

Abstract
Both genetic and biochemical data suggest that transcriptional activators with little sequence homology nevertheless function through interaction with a shared group of coactivators. Here we show that a series of peptidomimetic transcriptional activation domains interact under cell-fiee and cellular conditions with the metazoan coactivator CBP despite differences in the positioning and identity of the constituent functional groups. Taken together, these results suggest that a key activator binding site within CBP is permissive, accepting multiple arrangements of hydrophobic functional groups. Further, this permissiveness is also observed with a coactivator from S. cerevisiae. Thus, the design of small molecule mimics of transcriptional activation domains with broad function may be more straightforward than previously envisioned.
Keywords: transcriptional activator, CBP, KIX domain, Med15
Regulated transcription of specific genes is one of the fundamental processes that underlie all of human physiology. As a result, the mis-regulation of transcription is related to almost all of human pathophysiology as either a cause or a consequence.1–4 This realization has spurred intense interest in uncovering the fundamental characteristics of transcriptional regulation and, further, in the eventual development of molecules capable of regulating transcription in living systems and acting as transcription-based therapeutics.1,5 One of the greatest challenges has been the identification of molecules that can either inhibit or mimic the function of transcriptional activators and by doing so precisely regulate the expression of preselected genes.6,7
Transcriptional activators initiate transcription by binding to DNA and facilitating the assembly of the transcriptional machinery through one or more direct binding interactions with coactivator proteins within the RNA polymerase II holoenzyme.8 Activator-coactivator interactions thus play an essential role in the gene activation process, yet there are many unanswered questions surrounding these binding events.5 Activators utilize a transcriptional activation domain (TAD) to bind to the transcriptional machinery and the largest and most well-studied class is the amphipathic class, named for the interspersed polar and hydrophobic amino acids present in the TAD sequences. Several lines of evidence suggest that amphipathic transcriptional activators interact with a shared group of coactivators within the transcriptional machinery; in vitro crosslinking experiments, for example, have shown that the coactivators Med15, Tra1, and Taf12 are targeted by the activators Gcn4 and Gal4.9,10 There is also emerging evidence that coactivators may use a single binding site to interact with a diverse group of activators.11–17 This is surprising because the TADs of amphipathic activators have little or no sequence homology, suggesting that either these diverse sequences must be able to fold into similar structures or that the binding sites within the coactivators are permissive, accepting more than one arrangement of hydrophobic and polar functional groups. Here we use peptidomimetics to show that one of the primary activator binding sites within the coactivator CBP (CREB binding protein) can interact in cell-free and cellular systems with several amphipathic scaffolds and is thus quite permissive. Further, we demonstrate that this permissiveness is conserved from yeast through metazoans in the case of a common activator-binding motif.
The coactivator and histone acetyl transferase CBP assimilates signals from a variety of transcriptional activators, including p53,18–20 Hif1α,21 Tax,22 MLL17 and CREB11 and thereby plays an integral role in cellular processes as diverse as hypoxic response and memory formation. CBP is the founding member of the small but growing class of GACKIX coactivators that use a 3-helix KIX domain to interact with the TADs of activators and is one of the most structurally well-characterized eukaryotic coactivators (Figure 1a).23 The KIX domain of CBP interacts with the TADs of CREB,11 c-Myb,12 and others in addition to the artificial TADs KBP 1.66 and KBP 2.20 (Figure 1b).24 Despite significant differences in sequence, these TADs are proposed to interact with a single binding site within the KIX domain through the formation of an amphipathic helix with the hydrophobic residues indicated mediating the important contacts. The CBP KIX domain is thus an excellent coactivator in which to investigate the permissiveness of activator binding sites. To this end, peptidomimetic variants of KBP 1.66 and 2.20 were designed that would alter the presentation and spacing of the functional groups within the sequence; the d-peptide version would produce the enantiomeric arrangement of functional groups for interaction, the β-peptide would be predicted to form a 14-helix upon binding and thus change the spacing and the hydrophobic interface, and the peptoid analog would have the N-H hydrogen bonds removed (Figures 1c and 1d).
FIGURE 1.

The GACKIX domain of CBP is targeted by many eukaryotic activators. (a) Solution structure of CBP(586–666) in complex with the activation domain of CREB (PDB accession number 1KDX).11 (b) Sequences of four transcriptional activation domains that interact with the CBP GACKIX domain. CREB and c-Myb are natural activators whereas KBP 1.66 and 2.20 were isolated via phage display screen against KIX.24 (c) Helical representations of the natural, enantiomeric and β-peptide versions of the KBP 2.20 TAD; no structure of the peptoid version is provided as it is difficult to predict peptoid conformation. (d) Helical representations of the natural, enantiomeric, and β-peptide versions of the KBP 1.66 TAD.
The synthesis of each of the peptides and peptidomimetics was carried out on solid support using established protocols.25–27 Each of the ligands was fluorescently labeled at their amino termini and fluorescence polarization was used to determine their respective binding affinities (Table I) for an exogenously expressed murine CBP KIX domain (CBP(586–672)). Consistent with earlier observations, KBP 2.20 exhibits an ~3-fold lower dissociation constant for the KIX domain relative to KBP 1.66; this is likely due to the smaller hydrophobic interface of KBP 1.66, predicted to consist of only two residues rather than the three residues of KBP 2.20.24 Of the peptidomimetics examined, only the KBP 1.66-derived peptoid failed to interact with the target protein to a detectable extent. This may reflect the loss of one or more key hydrogen bonds relative to the parent peptide. Remarkably, the β-peptide KBP 1.66 exhibited a 3-fold enhancement in affinity relative to the parent peptide. Again assuming that the ligands interact with the KIX domain as amphipathic helices, the 14-helix of β-KBP 1.66 would have a hydrophobic surface of three residues (L-L-F) rather than two (Figure 1d).28
Table I.
Dissociation Constants for the Interaction of KBP 2.20 and 1.66 Derivatives with the GACKIX Domains of CBP and Med 15
| CBP(586–672) | Med15(1–345) | |
|---|---|---|
| KBP 2.20 | 34 ± 2 μM | 7 ± 1 μM |
| d-KBP 2.20 | 28 ± 3 μM | 4.5 ± .3 μM |
| β-KBP 2.20 | 20 ± 2 μM | 4.7 ± .5 μM |
| peptoid 2.20 | 5.5 ± .2 μM | >30 μM |
| KBP 1.66 | 76 ± 10 μM | 10.8 ± .6 μM |
| d-KBP 1.66 | 84 ± 11 μM | 12.4 ± .7 μM |
| β-KBP 1.66 | 20 ± 1 μM | 12 ± 1 μM |
| Peptoid 1.66 | N.B. | N.B. |
Fluorescein-labeled peptides or peptidomimetics at a constant concentration were individually incubated at 25°C with increasing concentrations of either CBP or Med15. The fluorescence polarization at each concentration was measured and the resulting data fit using the Levenberg-Marquardt least squares method to obtain the dissociation constants. Each experiment was performed in triplicate (R2 >0.98) with the error indicated. See Supporting Information for details and binding curves.
Coactivators often have two or more binding sites for transcriptional activation domains and the KIX domain is no exception, containing two distinct binding surfaces for activators; these sites can be simultaneously occupied.29 To test if the peptidomimetic versions of KBP 1.66 and 2.20 bound to the same site as the peptide versions, we carried out competitive inhibition experiments in HeLa cells (see Figure 2). In these experiments, a firefly luciferase reporter gene was placed under the control of Gal4(1–148)+KBP 2.20 that was constitutively expressed. Increasing concentrations of KBP 2.20, KBP 1.66, or the peptidomimetic analogs were added to the cells and the change in KBP 2.20-mediated transcription assessed through alterations in firefly luciferase activity. As illustrated in Figure 2, peptides KBP 2.20 and 1.66 showed little or no inhibitory activity. This was not unexpected since these short peptides should be rapidly degraded by proteolysis. However, the peptidomimetic TADs that bound to the KIX domain inhibited KBP 2.20-mediated transcription up to 75% at 20 μM concentrations; similar results were obtained with Gal4(1–148)+KBP 1.66 as the activator (data not shown). In contrast, a d-peptide in which two of the key hydrophobic residues of KBP 2.20 were replaced with Arg (SRAVRELLFGS) had no impact on KBP 2.20-mediated transcription despite exhibiting similar cellular uptake and nuclear localization as the most effective of the inhibitors, peptoid KBP 2.20 (see Supporting Information for details). Taken together, these results are consistent with the peptidomimetic versions of the KBP TADs interacting with the same binding site, the binding site that the KBP 2.20 and 1.66 TADs employ for transcriptional activation. Importantly, these data also suggest that the TAD binding site is permissive, interacting with several different presentations of hydrophobic functional groups.
FIGURE 2.
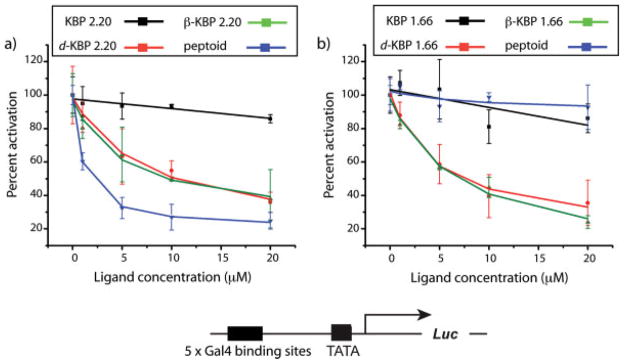
Results from luciferase assays in HeLa cell culture. (a) Activity of Gal4(1–148)+KBP 2.20 in the presence of increasing concentrations (0 → 20 μM) of KBP 2.20 (black line), d-KBP 2.20 (orange line), β-KBP 2.20 (green line), or peptoid KBP 2.20 (blue line) expressed as percent activation (activity of Gal4(1–148)+KBP 2.20 at each concentration of KBP 2.20 derivative relative to activity of Gal4(1–148)+KBP 2.20 alone). Compounds were added to cells as a solution in dimethyl-sulfoxide (DMSO)/EtOH (70:30 mixture) 3 h after transfection such that the final concentration of DMSO was <1% (vol/vol). Activity was measured 40 h after compound addition. (b) Activity of Gal4(1–148)+KBP 2.20 in the presence of increasing concentrations (0 → 20 μM) of KBP 1.66 (black line), d-KBP 1.66 (orange line), β-KBP 1.66 (green line), or peptoid KBP 1.66 (blue line) expressed as percent activation as described for a). In all of these experiments cell viability was unaffected by the addition of the peptides or the peptidomimetics as assessed by growth rate, cell number, and visual inspection. In addition, no impact on the expression of Renilla luciferase (included in each experiment as a control) was observed, an indication that the molecules are not general inhibitors of transcription in this concentration range. See Supporting Information for details.
As described earlier, the KIX domain has been identified in a growing number of coactivators including Med15, a coactivator that has no direct metazoan homolog.23 Since it has been proposed that this domain is a transcriptional activator-binding interface conserved throughout eukaryotes, we were interested to see if the permissive character of the putative activator binding sites was also conserved. To test this, we assessed the ability of each of the KBP 2.20 and KBP 1.66 variants to interact with Med15(1–345). As shown in Table I, all bound well to the protein with the exception of the peptoids. Importantly, this is a direct demonstration of a significantly conserved activator binding surface from yeast to metazoans. It is thus not surprising that amphipathic activators tend to function in all eukaryotes regardless of their species of origin.30–32
The observation that at least one key activator binding site is permissive has significant implications for the discovery of artificial transcriptional activators and of molecules designed to inhibit activator-coactivator interactions. Perhaps most important is that it suggests that it is not necessary to precisely reconstitute a three-dimensional array of amphipathic functional groups within a small molecule in order to target activator binding sites within coactivators. Thus the identification of new classes of small molecule activators may be more straightforward than previously envisioned.
Supplementary Material
Acknowledgments
Contract grant sponsor: National Institutes of Health
Contract grant number: GM65330
Contract grant sponsors: American Cancer Society
Contract grant number: RSG 05-195-01-CDD
Footnotes
This article contains supplementary material available via the Internet at http://www.interscience.wiley.com/jpages/0006-3525/suppmat.
This article was originally published online as an accepted preprint. The “Published Online” date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers editorial office at biopolymers@wiley.com
References
- 1.Pandolfi PP. Oncogene. 2001;20:3116–3127. doi: 10.1038/sj.onc.1204299. [DOI] [PubMed] [Google Scholar]
- 2.Chen X, Cheung ST, So S, Fan ST, Barry C, Higgins J, Lai KM, Ji J, Dudoit S, Ng IO, Van De Rijn M, Botstein D, Brown PO. Mol Biol Cell. 2002;13:1929–1939. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darnell JE., Jr Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 4.Warke RV, Xhaja K, Martin KJ, Fournier MF, Shaw SK, Brizuela N, de Bosch N, Lapointe D, Ennis FA, Rothman AL, Bosch I. J Virol. 2003;77:11822–11832. doi: 10.1128/JVI.77.21.11822-11832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mapp AK, Ansari AZ. ACS Chem Biol. 2007;2:62–75. doi: 10.1021/cb600463w. [DOI] [PubMed] [Google Scholar]
- 6.Ansari AZ, Mapp AK. Curr Opin Chem Biol. 2002;6:765–772. doi: 10.1016/s1367-5931(02)00377-0. [DOI] [PubMed] [Google Scholar]
- 7.Majmudar CY, Mapp AK. Curr Opin Chem Biol. 2005;9:467–474. doi: 10.1016/j.cbpa.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Ptashne M, Gann A. Genes & Signals. Cold Spring Harbor Laboratory; New York: 2001. [Google Scholar]
- 9.Fishburn J, Mohibullah N, Hahn S. Mol Cell. 2005;18:369–378. doi: 10.1016/j.molcel.2005.03.029. [DOI] [PubMed] [Google Scholar]
- 10.Reeves WM, Hahn S. Mol Cell Biol. 2005;25:9092–9102. doi: 10.1128/MCB.25.20.9092-9102.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 12.Zor T, De Guzman RN, Dyson HJ, Wright PE. J Mol Biol. 2004;337:521–534. doi: 10.1016/j.jmb.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 13.Liu YP, Chang CW, Chang KY. FEBS Lett. 2003;554:403–409. doi: 10.1016/s0014-5793(03)01200-6. [DOI] [PubMed] [Google Scholar]
- 14.Campbell KM, Lumb K. J Biochem. 2002;41:13956–13964. doi: 10.1021/bi026222m. [DOI] [PubMed] [Google Scholar]
- 15.Vendel AC, McBryant SJ, Lumb K. J Biochem. 2003;42:12481–12487. doi: 10.1021/bi0353023. [DOI] [PubMed] [Google Scholar]
- 16.Vendel AC, Lumb K. J Biochem. 2004;43:904–908. doi: 10.1021/bi035612l. [DOI] [PubMed] [Google Scholar]
- 17.De Guzman RN, Goto NK, Dyson HJ, Wright PE. J Mol Biol. 2006;355:1005–1013. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 18.Gu W, Shi XL, Roeder RG. Nature. 1997;387:819–823. doi: 10.1038/42972. [DOI] [PubMed] [Google Scholar]
- 19.Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Nature. 1997;387:823–827. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- 20.Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Cell. 1997;89:1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- 21.Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM. Proc Natl Acad Sci USA. 1996;93:12969–12973. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwok RP, Laurance ME, Lundblad JR, Goldman PS, Shih H, Connor LM, Marriott SJ, Goodman RH. Nature. 1996;380:642–646. doi: 10.1038/380642a0. [DOI] [PubMed] [Google Scholar]
- 23.Novatchkova M, Eisenhaber F. Curr Biol. 2004;14:R54–R55. doi: 10.1016/j.cub.2003.12.042. [DOI] [PubMed] [Google Scholar]
- 24.Frangioni JV, LaRiccia LM, Cantley LC, Montminy MR. Nat Biotechnol. 2000;18:1080–1085. doi: 10.1038/80280. [DOI] [PubMed] [Google Scholar]
- 25.Chan WCWPD. Fmoc Solid-Phase Peptide Synthesis: A Practical Approach. Oxford University Press; New York: 2000. [Google Scholar]
- 26.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 27.Olivos HJ, Alluri PG, Reddy MM, Salony D, Kodadek T. Org Lett. 2002;4:4057–4059. doi: 10.1021/ol0267578. [DOI] [PubMed] [Google Scholar]
- 28.Kritzer JA, Hodsdon ME, Schepartz A. J Am Chem Soc. 2005;127:4118–4119. doi: 10.1021/ja042933r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE. J Biol Chem. 2002;277:43168–43174. doi: 10.1074/jbc.M207660200. [DOI] [PubMed] [Google Scholar]
- 30.Fischer JA, Giniger E, Maniatis T, Ptashne M. Nature. 1988;332:853–856. doi: 10.1038/332853a0. [DOI] [PubMed] [Google Scholar]
- 31.Kakidani H, Ptashne M. Cell. 1988;52:161–167. doi: 10.1016/0092-8674(88)90504-1. [DOI] [PubMed] [Google Scholar]
- 32.Ma J, Przibilla E, Hu J, Bogorad L, Ptashne M. Nature. 1988;334:631–633. doi: 10.1038/334631a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.