

Abstract
The Critical Assessment of Small Molecule Identification (CASMI) contest was initiated in 2012 to evaluate manual and automated strategies for the identification of small molecules from raw mass spectrometric data. The authors participated in both category 1 (molecular formula determination) and category 2 (molecular structure determination) of the second annual CASMI contest (CASMI 2013) using slow but effective manual methods. The provided high resolution mass spectrometric data were interpreted manually using a combination of molecular formula calculators, fragment and neutral loss analysis, literature consultation, manual database searches, deductive logic, and experience. The authors submitted correct formulas as lead candidates for 16 of 16 challenges and submitted correct structure solutions as lead candidates for 14 of 16 challenges. One structure submission (Challenge 3) was very close but not exact (N2-acetylglutaminylisoleucinamide instead of the correct N2-acetylglutaminylleucinamide). A solution for one (Challenge 13) was not submitted due to an inability to reconcile the provided fragmentation pattern with any known structures with the provided molecular composition.
Keywords: CASMI, high resolution mass spectrometry, tandem mass spectrometry, small molecule identification, metabolite identification
INTRODUCTION
The Analytical Core of the UIC/NIH Center for Botanical Dietary Supplements Research is involved in the structure elucidation of natural products from terrestrial sources. Rapid identification of known natural products (dereplication) is an essential service provided by the Core. Such analyses are invaluable for natural product chemists to properly allocate resources to pursue novel and more promising structures and essential for the identification of active compounds within complex extracts screened against biological targets. Mass spectrometry coupled to a high performance separation technique such as ultra-high pressure liquid chromatography is the primary tool used to accomplish these tasks. A standard workflow for sample processing and analysis includes the determination of elemental composition by accurate mass measurement followed by acquisition of tandem mass spectra on a high resolution mass spectrometer such as ion trap/time-of-flight (IT-TOF) or quadrupole/time-of-flight (Q-TOF) instruments. Elemental compositions are searched in relevant databases containing known natural products such as SciFinder, Reaxys or KEGG as well as in tandem mass spectral databases such as MassBank and METLIN. The Core has also built and is constantly expanding an in-house database of tandem mass spectra of natural products based on the MassBank platform. The team entered the contest with the goal of assessing its interpretation skills on a set of unknown compounds from chemical spaces not normally encountered in routine activities. Another benefit of participation is an opportunity to learn and evaluate alternative approaches for identification of small molecules from mass spectrometric data. The team submitted molecular formulas for all challenges (Category 1) and structure solutions for 15 of 16 challenges (Category 2).
EXPERIMENTAL
For each challenge, the raw challenge data were copied and pasted from the CASMI 2013 website into a Microsoft Excel spreadsheet and then converted from tab delimited text into two separate columns. The mass spectra and tandem mass spectra were reconstructed using X–Y scatterplots. Additional columns for the exact masses of neutral losses and possible molecular formula for fragment ions and neutral losses were added. All molecular formula calculations from exact masses were performed using the molecular formula calculator from Shimadzu LCMS Solutions software version 3.6. ChemDraw Ultra 12.0 was used for importing chemical structures, structure editing, some fragmentation analysis, and generation of SMILES notation.
Molecular formula determination (Category 1)
Formula candidates were determined on a case by case basis using manual methods. The monoisotopic mass was determined from the MS data using adduct patterns such as protonated/sodiated molecules or when no obvious evidence for adducts was present the base peak was assumed to be a protonated/deprotonated molecule. Molecular formulas were provided for Challenges 7, 8, 13, and 14. Mass fragment ion and neutral loss analysis using a standard molecular formula calculator and a spreadsheet were used to help establish molecular formula. The molecular formula calculator was applied initially with wide limits for common organic elements C (0 to 100), H (0 to 200), O (0 to 100), and N (0 to 10), double bond equivalents from 0 to 100, application of the nitrogen rule, and mass error tolerance from 0 to 10 ppm or the measurement error of the instrument when given (e.g. LTQ Orbitrap, 3 ppm). In some cases, the formula could be determined strictly from the accurate mass and fragment ion information.
Formula determination for some cases was more challenging and required additional work. Since standard molecular formula calculators return all arithmetically possible matches to an exact mass with no filtration of absurd or unreasonable formulas, exact mass values were searched using the ChemSpider database within an error tolerance appropriate for the instrument performance (e.g. within 10 ppm for IT-TOF data) to generate a list of matching molecular formulas for real molecules. Most molecular formulas returned from ChemSpider could be ruled out based upon isotopic and fragmentation information. In some cases, the final formula candidate was determined by arriving at the Category 2 structure candidate. A single formula candidate was submitted for each of the 16 challenges.
Candidate structure determination (Category 2)
When it was specified that the compound was a natural product, the Reaxys database (www.reaxys.com) was used to search candidate molecular formulas with the constraint “isolation from natural product exists.” In some cases the in-home database of tandem mass spectra of natural products was also used. CAS SciFinder scholar was used to search for molecular formula for all challenges. Search results were sorted by the number of references. Candidates with very few references were regarded with very low priority for consideration since they are uncommon or unavailable compounds. SciFinder was used to retrieve references that may contain spectral information for specific candidate compounds. Once a small number of structural candidates was established, various online strategies were employed to attempt to locate a reference or literature tandem mass spectra for the candidate structures using resources on the World Wide Web. Other resources used included MassBank searches, PubChem queries, Google scholar searches of the literature, and Reaxys or SciFinder substance records containing links to the published mass spectra.
Since a ranking score for all candidate formula and structure entries is required for CASMI evaluation, a subjective and arbitrary confidence scale from 0.60 to 1.00 was used. Structures were placed on the scale based upon how “confident” we felt about the overall fit of the proposed structure to the challenge data. The confidence scale ranking brackets were defined as shown in Fig. 1.
Fig. 1. The arbitrary “confidence” scoring scale used to score structure and formula candidates for the contest along with factors which tend to increase or decrease confidence.
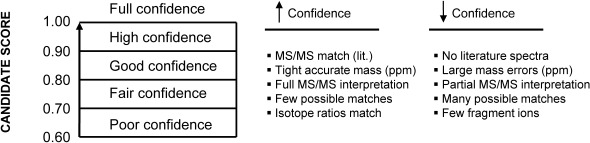
Where several possible structural isomers existed that matched the challenge data, isomers that were thought to be more likely were placed in a higher ranking bracket. In cases where there were many possible structures that could potentially match the challenge data, the confidence score was lowered accordingly. Structures placed in the same ranking bracket were regarded as equally likely. Structure candidates were submitted for all challenges except for Challenge 13.
RESULTS
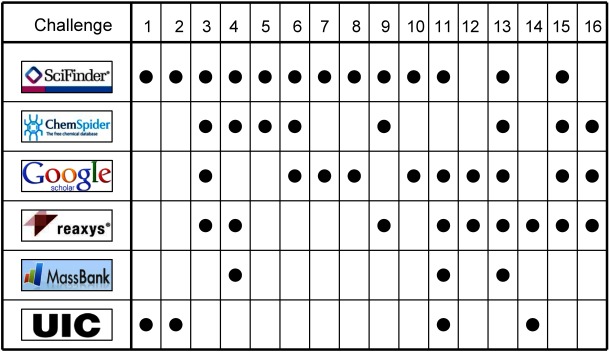
Table 1 summarizes the molecular formulas and lead structure candidates submitted by the team along with the confidence score assigned to the lead candidate and the total number of candidates submitted for each challenge. The four challenges where the formula was provided are noted. A single molecular formula was submitted per challenge and all with high (0.90+) or full (1.00) confidence. The chemical structures of the team’s leading Category 2 solutions are provided in Fig. 2. The lead candidate submitted was the correct solution for 12 of 12 formula submissions and for 14 of 15 structure submissions with the exception that our Challenge 3 candidate had an isoleucine (incorrect) rather than a leucine (correct) side chain. The lead candidate for Challenge 13 was correct but was not submitted. Figure 3 summarizes the resources employed which were of primary usefulness in arriving at a solution for each challenge. A detailed description of the various tools and thought processes used to arrive at the molecular formula and structure solutions for each challenge are provided herein.
Table 1. Summary of CASMI 2013 submissions (Newsome/Nikolic).
| No. | Category 1 | Score | N (cat 1)a | Category 2 | Score | N (cat 2)a |
|---|---|---|---|---|---|---|
| 1 | C18H19N1O4 | 1.00 | 1 | N-trans-Feruloyltyramine | 0.90 | 2 |
| 2 | C14H20N2O3 | 1.00 | 1 | N-trans-Feruloylputrescine | 0.90 | 2 |
| 3 | C13H24N4O4 | 0.90 | 1 | N2-Acetyl-glutaminylisoleucinamideb | 0.60 | 1 |
| 4 | C15H14O1 | 0.95 | 1 | Dihydrochalcone | 0.75 | 1 |
| 5 | C12H18O4S2 | 0.95 | 1 | Isoprothiolane | 0.90 | 3 |
| 6 | C49H93O14P1 | 1.00 | 1 | Phosphatidylglucoside (PtdGlc) | 1.00 | 1 |
| 7 | C75H62O30c | 1.00 | 1 | (Epi)catechin pentamer: B type | 0.90 | 7 |
| 8 | C45H38O20c | 1.00 | 1 | (E)GC-(E)GC-(E)C | 0.90 | 3 |
| 9 | C9H11Cl3N1O3P1S1 | 0.95 | 1 | Chlorpyrifos | 0.95 | 6 |
| 10 | C42H71N11O12 | 0.90 | 1 | Peptide VHLTPVEK | 0.90 | 2 |
| 11 | C20H18O5 | 1.00 | 1 | Desmethoxycurcumin | 0.95 | 3 |
| 12 | C15H10O5 | 1.00 | 1 | Baicalein | 0.91 | 3 |
| 13 | C17H30N2O5c | 1.00 | 1 | Aloxistatin | — | 0 |
| 14 | C21H24N2O3c | 1.00 | 1 | Almalicine | 0.90 | 1 |
| 15 | C10H5F17O1 | 0.90 | 1 | 8-2 Fluorotelomer alcohol (FTOH) | 0.90 | 3 |
| 16 | C18H20N3O4F1 | 0.90 | 1 | Ofloxacine | 0.85 | 1 |
a. Number of candidates submitted. b. The correct solution was leucinamide rather than isoleucinamide. The submitted was designated by the organizing team as a “highly commended” structure. c. The formula of the [M+H]+ ion was provided in the challenge.
Fig. 2. Chemical structures of leading solutions (Newsome/Nikolic) for CASMI 2013 Challenges 1 through 16 (Category 2).

Fig. 3. The databases or resources that were of primary use in arriving at the lead candidate for each (Category 2) structure solution.
Challenge 1. A search of the tandem mass data for Challenge 1 in the in-home database of tandem mass spectra returned several hits all matching ferulic and isoferulic acid amides. When the molecular mass was included in the search, hits were obtained for feruloyl and isoferuloyl tyramine. The elemental composition of these compounds fit the provided accurate mass data well. The only points of ambiguity were the identity of the acid moiety (feruloyl or isoferuloyl) and the cis or trans configuration of the double bond. Regarding the first issue, the provided information that the compound was isolated from Solanaceae was evidence in favor of the ferulic acid analog since this is the analog known from Solanaceae. However, it is worth discussing these isomers from a purely mass spectrometric point of view.
Over the years we have extensively worked with this compound class and have observed trends that can help distinguish ferulic from isoferulic acid amides.1) Ferulic acid amides tend to produce a higher abundance of the secondary fragment ions of m/z 149, 145, and 117 arising from the fragmentation of the primary acylium ion of m/z 177. In addition, a small but very diagnostic ion of m/z 163 is formed only during fragmentation of isoferulic acid amides. These trends were established for a Q-TOF instrument, but the challenge data were obtained using an IT-TOF. Therefore, we acquired tandem mass spectra of both feruloyl and isoferuloyl tyramine on a Shimadzu IT-TOF instrument. The only difference in the tandem mass spectra between the two analogs was a slightly higher intensity of the peak at m/z 145 in the spectrum of feruloyltyramine. Thus, a database search or computational approaches alone would likely produce both isomers as matches to the data. The second issue, the cis or trans configuration of the double bond, cannot be resolved by the provided data. Since only a few natural cis analogs have been isolated and nearly all of the natural analogs are trans, our top ranked submission was trans-N-feruloyltyramine.
We arrived at the same formula and structure candidates for Challenge 1 without consulting the in-house database and that effort is described here as well. The molecular mass of the neutral molecule was determined as 313 by the positive mode protonated/sodiated adduct pattern. Of the three molecular formulas (C, H, N, O atoms only) within 10 ppm, two were reasonable: C13H19N3O6 (5.6 ppm) and C18H19NO4 (7.3 ppm). Using the molecular compositions of the fragment ions as a filter, the formula C18H19NO4 was established as the better match. The other formula C13H19N3O6 could not be conclusively ruled out. Searching the formula C13H19N3O6 in SciFinder produced 287 substance records. One substance (a peptide) was categorized as naturally occurring but it was not isolated from Solanaceae and the tandem mass spectrum did not match so the formula was eliminated. Therefore, the formula was concluded as C18H19NO4.
The formula C18H19NO4 when searched in SciFinder returned almost 7000 records. These records were limited to natural products using the “by occurrence” category (56 records). The two compounds with the highest number of associated references (1500 and 300) were eliminated by comparing their predicted fragmentation pattern with the provided data. References were retrieved for the remaining 54 substances (about 1,000 references) and this reference set was refined using the keyword Solanaceae (12 references). Substances were retrieved from these 12 references using the “get substances” function (97 substances) and then refined by the molecular mass range 312 to 314 to give two geometrically isomeric substances: N-cis-Feruloyltyramine and N-trans-feruloyltyramine. The result was confirmed with an orthogonal SciFinder scholar search beginning with the literature search “Solanaceae and alkaloids,” retrieval of all substances for the references returned, refinement of the substances using “by occurrence,” and refinement by the molecular mass range from 312 to 314 to again give N-cis-feruloyltyramine and N-trans-feruloyltyramine.
Challenge 2. Challenge 2 was identified using the same approach described for Challenge 1. This compound was also in our database, but here we present only the process for arrival at solutions using online external databases. The data showed the protonated molecule of m/z 265.1524 with only two reasonable formula possibilities (C, H, N, O atoms only) within 10 ppm. The most abundant ion in the tandem mass spectrum of m/z 177.055 (C10H9O3) was in common with Challenge 1. The only possible formula within 100 ppm for the corresponding neutral loss of 88.098 was C4H12N2 (25.5 ppm). The established the molecular formula was C14H20N2O3 (8.6 ppm). A similar SciFinder search process as described for Challenge 1 was used to find molecules with the formula C14H20N2O3 isolated from the family Solanaceae. A SciFinder search of the molecular formula led to over 11,000 substance records but only 14 remained after refinement by occurrence. Retrieval of all references (∼400) for these 14 substances and refinement of these references using the keyword “Solanaceae” led to two references. Retrieval of all substances from these two references and refinement by molecular mass of 263 to 265 provided two compounds. One compound, N-feruloylputrescine, was consistent with having an ion of m/z 177 as the major fragment. Orthogonal SciFinder searches beginning with “Solanaceae and alkaloids” and “Solanaceae and constituents” (similar to Challenge 1) did not produce any additional structural possibilities. As with Challenge 1, both the cis and trans isomers were submitted as candidates with the trans isomer given the higher rank under the same rationale.
Challenge 3. The positive ion mode pattern for the protonated/sodiated molecule established a monoisotopic mass for the protonated molecule at m/z 301.1843 within 10 ppm. The provided information that the compound contained one or more amide bonds gave formula constraints of at least one double bond equivalent, one nitrogen, and one oxygen. The formula C13H24N4O4 (9.10 ppm) was the only matching C, H, N, O formula within 10 ppm. The major fragment ion of m/z 171.0774 (C7H11N2O3, 5.77 ppm) and minor ion of m/z 131.1189 (C6H15N2O, 7.76 ppm) further supported the formula. There was no evidence of other heteroatoms. A search of the formula C13H24N4O4 in SciFinder returned 232 substances. The tandem mass spectrum of the unknown showed a loss of ammonia and one major central cleavage site to form a major ion of m/z 171 (C7H11N2O3) with a neutral loss of 130 amu (C6H14N2O). One synthetic peptide derivative, N2-acetyl-glutaminylisoleucinamide, was found that could reasonably be expected to lose ammonia and also produce a major y-type ion of m/z 171. The structure was submitted as the candidate but with the lowest confidence score (0.60) because of the limited fragment information, the relative obscurity of the compound, and because a large number of possible synthetic substances could also cleave to provide the two fragments observed in the unknown. The correct structure solution was N2-acetyl-glutaminylleucinamide rather than N2-acetyl-glutaminylisoleucinamide. However, a leucine versus an isoleucine side chain could not be distinguished from the limited data information provided. This was acknowledged by the organizing team and the structure submission was given credit as highly commended.
Challenge 4. The positive ion mode pattern for protonated/sodiated molecule established a monoisotopic mass for the protonated molecule of 211.11214 within 3 ppm for which there was only one possible C, H, N, O formula: C15H14O (1.70 ppm). The formula was further supported by the accurate mass of the fragment ion of m/z 91 representing a tropylium ion (C7H7, 0.30 ppm) with a neutral loss of C8H8O (0.12 ppm), and also the fragment ion of m/z 105, C8H9 (1.18 ppm), with a neutral loss of C7H6O (1.76 ppm). A SciFinder search of the formula provided over 1500 registered substances. The simple fragmentation pattern was strongly suggestive of two benzene rings. Many structural elements such as acetyl, methoxy, and stilbene functionalities were unlikely and could be ruled out. The compound dihydrochalcone was concluded as the structure most consistent with the provided data. However, because of the limited structural information provided by the fragmentation data and large number of formula isomers, the submission was given a confidence score of “fair” (Score=0.75).
Challenge 5. The positive ion mode pattern for protonated/sodiated molecule established a monoisotopic mass for the protonated molecule at m/z 291.07187 within 3 ppm for which there were many formula options. However, accurate mass fragment and neutral loss analysis conclusively provided the formula C12H19O4S2 with a key fragment ion of m/z 189 having only one reasonable formula option of C6H5O3S2 (0.33 ppm). The formula was further supported by the 8% abundance of the m+2 peak consistent with two sulfur atoms. ChemSpider and SciFinder searches of the formula C12H18O4S2 returned 24 and 144 hits, respectively. The fragmentation pattern with losses of C3H6 and C3H8O and the loss of C6H14O to form the ion of m/z 189 was analogous to the fragmentation of dipropylphthalate which loses C6H14O to form the ion of m/z 149. The structure candidate most consistent with the fragmentation pattern was the pesticide isoprothiolane. This was submitted as the lead candidate with high confidence (Score=0.90) along with two equally consistent but less common alkyl isomers (assigned lower scores).
Challenge 6. The base peak at m/z 935.6224 was assumed to be the deprotonated molecule while keeping in mind a possibility of an [M+acetate]− adduct since ammonium acetate was used in the mobile phase. A table of the tandem MS fragmentation data was constructed to scrutinize formulas of fragment ions and neutral losses. The analysis along with an accurate mass formula calculator revealed the product ion formulas of C20H39O2 and C18H35O2 corresponding to probable saturated fatty acid chains. Also, the fragment ion of m/z 152.9958 yielded few if any sensible formula matches with the elements of C, H, O, and N. The compound having been isolated from animal tissue and containing lipid chains was suggestive of a phospholipid molecule. Adding phosphorus to the formula calculator provided the formula of C3H6O5P for the fragment ion of m/z 152.9958. Next, the literature was consulted and negative mode tandem mass spectra of phospholipids with similar fragmentation patterns and fragment ions in common with the unknown (m/z 153 and 419) were found. These searches established the molecule as a probable phospholipid. The formula calculator was constrained to require one phosphorus atom and 5 ppm accuracy (a safe assumption of performance for an FT-ICR instrument) to give C49H93O14P as the most logical formula. Other formula possibilities were not eliminated until the structure was solved. The formula C49H93O14P was searched in the SciFinder database which returned only one reference describing a neurophospholipid isolated and characterized from murine brain tissue. The tandem mass spectrum of the molecule in the reference matched the Challenge 6 data precisely.2) Normally, other minor isomers of the phospholipid molecule would be considered. However, in this case the accurate mass of the deprotonated molecule (m/z 935.62239) and the abundance of the product ions in the challenge data exactly matched the data in the original research article. Incidentally, the lone reference could be quickly retrieved by searching “935.62239” in Google Scholar. The compound, phosphatidyl-6-acetyl-glucose (18 : 0/20 : 0), was submitted with full confidence (Score=1.00).
Challenge 7. The molecular formula C75H63O30 for the protonated molecule was provided. Based upon the formula, the fragmentation pattern, and wine as the natural source, the compound was determined to be a condensed tannin. Sixteen condensed tannin entries were returned for the formula C75H62O30 and all were pentamers of (epi)-catechin. Consultation of the literature on the tandem mass fragmentation patterns of condensed tannins was very helpful.3–5) An A-type condensed tannin could be ruled out based on the formula and MS2 fragmentation. Based on the data, the tannin does not contain (epi)-gallocatechin units. The B-type condensed tannins may consist of both C4–C8 linkages and C4–C6 linkages. The most common pentamer consists of entirely C4–C8 linkages. However, C4–C6 linkages do occur, and tandem mass spectrometry thus far cannot distinguish between these two types of linkages. Therefore, the fully C4–C8 linked B-type (epi)-catechin pentamer was submitted as the lead candidate (Score=0.90) along with six less common B-type pentamers containing one or two C4–C6 linkages as lower ranked isomer candidates.
Challenge 8. The formula C45H39O20 for the protonated molecule was provided. The compositional monomers of the condensed tannin were determined by comparing the provided tandem mass spectrum with published data on sequencing of proanthocyanidins using collision induced dissociation in positive ion mode.5) The tandem mass spectrum was most consistent with a trimer consisting of one (epi)-catechin monomer and two (epi)gallocatechin monomers based on the characteristic fragment ions of m/z 609, 595, 443, and 317. The most likely sequence based upon the fragmentation data and the isolation from hops was (E)GC-(E)GC-(E)C and this structure was submitted as the lead candidate (Score=0.90). The two other sequences (E)C-(E)GC-(E)GC and (E)GC-(E)C-(E)GC were submitted with much lower confidence scores.
Challenge 9. In this case the formula of the compound was determined simultaneously with the structure. The provided mass spectrum showed a distinct m+2 and m+4 isotope pattern indicative of the presence of three chlorine atoms. Also, the best elemental composition match for the neutral loss of 152.0062 Da to give the product ion of m/z 197.9275 was C5H3NOCl3. Next a table of fragment ions and neutral losses was constructed and analyzed for possible formula pairs at each major fragment ion. The analysis produced a best logical formula of C8H10N3O2S2Cl3. However, the search of this formula and other candidates in SciFinder did not yield any feasible structural results. With initial approach a dead end, the possible presence of heteroatoms besides N, O, S, and Cl was suspected. Next, the accurate mass (349.9337±0.0010) Da was searched in ChemSpider. The search resulted in 12 hits. One of the hits, a pesticide chlorpyrifos C9H11Cl3NO3PS, was consistent with the trichlorinated isotope pattern and the molecular composition of the base peak in the tandem mass spectrum (C5H3NOCl3 at m/z 197.9275). With the incorporation of a phosphorous atom in the accurate mass formula calculations, the product ions and corresponding neutral losses were consistent with the structure of chlorpyrifos. The compound was submitted as the best matching candidate structure (Score=0.95) along with five closely related but unlikely structural isomers.
Challenge 10. An initial ChemSpider search of the presumed protonated molecule of m/z 922.53505 (within 3 ppm) returned three structure results. Two were peptides with the formula C42H71N11O12. Since the fragmentation data was suggestive of a peptide, this was very likely the correct formula and was finalized after reference spectrum was found for the Category 2 structure. The molecular formula C42H71N11O12 was searched in SciFinder and returned 32 different peptides. Two of the peptides, VHLTPVEK and VHLTPEVK, showed good consistency with the fragmentation data. The sequences were searched using the NIST peptide tandem mass spectral library (peptide.nist.gov/). The NIST spectrum for the human peptide VHLTPVEK was a very good match with the experimental challenge data. The reference spectrum for VHLTPEVK was not available. Both peptides were submitted with VHLTPVEK given the higher score (Score=0.90) because of the greater number of associated references. The remaining 30 peptide structures were not consistent with the provided tandem MS fragmentation data.
Challenge 11. A ChemSpider search of the presumed deprotonated molecule of m/z 337.1080 returned over 2000 results within a 10 ppm mass error tolerance. The vast majority had the formula C20H18O5. The formula calculator provided multiple C, H, N, O formula options within 10 ppm for the monoisotopic mass. Accurate mass analysis of the fragment ions and neutral losses eliminated the other possibilities to confirm the formula C20H18O5 (0.43 ppm). A SciFinder search of the formula C20H18O5 returned almost 2000 entries. The substances were sorted by descending number of references. The most referenced substance was the natural product desmethoxycurcumin. The formula was also searched in the Reaxys database. The Reaxys search led to a publication containing the negative ion tandem mass spectrum of desmethoxycurcumin.6) The literature spectrum was a very good match to the challenge data, with the fragment ions of m/z 217, 187, 173, 143, and 119 present with the same overall intensity pattern. Desmethoxycurcumin (Score=0.95) and two tautomeric structures thereof were submitted as the candidates. The assignment was further strengthened by comparison of the data with the in-house tandem mass spectrum of the analog curcumin. The submission was made with the apprehension that well over one hundred theoretical isomers could be drawn that would produce similar or identical fragmentation spectra from permutations of cis–trans isomerism, ring substitution patterns, and tautomeric forms. All three tautomers of desmethoxycurcumin were accepted as structure solutions by the CASMI organizers.
Challenge 12. Based on pattern recognition from experience with natural products, the negative mode mass of 269 and fragmentation pattern resembled that of a flavonoid. Formula calculation provided the formula C15H10O5 as the best match within 5 ppm. The formula became certain only after good agreement was found of the challenge data with published tandem mass spectra. A search of the formula in Reaxys produced 84 isolated natural product hits. Many of them were flavonoids. Literature review on the fragmentation patterns of different classes of flavonoids (isoflavones, chalcones, flavanols, etc.) was helpful in establishing a probable flavone subclass.7,8) A publication comparing fragmentation patterns of flavones with different hydroxyl group substitution patterns9) revealed that only flavones containing both a trihydroxy substituted A ring and unsubstituted B ring (baicalein and norwogonin) matched the major fragment ions of m/z 251, 241, 223, and 197. Only 3 such isomers are possible. The tandem mass spectra for the compounds was confirmed in a second reference.10) No other compounds of the flavonoid class were found with matching tandem mass spectra. Baicalein (5,6,7-trihydroxyflavone) and norwogonin (5,7,8-trihydroxyflavone) were submitted as the lead candidates with high confidence. The third isomer (5,6,8-trihydroxyflavone), an uncommon and apparently not naturally occurring substance, was submitted with a much lower score. The leading candidate, baicalein (Score=0.91), was the correct solution.
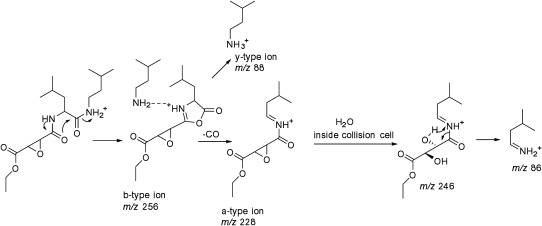
Challenge 13. The team could not conclusively solve this challenge. The molecular formula C17H31N2O5 for the protonated molecule was provided. The SciFinder search of this formula returned 364 substances. When the substances were sorted based on the number of references, the correct solution aloxistatin was the top hit. This choice was seriously considered as a possible solution, but the team decided against submitting it as a solution due to the inability to correctly interpret the provided tandem mass spectrum. The most intense peak in the spectrum (m/z 246) corresponded to a neutral loss of C6H11N. All possible structures that could directly produce such a loss were examined but none were consistent with the rest of the fragmentation data. The cleavage at the amide bond in aloxistatin would produce a fragment with 5 (not 6) carbon atoms. Since the major neutral loss of C6H11N could not be reconciled, aloxistatin was not submitted as a solution. Post mortem when solutions were revealed, the team purchased the standard and acquired tandem mass spectrum on a Q-TOF instrument. The spectrum contained a fragment ion at m/z 88.1130 with the molecular composition of C5H14N corresponding to the y type ion formed by cleavage of the terminal amide bond. This was the ion that the team had anticipated. This challenge is a good example of well-known problem in identification of small molecules using tandem mass spectrometry in that different instruments produce different tandem mass spectra which in this case provide different informational value. The team was intrigued by the fragmentation pattern of this molecule and are currently working on detailed elucidation of the origin of major fragment ions. The current working hypotheses for the origin of ion of m/z 246 is that it is formed by addition of water inside the collision chamber to the a-type ion of m/z 228 as shown in Fig. 4. This challenge also demonstrates a disadvantage of our approach and a challenge with tandem mass spectrometry in general. The occurrence of unusual fragmentation pathways such as rearrangements or secondary ion-molecule reactions often makes interpretation difficult or misleading.
Fig. 4. Proposed mechanism for formation of major ions in the tandem mass spectrum of aloxistatin. The ion of m/z 256 representing b-type fragment was not shown in the challenge data and it was a minor fragment in the Q-TOF instrument. The b-ion is likely held in a protonated dimer form with the neutral amine (C5H13N) from which hydrogen transfer can occur to form a y-type of m/z 88. This diagnostic ion was observed in the Q-TOF instrument but was not part of the challenge data. The a-type ion can be attacked by water present in the collision cell to produce ion of m/z 246.
Challenge 14. The formula C21H25N2O3 for the protonated molecule was provided. Since the challenge was described as a natural product, the provided formula C21H24N2O3 was searched in the Reaxys database. The ion of m/z 144 was also searched against our in-house database of tandem spectra of alkaloids. The presence of this ion suggested an indole type alkaloid. The ion of m/z 321 represents the loss of methanol from the precursor ion. With these constraints and based on the overall interpretation of the tandem mass spectrum the most likely structure was that of ajmalicine. The provided spectrum matched the published spectrum of ajmalicine.11) Ajmalicine was submitted with high confidence (Score=0.90) with the constraint that there are numerous epimers of this compound available and that in real life no definitive conclusion can be made regarding exact stereoisomer/epimer.
Challenge 15. The nominal mass for this challenge was provided. The accurate mass for the deprotonated molecule of m/z 462.99731 produced many formula possibilities. Although both unit resolution triple quadrupole MS/MS data and high resolution Orbitrap data were provided, this challenge was solved primarily using the accurate mass data. The ion of m/z 68.99463 represents a CF3− ion indicating that the compound contains fluorine. Careful analysis of the accurate mass and tandem mass fragmentation data revealed ion formulas and neutral losses consistent with a polyfluorinated substance. Exact mass differences between fragment ions were analyzed in detail and revealed mass differences corresponding to F2, HF, and C2F2. The formula C10H5F17O1 was concluded only after the reference spectra for the leading structure candidate was found as follows. A ChemSpider search of m/z 462.9973±0.005 with the constraint that the molecules contained fluorine but no other halogens provided 10 compounds. The results led to the investigation of the straight chain perfluorinated alcohols. A reference negative ion electrospray CID tandem mass spectrum for 8-2 fluorotelomer alcohol was found that matched the provided data very well.12) After a SciFinder search of the formula to investigate potential isomers, 8-2 fluorotelomer alcohol was concluded as the leading structure candidate with high confidence (Score=0.90) along with two uncommon structural isomers score with poor confidence.
Challenge 16. Similarly to Challenge 15, this challenge was solved primarily by using the accurate mass data obtained on the Orbitrap instrument. Accurate masses of all fragments were checked to determine what atoms were present in the molecule. A particularly diagnostic ion was that of m/z 122.04078. The accurate mass of this ion did not return any hits when standard atoms (C, H, N, O, S) were considered. However, when fluorine was added this ion had the best formula fit of C7H5NF. Thus, the compound contained at least one fluorine, one nitrogen, and also a minimum of two oxygen atoms (loss of CO2, m/z 318). When the search of the molecular formula was constrained to contain at least one fluorine and one nitrogen atom, the best fit to the challenge data was C18H20N3O4F. ChemSpider searches of the accurate mass of the protonated molecule of m/z 362.1525±0.002 and of the candidate formula revealed fluoroquinolone antibiotics such as ofloxacin and levofloxacin as hits with the highest number of associated references. The literature was searched for published tandem mass spectra of these fluoroquinolones and compared with the provided data with the best match to ofloxacin.13–15) The spectral match further supported the proposed molecular formula. Ofloxacin was submitted as the sole structure candidate with good confidence (Score=0.85). Isomeric levofloxacin could not be fully differentiated based on the provided data.
DISCUSSION
Mass spectrometry has become an indispensable tool for identification of small molecules, particularly when sample size is limited. The ever growing list of applications that rely almost exclusively on mass spectrometry includes such diverse areas as metabolomics, forensic science and archeological research. Correct identification of unknown compounds is certainly the most desirable result; however, in many applications even a partial structural characterization may be sufficient. For example, in natural products research, phytochemists are often interested in quickly obtaining dereplication information to determine common compounds and compound classes in order to focus further efforts on the most promising compounds. In drug metabolism studies researchers sometimes want to quickly identify soft spots in a drug candidate without necessarily determining the exact position of metabolic transformation. On the other side of the spectrum, the correct identification of a compound is paramount such as, for example, in forensic sciences. No one wants to accuse an athlete of cheating based on the misidentification of a prohibited substance.
Unfortunately, for all its power, mass spectrometry is not a de novo structure elucidation technique and is thus primarily used for identification of known compounds. This process is an exercise in comparative analysis in which data obtained from the unknown are compared with those from already identified and characterized compounds. Because of this inherent weakness, structural assignments by mass spectrometry always come with a degree of uncertainty. The degree of (un)certainty will vary depending on how much information obtained for the unknown can be matched to the available data for the known compound. Currently, the most commonly accepted nomenclature for metabolite annotation includes three levels of reporting.16) Identification at level 1 is established by comparing the retention time and fragmentation pattern of an unknown with those of an authentic standard. This level of evidence provides the highest degree of confidence in the assignment and is a widely accepted criterion for positive identification of compounds not only in the research domain but also in forensic and regulatory areas. Level 2 and level 3 represent putative identification whereby a compound (level 2) or compound class (level 3) is identified based on databases and spectral library searches but without having an authentic standard to compare under identical conditions. In a more recent commentary, Schymanski et al. argue for a finer scale that includes five levels of confidence.17) We believe that this finer scale is more appropriate as it better captures the reality in which there are many shades of gray as to how well the available data can differentiate between structural possibilities. We also propose that information on the origin of the sample should also be taken into account when evaluating uncertainty in the assignment. For example, in natural product research many compounds are uniquely present in a certain plant, thus even a molecular formula alone may provide very strong evidence for the proposed assignment.
When discussing how to best describe strength of analytical evidence, the terms “level of confidence” and “confidence” are often used interchangeably but there may be subtle difference between the two. Level of confidence is a specific descriptor as to what information is available to support the proposed assignment; confidence is more of a qualitative term that includes investigator’s assessment as to how well the available data fit the proposed structure. It is precisely this aspect that makes the CASMI contest a valuable exercise. The information on which the assignments in the contest are made would qualify all submissions as level 2 annotations. Thus, the contest in essence provides assessment as to how different investigators use the data to arrive at a conclusion and how reliable those conclusions are. For the purpose of this contest we devised an arbitrary scale that simply reflected how confident we felt that the proposed structure is right. While the scale at this point is more or less qualitative, in the future we plan to develop a more quantitative scale that will assign point value to each piece of information used to arrive at the proposed structure.
As evident from the above discussion, our approach takes into account all available information to arrive at the proposed structure. The key aspect of the approach is the investigator’s experience and skill in interpretation of tandem mass spectra. The overall approach is in essence a method of elimination of possibilities that obviously do not fit the data until only a handful of probable structures remains that can be further scrutinized in more detail. The approach works best when fragmentation pattern contains diagnostic ions that can be predicted from a candidate structure. As Challenge 13 demonstrated, if unusual rearrangements occur that are not easy to predict from looking at the structure, the method is not always successful. Furthermore, the approach is labor intensive and slow. Some structure solutions here required as little as ten minutes while others required hours of dedicated manual effort to arrive at a solution. As such, it is most suitable for applications that place a premium on accuracy over speed. Another side benefit of this approach is possibility to identify novel compounds. Since the available data are thoroughly scrutinized through all available sources of prior knowledge, it is possible to determine with great confidence whether the unknown at hand is a new compound and even identify a structure if the fragmentation pattern is similar to a known structure.1)
CONCLUSION
The manual methods described here are very effective at arriving at a small number of good structural candidates. Experienced mass spectrometrists can arrive at a small set of candidate structures often containing the correct structure because they are able to employ far more flexibility and customization in the overall manual search process than the current automated methods. Unlike current computer automated platforms, humans are able to employ the full use of more subtle contextual information by inference, recognition, experience, and logic. The approach is best suited for a small problem set because of the manual time and effort involved. A manual approach could be used in conjunction with and after application of an automated computerized method to greatly eliminate the number of structural candidates often generated by these approaches.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
We would like to thank the organizers and contributors of the CASMI 2013 contest for their efforts in making this contest possible. We also thank the Office of Dietary Supplements and the National Center for Complementary and Alternative Medicine for financial support (NIH grant P50 AT00155).
A. G. Newsome and D. Nikolic, CASMI 2013: Identification of Small Molecules by Tandem Mass Spectrometry Combined with Database and Literature Mining, Mass Spectrom (Tokyo) 2014; 3(3): S0034; DOI: 10.5702/massspectrometry.S0034
References
- 1) D. Nikolic, T. Godecke, S. N. Chen, J. White, D. C. Lankin, G. F. Pauli, R. B. van Breemen. Mass spectrometric dereplication of nitrogen-containing constituents of black cohosh (Cimicifuga racemosa L.). Fitoterapia 83: 441–460, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2) Y. Nagatsuka, Y. Horibata, Y. Yamazaki, M. Kinoshita, Y. Shinoda, T. Hashikawa, H. Koshino, T. Nakamura, Y. Hirabayashi. Phosphatidylglucoside exists as a single molecular species with saturated fatty acyl chains in developing astroglial membranes. Biochemistry 45: 8742–8750, 2006. [DOI] [PubMed] [Google Scholar]
- 3) L. Gu, M. A. Kelm, J. F. Hammerstone, Z. Zhang, G. Beecher, J. Holden, D. Haytowitz, R. L. Prior. Liquid chromatographic/electrospray ionization mass spectrometric studies of proanthocyanidins in foods. J. Mass Spectrom. 38: 1272–1280, 2003. [DOI] [PubMed] [Google Scholar]
- 4) Y. Hayasaka, E. J. Waters, V. Cheynier, M. J. Herderich, S. Vidal. Characterization of proanthocyanidins in grape seeds using electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 17: 9–16, 2003. [DOI] [PubMed] [Google Scholar]
- 5) H. J. Li, M. L. Deinzer. Tandem mass spectrometry for sequencing proanthocyanidins. Anal. Chem. 79: 1739–1748, 2007. [DOI] [PubMed] [Google Scholar]
- 6) R. Li, C. Xiang, M. Ye, H. F. Li, X. Zhang, D. A. Guo. Qualitative and quantitative analysis of curcuminoids in herbal medicines derived from Curcuma species. Food Chem. 126: 1890–1895, 2011. [DOI] [PubMed] [Google Scholar]
- 7) F. Cuyckens, M. Claeys. Mass spectrometry in the structural analysis of flavonoids. J. Mass Spectrom. 39: 1–15, 2004. [DOI] [PubMed] [Google Scholar]
- 8) N. Fabre, I. Rustan, E. de Hoffmann, J. Quetin-Leclercq. Determination of flavone, flavonol, and flavanone aglycones by negative ion liquid chromatography electrospray ion trap mass spectrometry. J. Am. Soc. Mass Spectrom. 12: 707–715, 2001. [DOI] [PubMed] [Google Scholar]
- 9) W. Wu, Z. Liu, F. Song, S. Liu. Structural analysis of selected characteristic flavones by electrospray tandem mass spectrometry. Anal. Sci. 20: 1103–1105, 2004. [DOI] [PubMed] [Google Scholar]
- 10) J. Han, M. Ye, M. Xu, J. Sun, B. Wang, D. Guo. Characterization of flavonoids in the traditional Chinese herbal medicine-Huangqin by liquid chromatography coupled with electrospray ionization mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 848: 355–362, 2007. [DOI] [PubMed] [Google Scholar]
- 11) F. Ferreres, D. M. Pereira, P. Valentao, J. M. Oliveira, J. Faria, L. Gaspar, M. Sottomayor, P. B. Andrade. Simple and reproducible HPLC-DAD-ESI-MS/MS analysis of alkaloids in Catharanthus roseus roots. J. Pharm. Biomed. Anal. 51: 65–69, 2010. [DOI] [PubMed] [Google Scholar]
- 12) B. Szostek, K. B. Prickett, R. C. Buck. Determination of fluorotelomer alcohols by liquid chromatography/tandem mass spectrometry in water. Rapid Commun. Mass Spectrom. 20: 2837–2844, 2006. [DOI] [PubMed] [Google Scholar]
- 13) D. A. Volmer, B. Mansoori, S. J. Locke. Study of 4-quinolone antibiotics in biological samples by short-column liquid chromatography coupled with electrospray ionization tandem mass spectrometry. Anal. Chem. 69: 4143–4155, 1997. [DOI] [PubMed] [Google Scholar]
- 14) M. L. Bandu, H. Desaire. The STEP method (statistical test of equivalent pathways): Application to pharmaceuticals. Analyst (Lond.) 131: 268–274, 2006. [DOI] [PubMed] [Google Scholar]
- 15) R. Diaz, M. Ibanez, J. V. Sancho, F. Hernandez. Building an empirical mass spectra library for screening of organic pollutants by ultra-high-pressure liquid chromatography/hybrid quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 25: 355–369, 2011. [DOI] [PubMed] [Google Scholar]
- 16) L. W. Sumner, A. Amberg, D. Barrett, M. H. Beale, R. Beger, C. A. Daykin, T. W. Fan, O. Fiehn, R. Goodacre, J. L. Griffin, T. Hankemeier, N. Hardy, J. Harnly, R. Higashi, J. Kopka, A. N. Lane, J. C. Lindon, P. Marriott, A. W. Nicholls, M. D. Reily, J. J. Thaden, M. R. Viant. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3: 211–221, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17) E. L. Schymanski, J. Jeon, R. Gulde, K. Fenner, M. Ruff, H. P. Singer, J. Hollender. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 48: 2097–2098, 2014. [DOI] [PubMed] [Google Scholar]