. 3-Methylglutaconic aciduria—lessons from 50 genes and 977 patients. J. Inherit. Metab. Dis. 2013;36:913–921. doi: 10.1007/s10545-012-9579-6. [DOI] [PubMed] [Google Scholar]
- 16.Wortmann S.B., Morava E. 3-methylglutaconic aciduria type IV: a syndrome with an evolving phenotype. Clin. Dysmorphol. 2011;20:168–169. doi: 10.1097/MCD.0b013e328345bea8. [DOI] [PubMed] [Google Scholar]
- 17.Tort F., García-Silva M.T., Ferrer-Cortès X., Navarro-Sastre A., Garcia-Villoria J., Coll M.J., Vidal E., Jiménez-Almazán J., Dopazo J., Briones P. Exome sequencing identifies a new mutation in SERAC1 in a patient with 3-methylglutaconic aciduria. Mol. Genet. Metab. 2013;110:73–77. doi: 10.1016/j.ymgme.2013.04.021. [DOI] [PubMed] [Google Scholar]
- 18.Bell C.J., Dinwiddie D.L., Miller N.A., Hateley S.L., Ganusova E.E., Mudge J., Langley R.J., Zhang L., Lee C.C., Schilkey F.D. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Transl. Med. 2011;3:ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saunders C.J., Miller N.A., Soden S.E., Dinwiddie D.L., Noll A., Alnadi N.A., Andraws N., Patterson M.L., Krivohlavek L.A., Fellis J. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 2012;4:ra135. doi: 10.1126/scitranslmed.3004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards C.S., Bale S., Bellissimo D.B., Das S., Grody W.W., Hegde M.R., Lyon E., Ward B.E., Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 21.Zerbetto E., Vergani L., Dabbeni-Sala F. Quantification of muscle mitochondrial oxidative phosphorylation enzymes via histochemical staining of blue native polyacrylamide gels. Electrophoresis. 1997;18:2059–2064. doi: 10.1002/elps.1150181131. [DOI] [PubMed] [Google Scholar]
- 22.Roberts A.E., Nixon C., Steward C.G., Gauvreau K., Maisenbacher M., Fletcher M., Geva J., Byrne B.J., Spencer C.T. The Barth Syndrome Registry: distinguishing disease characteristics and growth data from a longitudinal study. Am. J. Med. Genet. A. 2012;158A:2726–2732. doi: 10.1002/ajmg.a.35609. [DOI] [PubMed] [Google Scholar]
- 23.Pedersen C.B., Zolkipli Z., Vang S., Palmfeldt J., Kjeldsen M., Stenbroen V., Schmidt S.P., Wanders R.J., Ruiter J.P., Wibrand F. Antioxidant dysfunction: potential risk for neurotoxicity in ethylmalonic aciduria. J. Inherit. Metab. Dis. 2010;33:211–222. doi: 10.1007/s10545-010-9086-6. [DOI] [PubMed] [Google Scholar]
- 24.van Ekeren G.J., Stadhouders A.M., Smeitink J.A., Sengers R.C. A retrospective study of patients with the hereditary syndrome of congenital cataract, mitochondrial myopathy of heart and skeletal muscle and lactic acidosis. Eur. J. Pediatr. 1993;152:255–259. doi: 10.1007/BF01956157. [DOI] [PubMed] [Google Scholar]
- 25.Spiegel R., Khayat M., Shalev S.A., Horovitz Y., Mandel H., Hershkovitz E., Barghuti F., Shaag A., Saada A., Korman S.H. TMEM70 mutations are a common cause of nuclear encoded ATP synthase assembly defect: further delineation of a new syndrome. J. Med. Genet. 2011;48:177–182. doi: 10.1136/jmg.2010.084608. [DOI] [PubMed] [Google Scholar]
- 26.Di Fonzo A., Ronchi D., Lodi T., Fassone E., Tigano M., Lamperti C., Corti S., Bordoni A., Fortunato F., Nizzardo M. The mitochondrial disulfide relay system protein GFER is mutated in autosomal-recessive myopathy with cataract and combined respiratory-chain deficiency. Am. J. Hum. Genet. 2009;84:594–604. doi: 10.1016/j.ajhg.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atay Z., Bereket A., Turan S., Haliloglu B., Memisoglu A., Khayat M., Shalev S.A., Spiegel R. A novel homozygous TMEM70 mutation results in congenital cataract and neonatal mitochondrial encephalo-cardiomyopathy. Gene. 2013;515:197–199. doi: 10.1016/j.gene.2012.11.044. [DOI] [PubMed] [Google Scholar]
- 28.Kratochvílová H., Hejzlarová K., Vrbacký M., Mráček T., Karbanová V., Tesařová M., Gombitová A., Cmarko D., Wittig I., Zeman J., Houštěk J. Mitochondrial membrane assembly of TMEM70 protein. Mitochondrion. 2014;15:1–9. doi: 10.1016/j.mito.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 29.Mayr J.A. Lipid metabolism in mitochondrial membranes. J. Inherit. Metab. Dis. 2014 doi: 10.1007/s10545-014-9748-x. Published online August 1, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Mazurová S., Tesařová M., Magner M., Houšt'ková H., Hansíková H., Augustínová J., Tomek V., Vondráčková A., Zeman J., Honzík T. Novel mutations in the TAZ gene in patients with Barth syndrome. Prague Med. Rep. 2013;114:139–153. doi: 10.14712/23362936.2014.16. [DOI] [PubMed] [Google Scholar]
- 31.Ensenauer R., Müller C.B., Schwab K.O., Gibson K.M., Brandis M., Lehnert W. 3-Methylglutaconyl-CoA hydratase deficiency: a new patient with speech retardation as the leading sign. J. Inherit. Metab. Dis. 2000;23:341–344. doi: 10.1023/a:1005670911799. [DOI] [PubMed] [Google Scholar]
- 32.Fernandes C.G., da Rosa M.S., Seminotti B., Pierozan P., Martell R.W., Lagranha V.L., Busanello E.N., Leipnitz G., Wajner M. In vivo experimental evidence that the major metabolites accumulating in 3-hydroxy-3-methylglutaryl-CoA lyase deficiency induce oxidative stress in striatum of developing rats: a potential pathophysiological mechanism of striatal damage in this disorder. Mol. Genet. Metab. 2013;109:144–153. doi: 10.1016/j.ymgme.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 33.von Janowsky B., Major T., Knapp K., Voos W. The disaggregation activity of the mitochondrial ClpB homolog Hsp78 maintains Hsp70 function during heat stress. J. Mol. Biol. 2006;357:793–807. doi: 10.1016/j.jmb.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Weibezahn J., Tessarz P., Schlieker C., Zahn R., Maglica Z., Lee S., Zentgraf H., Weber-Ban E.U., Dougan D.A., Tsai F.T. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119:653–665. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 35.Leidhold C., von Janowsky B., Becker D., Bender T., Voos W. Structure and function of Hsp78, the mitochondrial ClpB homolog. J. Struct. Biol. 2006;156:149–164. doi: 10.1016/j.jsb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 36.Schmitt M., Neupert W., Langer T. Hsp78, a Clp homologue within mitochondria, can substitute for chaperone functions of mt-hsp70. EMBO J. 1995;14:3434–3444. doi: 10.1002/j.1460-2075.1995.tb07349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmitt M., Neupert W., Langer T. The molecular chaperone Hsp78 confers compartment-specific thermotolerance to mitochondria. J. Cell Biol. 1996;134:1375–1386. doi: 10.1083/jcb.134.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Germaniuk A., Liberek K., Marszalek J. A bichaperone (Hsp70-Hsp78) system restores mitochondrial DNA synthesis following thermal inactivation of Mip1p polymerase. J. Biol. Chem. 2002;277:27801–27808. doi: 10.1074/jbc.M201756200. [DOI] [PubMed] [Google Scholar]
- 39.Moczko M., Schönfisch B., Voos W., Pfanner N., Rassow J. The mitochondrial ClpB homolog Hsp78 cooperates with matrix Hsp70 in maintenance of mitochondrial function. J. Mol. Biol. 1995;254:538–543. doi: 10.1006/jmbi.1995.0636. [DOI] [PubMed] [Google Scholar]
- 40.Squires C.L., Pedersen S., Ross B.M., Squires C. ClpB is the Escherichia coli heat shock protein F84.1. J. Bacteriol. 1991;173:4254–4262. doi: 10.1128/jb.173.14.4254-4262.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mogk A., Schlieker C., Strub C., Rist W., Weibezahn J., Bukau B. Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. J. Biol. Chem. 2003;278:17615–17624. doi: 10.1074/jbc.M209686200. [DOI] [PubMed] [Google Scholar]
- 42.Baker M.J., Frazier A.E., Gulbis J.M., Ryan M.T. Mitochondrial protein-import machinery: correlating structure with function. Trends Cell Biol. 2007;17:456–464. doi: 10.1016/j.tcb.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 43.Bross P., Magnoni R., Bie A.S. Molecular chaperone disorders: defective Hsp60 in neurodegeneration. Curr. Top. Med. Chem. 2012;12:2491–2503. doi: 10.2174/1568026611212220005. [DOI] [PubMed] [Google Scholar]
- 44.Magen D., Georgopoulos C., Bross P., Ang D., Segev Y., Goldsher D., Nemirovski A., Shahar E., Ravid S., Luder A. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am. J. Hum. Genet. 2008;83:30–42. doi: 10.1016/j.ajhg.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Christensen J.H., Nielsen M.N., Hansen J., Füchtbauer A., Füchtbauer E.M., West M., Corydon T.J., Gregersen N., Bross P. Inactivation of the hereditary spastic paraplegia-associated Hspd1 gene encoding the Hsp60 chaperone results in early embryonic lethality in mice. Cell Stress Chaperones. 2010;15:851–863. doi: 10.1007/s12192-010-0194-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raturi A., Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM) Biochim. Biophys. Acta. 2013;1833:213–224. doi: 10.1016/j.bbamcr.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 47.Cheng M.Y., Hartl F.U., Martin J., Pollock R.A., Kalousek F., Neupert W., Hallberg E.M., Hallberg R.L., Horwich A.L. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature. 1989;337:620–625. doi: 10.1038/337620a0. [DOI] [PubMed] [Google Scholar]