

Summary
Background
Familial cold autoinflammatory syndrome (FCAS) is an autosomal dominant disorder characterised by recurrent episodes of rash, arthralgia, and fever after cold exposure. The genetic basis of this disease has been elucidated. Cryopyrin, the protein that is altered in FCAS, is one of the adaptor proteins that activate caspase 1, resulting in release of interleukin 1.
Methods
An experimental cold challenge protocol was developed to study the acute inflammatory mechanisms occurring after a general cold exposure in FCAS patients and to investigate the effects of pretreatment with an antagonist of interleukin 1 receptor (IL-1Ra). ELISA, real-time PCR, and immunohistochemistry were used to measure cytokine responses.
Findings
After cold challenge, untreated patients with FCAS developed rash, fever, and arthralgias within 1–4 h. Significant increases in serum concentrations of interleukin 6 and white-blood-cell counts were seen 4–8 h after cold challenge. Serum concentrations of interleukin 1 and cytokine mRNA in peripheral-blood leucocytes were not raised, but amounts of interleukin 1 protein and mRNA were high in affected skin. IL-1Ra administered before cold challenge blocked symptoms and increases in white-blood-cell counts and serum interleukin 6.
Interpretation
The ability of IL-1Ra to prevent the clinical features and haematological and biochemical changes in patients with FCAS indicates a central role for interleukin 1β in this disorder. Involvement of cryopyrin in activation of caspase 1 and NF-κB signalling suggests that it might have a role in many chronic inflammatory diseases.
Relevance to practice
These findings support a new therapy for a disorder with no previously known acceptable treatment. They also offer insights into the role of interleukin 1β in more common inflammatory diseases.
Introduction
Familial cold autoinflammatory syndrome (FCAS), commonly known as familial cold urticaria, is an autosomal dominant inflammatory disease that has recently been included in the group of hereditary periodic-fever disorders. As in the more widely known familial Mediterranean fever and other inherited autoinflammatory syndromes, FCAS is characterised by recurrent bouts of fever, rash, and joint pain. However, in contrast to the other inherited autoinflammatory disorders, acute attacks in FCAS are consistently triggered by general cold exposure. Typically, FCAS patients report the development of general rash, low-grade fever, polyarthralgia, and in some cases conjunctivitis beginning 1–2 h after mild exposures such as air-conditioned rooms or cool breezes. These episodes generally resolve spontaneously within 24 h. Many patients with FCAS also show evidence of chronic inflammation between attacks, particularly a daily pattern of rash developing in the afternoon that can be associated with headaches, myalgia, and fatigue by the evening. The atypical urticarial rash in FCAS does not necessarily occur on exposed areas of skin, unlike the classic urticarial rash in the more common acquired cold urticaria, in which direct contact with cold objects causes pruritic hives at the site of exposure. Although colchicine prevents attacks in familial Mediterranean fever, there has been no known effective treatment for FCAS.1
There have been substantial advances in understanding of the genetic basis of the periodic-fever disorders in the past decade. The gene that causes familial Mediterranean fever was identified in 1997; it encodes a protein called pyrin,2,3 which interacts with and perhaps inhibits an intracellular adaptor protein called ASC that is involved in release of interleukin 1.4,5 In 2001, we identified the CIAS1 gene, which is mutated in patients with FCAS. Our group and others also identified CIAS1 mutations in two related inflammatory diseases, Muckle-Wells syndrome6-9 and neonatal-onset multisystem inflammatory disease (NOMID).10,11 CIAS1 codes for cryopyrin, which shares a similar N-terminal domain (PYD) with pyrin and ASC proteins.6 Interactions between these domains from different proteins are thought to mediate downstream signalling events.12 Cryopyrin interacts with ASC, leading to activation of caspase 1 and subsequent release of interleukin 1, as well as activation of nuclear factor κB (NF-κB) which results in the release of many proinflammatory cytokines such as interleukin 6 (figure 1).13,14 Because cryopyrin is positioned at a crucial convergence point in signal transduction, it could have a central role in the pathogenesis of many diseases that involve translocation of NF-κB and release of proinflammatory cytokines. Elucidation of the pathobiology of cryopyrin defects in FCAS might therefore provide clues to the mechanisms of more common diseases.
Figure 1. Proposed mechanism of cryopyrin function.
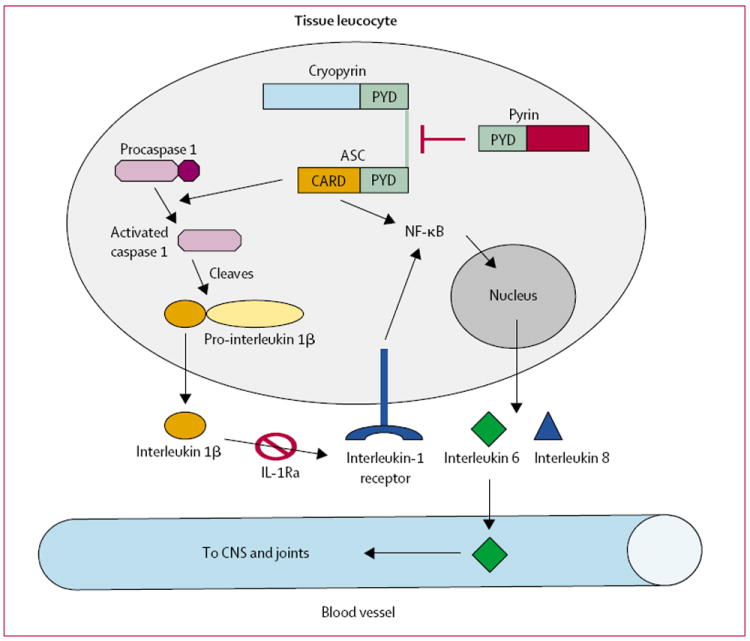
Cryopyrin interacts with ASC resulting in activation of caspase 1 or NF-κB, leading to the release of proinflammatory cytokines. Pyrin inhibits the interaction between cryopyrin and ASC. IL-1Ra competes with interleukin 1, for its receptor. Activation of interleukin-1 receptor also leads to NF-κB activation. Interleukin 6 is released into the blood, leading to fever and arthralgia.
Anakinra, a recombinant selective interleukin-1 receptor antagonist (IL-1Ra), has shown clinical efficacy in terms of symptoms and radiological progression in patients with rheumatoid arthritis. The proposed function of cryopyrin in the interleukin-1 pathway and the finding that cultured monocytes from a patient with NOMID had constitutively increased interleukin-1 production11 implied a potential therapeutic role for IL-1Ra. Hawkins and colleagues gave IL-1Ra to five patients with Muckle-Wells syndrome over a period of several months; the clinical efficacy was striking.15,16 In this study, we used a novel cold-challenge model to investigate the inflammatory responses in patients with FCAS and the effectiveness of IL-1Ra at preventing acute inflammation.
Methods
Patients
All participants in this study were recruited from a family affected by FCAS that has been reported and previously studied at the University of California at San Diego. Diagnosis of FCAS was confirmed by history and direct sequencing of CIAS1. All participants in this study with FCAS have an L353P mutation, which is the most common CIAS1 mutation in North America and is associated with a founder family that accounts for up to 90% of cases of FCAS worldwide.17 Disease severity was assessed by questionnaire and based on self-reports of frequency and severity of attacks and number and severity of symptoms. The Institutional Review Board of the University of California at San Diego approved this study (number 020732, 030381), and written informed consent was obtained from all participants. Other than FCAS, no participant had significant medical conditions. The ages ranged from 49 years to 74 years.
Procedures
The participants were admitted to the the University of California at San Diego General Clinical Research Center, and baseline samples of peripheral blood, skin by biopsy, and urine were obtained before cold challenge. The participants (four with FCAS and three controls) were exposed for 45 min to a room with an ambient temperature of 4°C (forced cool air) and humidity greater than 80%. All of the challenges were done between 1000 h and 1300 h. After the cold challenge, the participants returned to a room with ambient temperature above 25°C for the rest of the study. They were monitored except while asleep with hourly vital signs, symptom ratings (arthralgia), and physical examinations including affected skin surface area. Peripheral-blood samples were taken 1 h, 4 h, 8 h, and 18 h after cold challenge, and skin biopsies were done at the time of initial development of rash, which was within 2 h of the cold challenge. Three of the affected participants returned 6 months or 12 months after the initial challenge for a therapeutic trial with IL-1Ra. The protocol for the treatment trial was the same as the non-treatment cold challenge, except that the participants were given 100 mg subcutaneous doses of IL-1Ra 24 h and 1 h before the cold challenge.
Concentrations of serum amyloid A were measured in duplicate by ELISA (Biosource, Camarillo, CA, USA). Serum concentrations of cytokines (interleukins 6, 1β, and 8, and tumour necrosis factor α [TNF α]) were measured in duplicate by ELISA (R&D systems, Minneapolis, MN, USA). Peripheral-blood leucocytes were separated by centrifugation with Percoll according to the manufacturer’s protocol (Amersham Biosciences, Piscataway, NJ, USA). The granulocyte fraction was obtained by red-blood-cell lysis of the lower layer, and peripheral-blood mononuclear cells were collected from the upper layer. Purity (>98%) was confirmed by cytospins stained with Wright-Giemsa, and cell fractions were pelleted and snap frozen.
Total RNA was isolated from leucocytes by the Trizol Reagent protocol (Invitrogen, Carlsbad, CA, USA) and that from skin biopsy tissue by RNA Stat-60 (Tel-Test, Friendswood, TX, USA). RNA was reverse transcribed, and quantitative PCR (Applied Biosystems, Foster City, CA, USA) was done with a standard curve constructed from serial dilutions of cDNA from control peripheral-blood mononuclear cells as previously described.18 Data are expressed as relative expression units (REU) normalised to glyceraldehyde-3-phosphate dehydroge-nase.18
Other skin biopsy samples were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned, mounted on slides, and dehydrated before analysis. Sections were stained with haematoxylin and eosin for histology. Tissue cytokine staining was done with primary antibodies at 0·01 g/L (R&D systems) to interleukin 6 (AF-206-NA), interleukin 1β (AF-201-NA), interleukin 8 (AF-208-NA), caspase-processed interleukin 1β (D116) at 1 in 50 dilution (Cell Signaling, Beverly, MA, USA), secondary biotinylated rabbit antibody to goat IgG or goat antibody to rabbit IgG at 1 in 200, and peroxidase-labelled avidin detection (Vector ABC CPK-6105, Vector Laboratories, Burlingame, CA, USA). Haematoxylin was used for counter-staining. Skin samples from controls and isotype-matched antibodies were used in control experiments.
Statistical analysis
All statistical analysis was done with JMP statistical software (SAS, version 4.0.4). Repeated-measures ANOVA followed by appropriate post-hoc tests were used to analyse time series data. mRNA concentrations in skin were compared by single ANOVA on log-transformed data to obtain homogeneity of variance. All statistical tests were two-tailed.
Role of the funding source
The sponsors of the study had no role in the study design; collection, analysis, or interpretation of data; or the writing of this report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
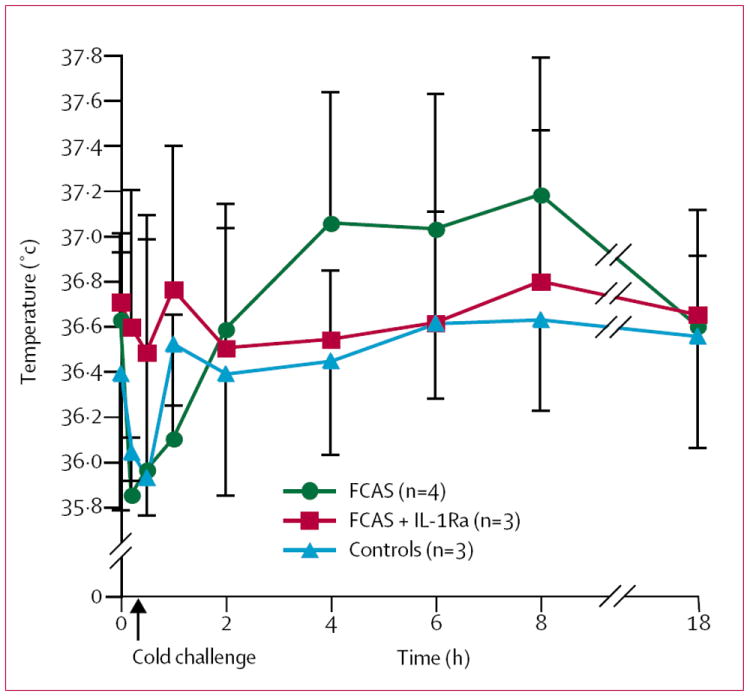
Disease severity was similar among the individuals with FCAS. There are no reported cases of renal amyloidosis in this family, and there was no evidence of proteinuria in the affected individuals. There was no significant difference in baseline erythrocyte sedimentation rate or concentrations of C-reactive protein, haptoglobin, or serum amyloid A between FCAS and control participants. One FCAS participant had a high concentration of serum amyloid A (table). There were no significant changes in these inflammatory markers after the cold challenge (data not shown). There was no significant difference between patients with FCAS (treated or untreated) and controls in oral (figure 2) or skin surface (data not shown) temperature at baseline or during cold exposure. The patients with FCAS, when subjected to cold challenge without treatment, developed low-grade fever beginning 2 h after the challenge, peaking at about 8 h, and abating by the next morning (figure 2). Neither controls nor IL-1Ra-treated FCAS patients patients developed fever.
Table.
Characteristics of participants at baseline
| Participant | Age, years | Sex | Number of affected joints | Severity score* | WBC, × 109/L | ESR, mm/h† | CRP, mg/L† | Haptoglobin, g/L† | SAA, mg/L |
|---|---|---|---|---|---|---|---|---|---|
|
FCAS
| |||||||||
| 1 | 74 | F | 4 | 7·5 | 8·8 | 25 | 37 | 1·99 | 293 |
| 2 | 72 | M | 2 | 5 | 9·9 | 18 | 9 | 1·58 | 59 |
| 3 | 55 | F | 4 | 6 | 8·5 | 30 | 16 | 2·30 | 98 |
| 4 | 49 | F | 2 | 8 | 4·2 | 20 | 18 | 1·44 | 57 |
|
| |||||||||
|
Control
| |||||||||
| 5 | 74 | M | 0 | 0 | 5·9 | 2 | <5 | 0·78 | 20 |
| 6 | 54 | M | 0 | 0 | 5·2 | 15 | 22‡ | 1·68 | 79 |
| 7 | 49 | M | 0 | 0 | 6·2 | 34 | 35‡ | 2·56 | 63 |
WBC=white-blood-cell count; ESR=erythrocyte sedimentation rate; CRP=C-reactive protein; SAA=serum amyloid A.
Based on self-reported analogue scale (1–10).
Normal range: ESR 0–30 mm/h, CRP 0–10 mg/L, haptoglobin 0·16–2·00 g/L.
No history of inflammatory disease, but both controls were slightly obese.
Figure 2. Mean oral temperature before, during, and after cold challenge.
Rash and arthralgia followed a pattern similar to the fever in patients with FCAS when untreated, with evidence of rash beginning 1 h after cold challenge, joint pain beginning after 2 h, and both symptoms peaking at 8 h (data not shown). Rash developed mainly on the trunk and limbs and, as previously described, was no more apparent on skin surfaces exposed during the cold challenge than on covered surfaces (figure 3). There was no joint swelling or effusion, but there was some evidence of mild general hand swelling and non-articular tenderness in two of the untreated FCAS patients about 8 h after cold challenge. One untreated FCAS patient developed acute painful bilateral conjunctivitis 8 h after the cold challenge. Rash, arthralgia, and conjunctivitis resolved completely by the next morning, as is commonly reported by patients with FCAS.
Figure 3. Appearance of skin (upper arm) of a patient with FCAS after cold challenge.
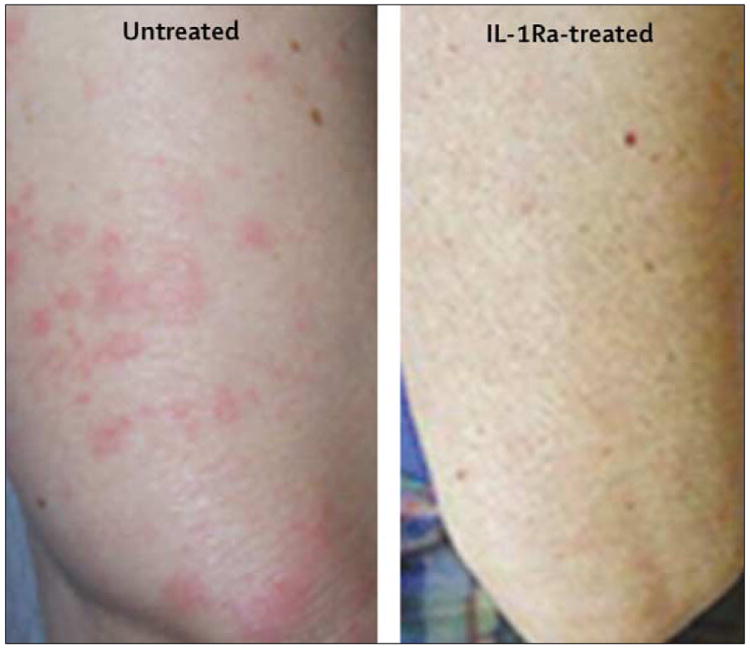
The three patients with FCAS treated with IL-1Ra before cold challenge did not develop any of the signs or symptoms that they developed during their previous cold challenge despite the same exposure and similar baseline degree of inflammation (figure 3). These participants did develop their usual mild evening rash after the initial dose of IL-1Ra but had no symptoms at all after the second dose. They remained symptom free for 24–48 h after the second dose, and the regular cold-induced symptoms and daily rash then returned. The participants also described having less fatigue and a general sense of well-being that lasted for 48–72 h after the second IL-1Ra dose.
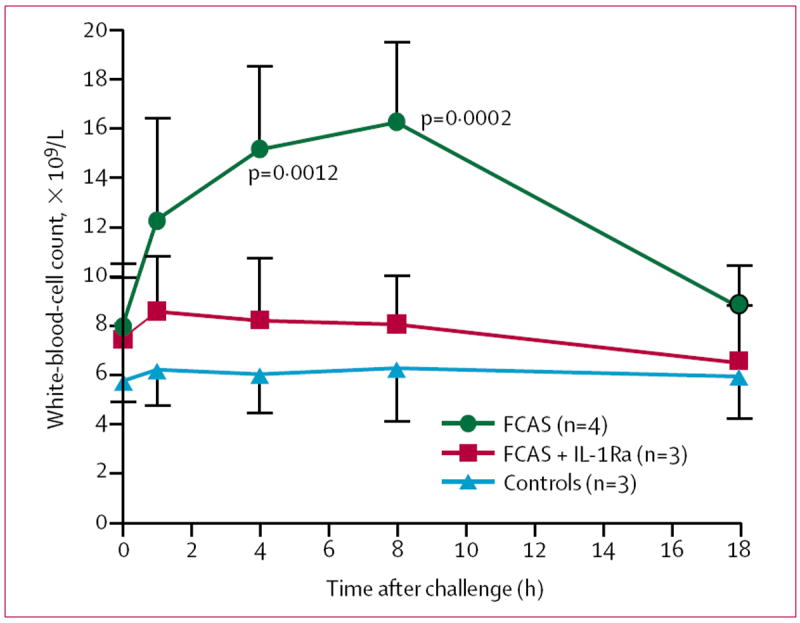
Before cold challenge, total white-blood-cell counts were slightly higher in patients with FCAS than in controls. Mirroring signs and symptoms, white-blood-cell counts of untreated FCAS participants increased within 1 h of cold challenge, peaked at 8 h, and returned to baseline by the next morning (figure 4). The increase in total count was due primarily to an increase in circulating neutrophils, with increases of more than 100% over baseline in absolute neutrophil counts in all FCAS participants 8 h after cold challenge. At 4 h and 8 h, the mean white-blood-cell count was significantly higher in untreated FCAS participants than in controls. There was no significant change in total white-blood-cell counts after cold challenge in IL-1Ra-treated FCAS participants or in controls (figure 4).
Figure 4. Mean white-blood-cell counts before and after cold challenge.
To elucidate the mechanisms underlying the systemic inflammatory response in FCAS, we measured serum cytokine concentrations in all participants at each time point. Serum concentrations of interleukin 6 at baseline did not differ between untreated FCAS participants, controls, and IL-1Ra treated FCAS patients. In untreated FCAS participants, serum concentrations of interleukin 6 increased substantially within 1 h of cold challenge, peaked at 4 h, and returned to baseline values by the next morning (figure 5). No significant change in serum concentrations of interleukin 6 was observed after cold challenge in the FCAS participants who received IL-1Ra or in controls. Serum concentrations of interleukin 1β were below the level of detection (2 ng/L) at all time points. There were low, but detectable serum concentrations of interleukin 8 and TNFα, but there were no differences between FCAS and control participants or between baseline and post-challenge concentrations in FCAS or control participants.
Figure 5. Mean change from baseline in serum concentrations of interleukin 6 after cold challenge.
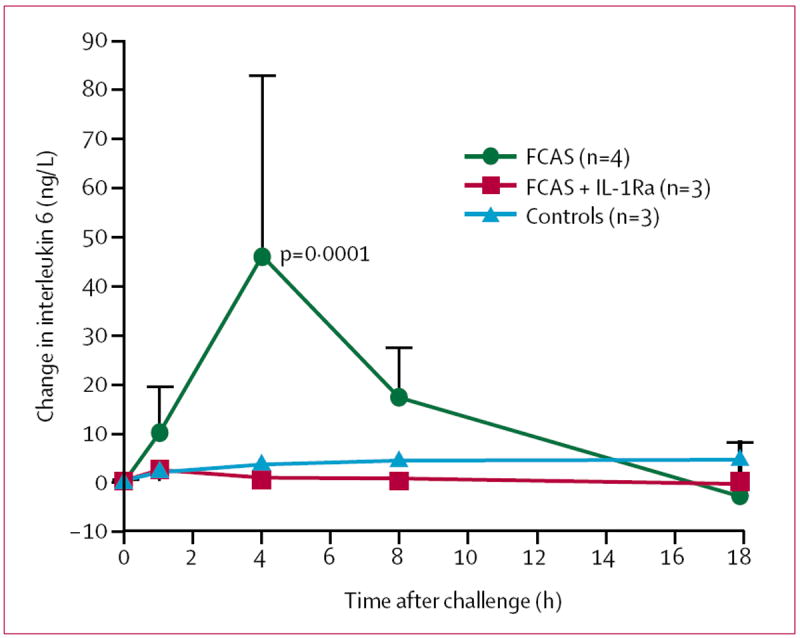
Error bars=SD.
To identify the source of serum interleukin 6 and to assess gene expression of other proinflammatory cytokines in leucocytes, we measured expression of cytokine mRNA in peripheral-blood leucocyte fractions from participants after cold challenge by quantitative real-time PCR. There was no detectable mRNA for interleukin 6 in granulocyte fractions and little mRNA expression in mononuclear-cell fractions, even when serum concentrations of interleukin 6 protein were highest. We could detect basal mRNA expression of interleukin 1β and TNFα in leucocyte fractions from some of the FCAS participants and controls, but there was no significantly increased expression after cold challenge.
We also examined cytokine gene expression in skin before and after cold exposure. There were increases of more than 200 times in expression of mRNA for interleukins 6, 1β, and 8 in affected skin from patients with FCAS compared with unaffected skin before cold challenge and compared with controls before and after challenge (figure 6). These findings raised the possibility that the skin, rather than circulating leucocytes, is the source of cytokines in this genetic disorder. To confirm this idea and to identify the cellular source of proinflammatory cytokines in skin tissue, we used immunohistochemistry with antibodies to interleukins 6, 1β, and 8. Affected skin from patients with FCAS after cold challenge showed the typical FCAS pathology, including neutrophilic infiltrate in the perivascular tissue and dermal oedema (figure 7, A and B). FCAS affected skin, but not unaffected skin from FCAS participants or controls, showed prominent staining for interleukin 6 (figure 7, C and D) and interleukin 1β (figure 7, E and F) of infiltrating leucocytes and resident cells. A similar pattern of staining was observed with an antibody to interleukin 1β (D116) that binds only to the processed protein not to its proform, indicating that active cytokine is present in the skin lesions (data not shown). Staining for interleukin 8 was was not seen in unaffected skin from FCAS and control participants (figure 7, G) and was limited to infiltrating leucocytes in affected skin samples (figure 7, H). Overall, interleukin 6 was the most prominent cytokine present in FCAS affected skin, which is consistent with the serum protein concentrations in FCAS participants.
Figure 6. Mean cytokine gene expression in skin tissue before and after cold challenge.
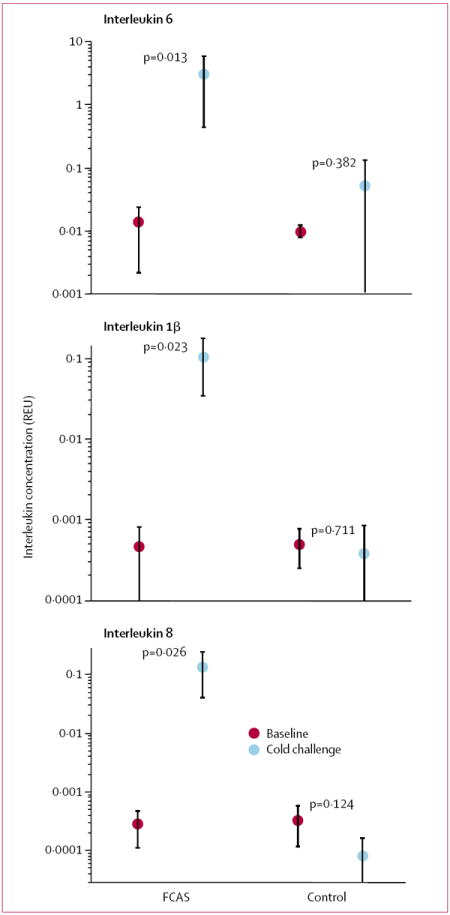
Cytokine mRNA concentrations in skin tissue are normalised to those of mRNA for glyceraldehyde-3-phosphate dehydrogenase and expressed as relative expression units (REU). Error bars=SD.
Figure 7. Histopathology and cytokine immunohistochemistry of FCAS skin.
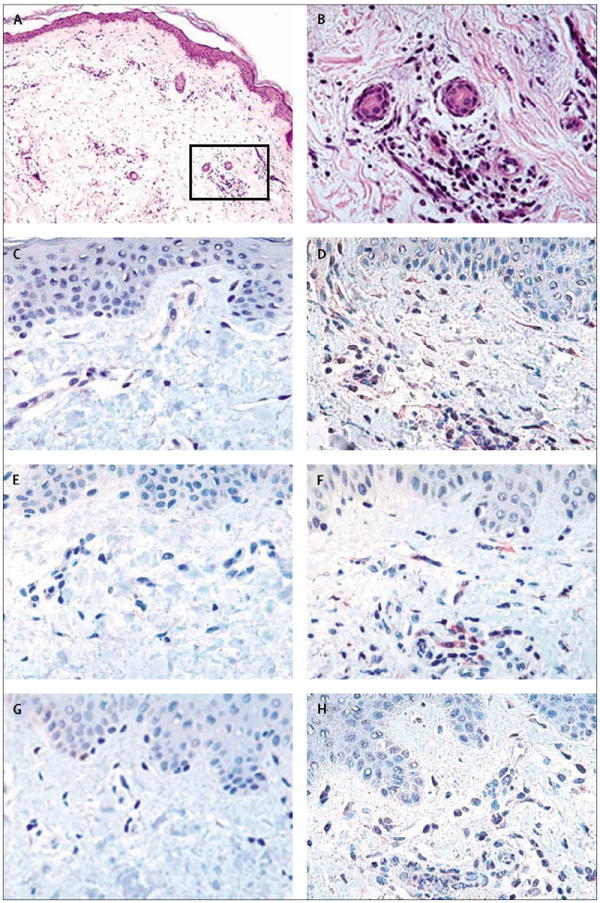
A: haematoxylin and eosin staining of skin from affected area (×10 magnification) showing dermal oedema and prominent leucocyte infiltrate. B: high-power (×40 magnification) showing predominant neutrophilic infiltrate concentrated in the perivascular and sweat gland areas. C: staining of unaffected skin for interleukin 6 before cold challenge. D: staining of affected skin for interleukin 6 after cold challenge; there is intracellular (red) staining of infiltrating leucocytes and resident spindle-shaped cells. E: staining of unaffected skin for interleukin 1β before cold challenge. F: staining of affected skin for interleukin 1β after cold challenge showing staining (red) of infiltrating leucocytes and resident cells. G: staining of unaffected skin for interleukin 8 before cold challenge. H: staining of affected skin for interleukin 8 after cold challenge showing staining (red) of infiltrating leucocytes.
Discussion
This study shows how the application of genomics to the study of human disease has led to an increased understanding of the underlying mechanisms and improved treatment for patients with FCAS. The discovery of CIAS1 and its protein product cryopyrin, the subsequent elucidation of interacting proteins and inflammatory pathways involved, and the recent advances in targeted cytokine therapy have resulted in a new therapy for a disorder with substantial morbidity and no previously known effective treatment. Although rare, FCAS allows insight into the function of a regulatory protein that probably is involved in the causation of more common inflammatory diseases.
Cryopyrin could contribute to pathogenetic events in more common inflammatory diseases. We have observed that expression of cryopyrin mRNA is three times higher in rheumatoid arthritis synovium than in osteoarthritis tissue.19 Several recent reports have suggested a central role for cryopyrin in activation of caspase 1 and NF-κB signalling,20-22 two pathways that are implicated in several common inflammatory diseases such as rheumatoid arthritis. Agostini and colleagues lately showed increased processing of interleukin 1β in macrophages isolated from a patient with Muckle-Wells syndrome.21 Further studies are needed to elucidate the regulatory functions of cryopyrin in inflammation.
The most prominent cytokine finding in the blood from FCAS patients was a striking increase in serum concentrations of interleukin 6; however, peripheral-blood leucocytes were not the source of this cytokine, as shown by sensitive real-time PCR. Interleukin 1β was not detected in blood, but it rarely is detected in serum because of its primary intracellular localisation and binding to plasma proteins and soluble receptor.23 Amounts of both protein and mRNA of interleukins 6, 1β, and 8 were high in affected skin. These cytokines seem to be produced by either resident skin macrophages or recruited inflammatory cells that have become activated in the skin, although other sources such as endothelial cells could also contribute.
Sensitive immunohistochemical assays of tissue production of cytokines clearly showed that active interleukin 1β is available at the disease site in affected FCAS skin. Interleukin 1 could be a crucial mediator early in an amplifying inflammatory cascade beginning in the tissue and leading to circulating concentrations of interleukin 6 that affect the central nervous system and joints, producing fever and arthralgia, respectively (figure 1). This pattern accords with the progression of inflammation in patients with FCAS; rash is the first symptom to appear during episodes. The hypothesis that interleukin 1 acts locally to stimulate the release of interleukin 6, a secondary circulating mediator, was proposed by Luheshi.24 This hypothesis is supported by the attenuation of increases in serum interleukin 6 with the administration of IL-1Ra in rats in the air pouch model23 and in patients with FCAS in this study. A rapid decrease in serum concentrations of interleukin 6 has also been shown in patients with sepsis treated with IL-1Ra.23
We designed a pilot study assessing the efficacy of IL-1Ra in FCAS patients based on evidence supporting a role for interleukin 1 in FCAS. We had observed prominent induced expression of interleukin 1 in FCAS skin in the initial cold challenges, and recent studies have implicated cryopyrin in activation of caspase 1 (figure 1). In the study, virtually all clinical and laboratory sequelae of systemic cold exposure were prevented by IL-1Ra, including complete suppression of systemic cytokine release. The modest efficacy of IL-1Ra in rheumatoid arthritis,25 a complex disease with multiple causes, is probably due to the involvement of other inflammatory pathways. The sensitivity of our patients with FCAS to IL-1Ra supports a central role for interleukin 1 in the pathogenesis of this single-gene disorder.
Although the small number of participants is a limitation of this study, the clinical presentation of these four patients is representative of that in more than 100 FCAS patients we have assessed during the past 7 years.1,26 These individuals are also representative in that they have the L353P CIAS1 mutation, which is present in more than 90% of our North American FCAS cohort, most of whom share a common ancestor with the participants in this study.26 The striking and consistent acute inflammatory responses seen and the effect of IL-1Ra on symptoms and laboratory markers of acute inflammation were statistically significant despite the small number of participants. The focus of future FCAS research should be the study of IL-1Ra or other interleukin-1-regulating approaches in a placebo-controlled trial with a larger and more genetically diverse group of patients. The demonstrated clinical effectiveness of IL-1Ra in Muckle-Wells syndrome15,16 and FCAS also supports its evaluation in NOMID, the most severe of the cryopyrin-associated periodic syndromes. Further study of the molecular defects and subsequent pathogenetic events in the autoinflammatory syndromes will probably lead to greater understanding of basic inflammatory mechanisms and could result in targeted therapies for other inflammatory disorders.
Acknowledgments
We thank the FCAS family members who took part in this study; David Broide, Richard Kolodner, and Arthur Kavanaugh (CIT), and Charles Dinarello for helpful advice; Alex Greiner, Seema Aceves, Suzan Pae, and James Bergman for clinical assessments and skin biopsies; David Enkelis for preparation of skin samples; Terence O’Grady for pathological consultation; and Sherry Soefje for editorial support. Funding for this study was provided by National Institute of Allergy and Infectious Diseases grant RO1 AI52430, the National Institutes of Health funded UCSD General Clinical Research Center M01 RR00827, and Rheumatic Diseases Core Center 5 P30 AR47360, and the Ludwig Institute of Cancer Research San Diego Branch.
Glossary
- Familial Mediterranean fever
An autosomal recessive inflammatory disease characterised by recurrent episodes of fever, arthralgia, rash, and abdominal and chest pain, that is found more commonly in people of Mediterranean origin
- Autoinflammatory syndromes
Diseases characterised by recurrent episodes of inflammation in the absence of infection, autoantibodies, or antigen-specific T cells—eg, hereditary periodic-fever disorders and Crohn’s disease
- ASC
Apoptosis-associated speck-like protein with a caspase activation and recruitment domain (CARD), an intracellular adaptor protein involved in activation of caspase 1. Also known as PYCARD, CARD5, and TMS1
- CIAS1
Cold-induced autoinflammatory syndrome 1, the gene that encodes cryopyrin and is mutated in the human diseases FCAS, Muckle-Wells syndrome, and NOMID. Also known as PYPAF1, NALP3, and CATERPILLAR 1.1
- Muckle-Wells syndrome
An autosomal dominant disease due to mutations in CIAS1 characterised by recurrent episodes of rash, fever, and arthralgia, progressive neurosensory hearing loss, and a risk of late-onset renal amyloidosis
- NOMID
A predominantly sporadic syndrome, with occasional autosomal dominant inheritance that is due to mutations in CIAS1 and is characterised by chronic meningitis, rash, and joint malformation. Also known as chronic infantile neurological, cutaneous, articular (CINCA) syndrome
- Cryopyrin
The intracellular protein encoded by CIAS1, which appears to be involved in interleukin-1 signalling pathways by interacting with ASC. Also known as PYPAF1, NALP3, and CATERPILLAR 1.1
- IL-1Ra
Recombinant interleukin-1 receptor antagonist protein (Anakinra, Kineret) that is currently used in the treatment of rheumatoid arthritis
Footnotes
Contributors
H M Hoffman oversaw the design and execution of the study, recruited and assessed the participants, prepared all drafts of the paper, and is the principal grant holder for the project. S Rosengren did the real-time PCR and statistical analysis and prepared data for presentation.
D L Boyle and G S Firestein participated in the initial study design, data analysis, preparation of the report, and presentation. J Y Cho carried out immunohistochemistry and prepared data for presentation. J Nayar, J L Mueller, and J P Anderson provided experimental support in the design and execution of the study. A A Wanderer referred the patients and participated in initial study design and clinical assessment. All of the investigators were involved in interpretation of the data and revision of the report and agreed to its final form.
Conflict of interest statement
We declare that we have no conflict of interest.
References
- 1.Hoffman HM, Wanderer AA, Broide DH. Familial cold autoinflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol. 2001;108:615–20. doi: 10.1067/mai.2001.118790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90:797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 3.French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 4.Richards N, Schaner P, Diaz A, et al. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J Biol Chem. 2001;9:9. doi: 10.1074/jbc.M104730200. [DOI] [PubMed] [Google Scholar]
- 5.Dowds TA, Masumoto J, Chen FF, Ogura Y, Inohara N, Nunez G. Regulation of cryopyrin/Pypaf1 signaling by pyrin, the familial Mediterranean fever gene product. Biochem Biophys Res Commun. 2003;302:575–80. doi: 10.1016/s0006-291x(03)00221-3. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein cause familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–05. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dode C, Le Du N, Cuisset L, et al. New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet. 2002;70:1498–506. doi: 10.1086/340786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum. 2002;46:2445–52. doi: 10.1002/art.10509. [DOI] [PubMed] [Google Scholar]
- 9.Muckle TJ, Wells M. Urticaria, deafness and amyloidosis: a new heredo-familial syndrome. QJM. 1962;31:235–48. [PubMed] [Google Scholar]
- 10.Feldmann J, Prieur AM, Quartier P, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46:3340–48. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertin J, DiStefano PS. The PYRIN domain: a novel motif found in apoptosis and inflammation proteins. Cell Death Differ. 2000;7:1273–74. doi: 10.1038/sj.cdd.4400774. [DOI] [PubMed] [Google Scholar]
- 13.Manji GA, Wang L, Geddes BJ, et al. PYPAF1: A PYRIN-containing Apaf1-like protein that assembles with ASC and regulates activation of NF-κB. J Biol Chem. 2002;277:11570–75. doi: 10.1074/jbc.M112208200. [DOI] [PubMed] [Google Scholar]
- 14.Grenier JM, Wang L, Manji GA, et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett. 2002;530:73–78. doi: 10.1016/s0014-5793(02)03416-6. [DOI] [PubMed] [Google Scholar]
- 15.Hawkins PN, Lachmann HJ, McDermott MF. Interleukin-1-receptor antagonist in the Muckle-Wells syndrome. N Engl J Med. 2003;348:2583–84. doi: 10.1056/NEJM200306193482523. [DOI] [PubMed] [Google Scholar]
- 16.Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50:607–12. doi: 10.1002/art.20033. [DOI] [PubMed] [Google Scholar]
- 17.Hoffman HM, Gregory SG, Mueller JL, et al. Fine structure mapping of CIAS1: identification of an ancestral haplotype and a common FCAS mutation, L353P. Hum Genet. 2003;112:209–16. doi: 10.1007/s00439-002-0860-x. [DOI] [PubMed] [Google Scholar]
- 18.Boyle DL, Rosengren S, Kavanaugh A, Bugbee W, Firestein GS. Quantitative biomarker analysis of synovial gene expression by real-time PCR. Arthritis Res Ther. 2003;5:R352–60. doi: 10.1186/ar1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosengren S, Hoffman H, Bugbee W, Boyle DL. Expression and regulation of cryopyrin and related proteins in rheumatoid arthritis synovium. Ann Rheum Dis. 2004 doi: 10.1136/ard.2004.025577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Connor W, Jr, Harton JA, Zhu X, Linhoff MW, Ting JP. Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/ CATERPILLAR 1.1 is an inducible inflammatory mediator with NF-kappaB suppressive properties. J Immunol. 2003;171:6329–33. doi: 10.4049/jimmunol.171.12.6329. [DOI] [PubMed] [Google Scholar]
- 21.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an interleukin-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 22.Dowds TA, Masumoto J, Zhu L, Inohara N, Nunez G. Cryopyrin induced interleukin 1beta secretion in monocytic cells: enhanced activity of disease-associated mutants and requirement for ASC. J Biol Chem. 2004;279:21924–28. doi: 10.1074/jbc.M401178200. [DOI] [PubMed] [Google Scholar]
- 23.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- 24.Luheshi GN. Cytokines and fever: mechanisms and sites of action. Ann NY Acad Sci. 1998;856:83–89. doi: 10.1111/j.1749-6632.1998.tb08316.x. [DOI] [PubMed] [Google Scholar]
- 25.Taylor PC. Anti-cytokines and cytokines in the treatment of rheumatoid arthritis. Curr Pharm Des. 2003;9:1095–106. doi: 10.2174/1381612033454991. [DOI] [PubMed] [Google Scholar]
- 26.Johnstone RF, Dolen WK, Hoffman HM. A large kindred with familial cold autoinflammatory syndrome. Ann Allergy Asthma Immunol. 2003;90:233–37. doi: 10.1016/S1081-1206(10)62147-3. [DOI] [PubMed] [Google Scholar]