

Abstract
In heart failure (HF), T-tubule (TT) disruption contributes to dyssynchronous calcium (Ca) release and impaired contraction, but its role in arrhythmogenesis remains unclear. In this study, we investigate the mechanisms of TT disruption and other HF remodeling factors on Ca alternans in ventricular myocytes using computer modeling. A ventricular myocyte model with detailed spatiotemporal Ca cycling modeled by a coupled Ca release unit (CRU) network was used, in which the L-type Ca channels and the ryanodine receptor (RyR) channels were simulated by random Markov transitions. TT disruption, which removes the L-type Ca channels from the associated CRUs, results in “orphaned” RyR clusters and thus provides increased opportunity for spark-induced Ca sparks to occur. This effect combined with other HF remodeling factors promoted alternans by two distinct mechanisms: 1) for normal sarco-endoplasmic reticulum Ca ATPase (SERCA) activity, alternans was caused by both CRU refractoriness and coupling. The increased opportunity for spark-induced sparks by TT disruption combined with the enhanced CRU coupling by Ca elevation in the presence or absence of increased RyR leakiness facilitated spark synchronization on alternate beats to promote Ca alternans; 2) for down-regulated SERCA, alternans was caused by the sarcoplasmic reticulum (SR) Ca load-dependent mechanism, independent of CRU refractoriness. TT disruption and increased RyR leakiness shifted and steepened the SR Ca release-load relationship, which combines with down-regulated SERCA to promote Ca alternans. In conclusion, the mechanisms of Ca alternans for normal and down-regulated SERCA are different, and TT disruption promotes Ca alternans by both mechanisms, which may contribute to alternans at different stages of HF.
Keywords: heart failure, remodeling, T-tubule disruption, arrhythmias, alternans
1. Introduction
In normal ventricular myocytes, the transverse-tubule (TT) network exists to facilitate effective excitation-contraction coupling by allowing synchronous calcium (Ca) release from the deep within the interior as well as the surface of the cell [1, 2]. Voltage gated L-type Ca channels (LCCs) are distributed in clusters in the TT membrane in close proximity to clusters of ryanodine receptor (RyR) channels in the sarcoplasmic reticulum (SR) membrane, forming a Ca release unit (CRU) network. Ca entry from the opening of one or more LCCs in a CRU can trigger the opening of its associated RyRs to release SR Ca into the cytosol via Ca-induced Ca release (CICR). During an action potential, the activation of LCCs in the TT network allows simultaneous Ca release of the CRUs throughout the cell and thus facilitates synchronous contraction of the myofilaments, a key feature promoting effective excitation-contraction coupling. In heart failure (HF), the TT network becomes disorganized and disrupted [3–11], causing some CRUs to lose their corresponding LCCs and resulting in so-called orphaned RyR clusters [4]. Lacking associated LCCs, the RyRs in the orphaned RyR clusters cannot be directly activated by LCCs, but have a probability of being activated later by Ca released from the neighboring CRUs. This leads to dyssynchronous Ca release and a reduced Ca transient amplitude [5, 8], which impairs contractile function. Besides causing dyssynchronous Ca release, TT disruption can also have consequences on the myocyte’s action potential properties [10, 12], which may contribute to arrhythmogenesis [13, 14].
In addition to TT disruption, HF remodeling involves many other proteins [15–18], including increased RyR leakiness, down-regulated sarcoplasmic/endoplasmic reticulum Ca ATPase (SERCA), and altered ionic currents, as well as structural changes, which depend on the stage of HF development. In addition to contractile dysfunction, these changes also promote arrhythmogenesis in HF, notably pulsus alternans and T-wave alternans (TWA), which have been known for more than a century to increase arrhythmia risk [19, 20]. TWA is currently used clinically as a risk prognosticator for ventricular arrhythmias in patients with HF [21–23]. Experimental studies have shown that pacing-induced HF remodeling promotes action potential duration (APD) and Ca alternans [24–27], with Ca alternans being the primary origin. Two mechanisms of Ca alternans have been described and analyzed both experimentally and computationally: 1) SR Ca load-dependent alternans, in which SR Ca content alternates in phase with cytoplasmic free Ca, requiring a steep SR Ca release-load relationship (or fractional SR Ca release curve) [28–34]; 2) refractoriness-dependent alternans, in which refractoriness of RyRs is required for Ca to alternate, while SR Ca load alternans is not required [35–40]. However, how TT disruption interacts with other remodeling factors to promote Ca alternans in HF, and which specific mechanisms are involved, are still poorly understood.
Accordingly, in this study, we investigated the effects of TT disruption and other HF remodeling factors on the susceptibility to Ca alternans in computer simulations, using a ventricular myocyte model with detailed spatiotemporal Ca cycling. The model contains a CRU network of spatially-distributed LCC and RyR clusters, in which TT disruption was simulated by random removal of LCC clusters and re-distribution of the corresponding LCCs to other CRUs in the cell. Since the electrophysiological remodeling in HF is complex and depends on the stage of HF as well as species, for simplicity, we carried out simulations under the following conditions: 1) normal Ca cycling proteins; 2) remodeled Ca cycling proteins; and 3) remodeled Ca cycling proteins and other remodeled ionic currents. We show that: 1) for normal SERCA activity, TT disruption without ICa,L reduction, ICa,L reduction alone, or both together can promote Ca alternans. The mechanism of Ca alternans is refractoriness-dependent, in which randomness of CRU activation, CRU refractoriness, and CRU recruitment interact to cause alternans, as described by the 3R theory developed by us previously [37, 41]. TT disruption results in orphaned RyR clusters, increasing the opportunity for spark-induced sparks to occur and facilitating spark synchronization to promote alternans; 2) for down-regulated SERCA, Ca alternans is caused by a steep SR load-release relationship. Alternans due to this mechanism can occur at very slow pacing rates. Increased RyR leakiness and TT disruption shift and steepen the fractional SR Ca release curve to promote alternans. We conclude that the mechanisms of Ca alternans for normal and down-regulated SERCA are different, and TT disruption promotes Ca alternans by both mechanisms, which may contribute to arrhythmogenic alternans in different stages of HF.
2. Methods
2.1. Ventricular cell model
The ventricular cell model is an action potential model with detailed spatiotemporal Ca cycling which includes a diffusively-coupled network of 100×20×10 CRUs, forming a three-dimensional cell (Fig. 1A). Details of the model were presented in our previous publication [42], and the parameters were the same unless stated in the present study. Briefly, each CRU contains 4 sub-spaces: a network SR space (NSR), a junctional SR space (JSR), a dyadic space (DS), and a cytosolic space. Ca diffuses freely between the NSR and the JSR, and between the DS and the cytosolic space. Under normal conditions, each CRU contains one cluster of 10 LCCs and one cluster of 100 RyRs. Ca enters the DS via the LCCs, which were simulated with stochastic Markov transitions. Ca is released from the JSR to DS via the RyRs which were also simulated by stochastic Markov transitions. Ca is extruded from cytosol via Na-Ca exchange (NCX) and taken up from cytosol via the SERCA pump. The CRUs are locally coupled by Ca diffusion in the cytosol and in the NSR. Fig. 1B shows an action potential and a line scan of cytosolic Ca of the normal control model at a pacing cycle length (PCL) of 500 ms. The cell model was periodically paced with this normal action potential wave form (action potential clamp) for all simulations except the ones in Fig. 7.
Figure 1. Computational model.
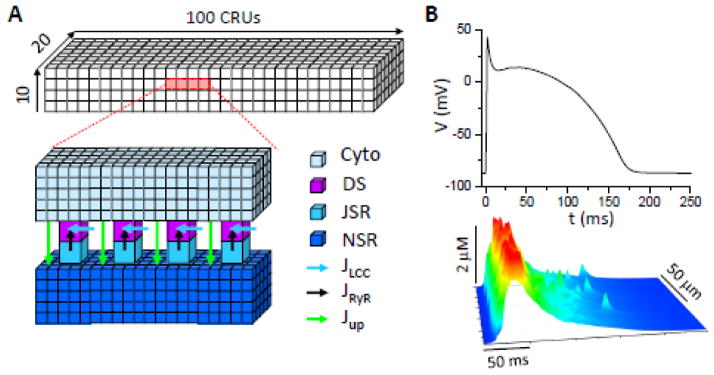
A. Schematic plot of the CRU network model. A full model contains 10×20×100 CRUs. A CRU contains a network SR (NSR) space, a junctional SR (JSR) space, a dyadic space (DS), and a cytosolic (Cyto) space. These spaces are coupled via SR Ca release (Jrel), uptake (Jup), and Ca diffusion. The CRU network is coupled via Ca diffusion in cytosol and NSR. B. An action potential and a line scan of cytoplasmic free [Ca]i under control conditions at PCL=500 ms. We set [Na]i= 9 mM as normal [Na]i. This action potential was used as the voltage wave form for all simulations under action potential clamp conditions.
Figure 7. APD and Ca alternans in HF under free running condition (current clamp).

APD (A), peak [Ca]i (B), and SR load (C) versus PCL; and V (D), [Ca]i (C), and [Ca]i line scan (F) versus time. [Na]i=10 mM and pLCC=30%.
2.2. TT disruption
TT disruption was modeled by removing LCC clusters from the CRU network, causing the corresponding RyR clusters to become orphaned. Since no quantitative data is available on TT disruption, we randomly chose CRUs with a probability pLCC and removed their corresponding LCC clusters, i.e., pLCC is the percentage of LCC clusters removed. Experimental studies have shown that the whole-cell ICa,L in HF is either reduced or unchanged [15, 18]. In most of the simulations, we retained the same whole-cell ICa,L by randomly re-distributed the removed LCCs back into the CRUs with LCC clusters. Therefore, with the exception of the simulations in Fig. 3, TT disruption for all other simulations in which a percentage (pLCC) of LCC clusters were randomly removed, the total number of LCCs remained constant.
Figure 3. Effects of LCC distribution on Ca alternans.

Shown are whole-cell cytosolic free [Ca]i (left) and whole-cell ICa,L (right). Black trace in each panel corresponds to normal LCC conductance and distribution, and purple traces are those after LCC conductance or distribution are altered. A. LCC single channel conductance reduced by 30%. B. 30% of the LCC clusters removed from the cell. C. 30% of the LCC clusters removed, but their LCCs re-distributed to the other CRUs with LCC clusters such that the total number of LCCS remain the same. [Na]i=14 mM.
2.3. Other HF remodeling
HF remodeling is a complex process which depends on the stage and etiology of HF. Here we choose a set of parameters to simulate other HF remodeling alterations largely based on the two references [17, 43] as follows: 1) RyR leakiness was increased by increasing the transition rate from the closed state to the open state by 30%; 2) the maximum SERCA activity was reduced by 50% and Kd was reduced from 1 μM to 0.5 μM to simulate increased phospholamban phosphorylation [44]; 3) INCX was doubled; and 4) IKs, Ito, and IK1 were reduced by 50%. In this study, intracellular sodium concentration ([Na]i) was used as a parameter instead of a variable, which is elevated in HF.
2.4. Numerical methods
The details of the computational methods have been presented previously [42]. Briefly, the NSR and cytosolic space were discretized into three-dimensional spatial grids using a 0.2×0.2×0.2 μm3 spatial resolution, such that each CRU contains 250 computational voxels, a JSR, and a DS. Therefore, a 20,000-CRU cell model contains more than 2 million randomly simulated ion channels and 5 million computational voxels. The equations were simulated using an operator splitting method with a time step of 0.01 ms. Although our code can simulate 1 s of heart time in 10 minutes of real time with our advanced computational methods [42], it is still computationally expensive for large-scale simulations. To perform systematic studies, we used a smaller CRU network which contains 20×10×5 CRUs (for simulations in Fig. 4 and Fig. 5). As we have shown before [38] (and confirmed in present study), the smaller grid gives almost the same result as the full model with slightly larger random fluctuations. All computations were performed on an Intel Xeon 2.53 GHz processor using Graphical Processing Unit parallel computing with an NVIDIA Tesla C2050.
Figure 4. TT disruption promotes Ca alternans when RyR and SERCA are normal.

A. Alternans amplitude (ΔCaALT= peak [Ca]i of the large beat minus the peak [Ca]i of the small beat) versus different combinations of [Na]i and LCC cluster removal and re-distribution (pLCC). PCL=500 ms. The map contains 42 parameter combinations (boxes), composed of 7 [Na]i values (8, 9, 10,11,12,13, and 14) and 6 pLCC values (0, 0.1, 0.2, 0.3, 0.4, and 0.5). B. Peak [Ca]i and SR load versus pLCC for [Na]i=11 mM and PCL=500 ms. C. Peak [Ca]i and SR load versus PCL for pLCC=30% and [Na]i=11 mM. D. Same as C but with SR load clamped at 700 μM.
Figure 5. TT disruption promotes Ca alternans when SERCA is down-regulated with other HF remodeling.

A. Alternans amplitude (ΔCaALT= peak [Ca]i of the large beat minus the peak [Ca]i of the small beat) versus different values of [Na]i and pLCC, for PCL=500 ms. B. Peak [Ca]i and SR Ca load versus pLCC for [Na]i=10 mM and PCL=500 ms. C. Peak [Ca]i and SR Ca load versus PCL for pLCC=30% and [Na]i=10 mM. D. Peak [Ca]i versus clamped SR Ca load for PCL=1000 ms, pLCC=30%, and [Na]i=10 mM. E. Same as C but with SERCA activity doubled.
3. Results
3.1. Effects of TT disruption on the Ca transient, SR load, and fractional SR Ca release
Figures 2 A–C compare SR Ca concentration ([Ca]SR), the whole-cell free cytosolic Ca ([Ca]i) and line scans of free cytosolic Ca for three conditions: 1) normal control (panel A); 2) RyR, SERCA, and NCX remodeled without TT disruption (panel B); and 3) RyR, SERCA, and NCX remodeled with TT disruption (panel C). Remodeling without TT disruption reduced SR Ca load, increased the rise time of [Ca]i, reduced peak [Ca]i, and made Ca release more dyssynchronous (see the line scans). The addition of TT disruption exacerbated the alterations to the Ca transient and dyssynchronous Ca release, but modestly increased the SR Ca load. The reason is straightforward: the efficiency of SR Ca release was reduced by orphaned RyR clusters, such that more Ca was retained in the SR and less was extruded by NCX, increasing the SR Ca load. Therefore, while TT disruption enhances dyssynchronous Ca release, it can also increase SR load if the whole-cell ICa,L is not reduced.
Figure 2. Effects of HF remodeling on Ca transient, SR load, and fractional SR Ca release.

Shown are whole-cell SR Ca, cytosolic free Ca, and a line scan of the cytosolic free Ca. SR load and cytosolic Ca peak time are marked. A. Normal control. B. RyR, SERCA and NCX remodeled without TT disruption. C. RyR, SERCA and NCX remodeled with TT disruption (pLCC=30%). PCL=500 ms and [Na]i=9 mM. D. Effects of remodeling and TT disruption on fractional SR Ca release curves under the same conditions as in A–C. The fractional release curves were calculated as described in a previous study [37].
Figure 2D compares the fractional SR Ca release under the three conditions as above. HF-remodeling changes in SERCA, RyR and NCX increased the steepness of the SR Ca fractional release-load relationship and shifted the curve to the lower SR Ca. TT disruption further steepened the fractional release curve with small right shift. The rightward shift occurs because after TT disruption, the orphaned RyR clusters do not fire at low SR Ca load, and thus the total number of CRUs activated to release Ca is less than that in the case of no TT disruption. However, once the SR Ca load reaches a certain level, sparks from the normal CRUs with LCCs recruit the orphaned RyR clusters to fire, resulting in a steep increase in the total number of firing CRUs and a steeper SR fractional release curve. The left shift and steepening of the fractional release curve by HF remodeling agrees with the experimental results observed in failing myocytes [45].
3.2. Effects of the TT disruption on Ca alternans when RyR and SERCA properties are normal
To analyze the effects of TT disruption on alternans, we first studied the condition in which NCX, RyR properties and SERCA activity are normal. To enhance CICR, we increased [Na]i to augment cellular Ca loading, as occurs in HF. With normal ICa,L, increasing [Na]i from the control value of 9 mM to 14 mM did not produce alternans at PCL=500 ms (black traces in Fig. 3), despite a doubling of peak [Ca]i (compare to Fig. 1B).
As shown in our previous studies [37, 38], reducing ICa,L promotes Ca alternans by reducing the number of primary sparks activated by LCC openings, thereby making more CRUs available to be recruited as secondary (spark-induced) sparks. By reducing the single channel conductance gLCC by 30%, peak ICa,L decreased from 6 pA/pF to 4 pA/pF, and Ca alternans now occurred at the same PCL=500 ms (purple traces in Fig. 3A). In the second case, 30% of the LCC clusters were randomly removed from the myocyte (resulting in 30% of the RyR clusters being orphaned), which also caused a 30% reduction in whole-cell ICa,L and promoted Ca alternans (purple traces in Fig. 3B). In the third case, instead of removing the LCCs from the myocytes, these (30%) LCCs were randomly redistributed to other CRUs (so that the total number of LCCs remained the same and the whole cell ICa,L was not decreased, and in fact became slightly larger due to a weaker Ca-dependent inactivation of the LCCs that were no longer sensing the higher Ca in dyadic space), Ca alternans still developed (purple traces in Fig. 3C). These findings show that not only is the amplitude of whole-cell ICa,L important for Ca alternans, but also the spatial distribution of LCCs.
To systematically study the effects of the spatial distribution of LCCs on Ca alternans, we carried out simulations using different levels of Ca load (by changing [Na]i) and varying the percentage of TT disruption (with total LCCs remaining the same by re-distributing the LCCs from the removed LCC clusters to other CRUs). The results at a fixed PCL=500 ms are shown in Fig. 4A. With no TT disruption or [Na]i<10 mM, alternans did not occur. Both [Na]i elevation and TT disruption were required to induce Ca alternans at this PCL. The amplitude of alternans increased as [Na]i or the percentage of TT disruption increased. Fig. 4B shows that for a fixed [Na]i of 11 mM, increasing the percentage of TT disruption promoted alternans and increased SR load.
The dependence of Ca alternans on PCL is shown in Fig. 4C. For [Na]i=11 mM and pLCC=30%, alternans started between PCL=800 ms and 700 ms, and its amplitude increased together with SR load as PCL was decreased further. SR load alternated together with peak [Ca]i. However, if [Ca]SR was clamped at a constant load (700 μM) and other conditions were the same as in Fig. 4C, alternans still occurred, although its onset started at a shorter PCL (between 500 ms and 400 ms), as shown in Fig. 4D. Since alternans occurred even when SR Ca was clamped, this indicates that refractoriness of SR Ca release was primarily responsible for alternans. However, SR load alternans also amplified Ca alternans under these conditions, since the onset of alternans occurred at a longer PCL when the SR load was allowed to vary freely.
3.3. Effects of TT disruption on Ca alternans when SERCA is down-regulated
We next studied the condition in which SERCA is down-regulated with increased RyR leakiness and INCX as described in Methods. Fig. 5A shows how [Na]i and the percentage of TT disruption affected the alternans amplitude (ΔCaALT). TT disruption promoted Ca alternans, but, unlike the case in Fig. 4A, TT disruption was not obligatory, since at 0% TT disruption, Ca alternans still occurred, albeit at a higher [Na]i. Fig. 5B shows peak [Ca]i and SR load versus pLCC for [Na]i=10 mM at PCL=500 ms. Fig. 5C shows peak [Ca]i and SR load at different PCL for pLCC=30% and [Na]i=10 mM. Under these conditions, alternans occurred at very slow pacing rates between PCL=4 s and 5 s. As PCL shortened, the amplitude of both the Ca transient and ΔCaALT decreased modestly. SR load also decreased until PCL=1 s, beyond which the SR Ca load increased. Alternans disappeared when SR Ca was clamped (Fig. 5D), indicating that Ca alternans depended on SR load alternans. Moreover, at these slow pacing rates, the SR and RyRs were fully recovered. Therefore, the refractoriness of SR Ca release due to SR refilling or RyR recovery played no role here. Interestingly, if the magnitude of the SERCA was restored to the normal value, the onset of alternans now occurred at a much shorter PCL (Fig. 5E), in the same range as in Fig. 4C, indicating the importance of decreased SERCA pump activity in promoting alternans. Note that Ca alternans at slow heart rates (≤0.5 Hz) have been observed in experimental studies [29, 33, 35].
Figure 6 compares traces of [Ca]i, [Ca]SR, INCX, ICa,L and the percentage of RyR available for opening during alternans when RyR, SERCA and NCX properties were normal (panel A) or remodeled (panel B). A major difference is that for normal SERCA activity, Ca uptake by the SR was much greater following the large release beat. However, when SERCA was down-regulated, Ca uptake by the SR was less following the large release beat than that following the small release beat (see Discussion).
Figure 6. Sample recordings of [Ca]i, [Ca]SR, INCX, ICa,L, and percentage of the RyR availability for opening.

A. From a simulation in Fig. 4. PCL=500 ms, [Na]i=11 mM, and pLCC=30%. B. From a simulation in Fig. 5. PCL= 1000 ms, [Na]i=10 mM, and pLCC=30%. The amount of Ca uptaken by SR for each of two alternating beats for the two conditions is marked.
3.4. Effects of HF remodeling on both APD and Ca alternans
Besides remodeling of Ca cycling proteins, many ion channels are also remodeled in HF. Therefore, we also simulated a third condition in which we added these ionic current alterations (see Methods) to the Ca cycling protein remodeling to study the interaction between voltage and Ca on alternans. We paced the model with voltage running freely (equivalent to current clamp conditions). Under the normal control condition, decreasing the PCL from 500 to 200 ms did not induce alternans. When Ca cycling properties and ionic currents were modified to simulate HF remodeling, with 30% TT disruption and elevated [Na]i from 9 to 10 mM, APD prolonged from 160 ms (Fig. 1B) to 230 ms (Fig. 7A). APD and Ca alternans now both occurred at very slow heart rates, beginning at PCL=5 s. If, instead of allowing voltage to run freely, we paced the myocyte with the action potential waveform corresponding to that immediately before the onset of alternans, Ca alternans still occurred at the same PCL with roughly the same magnitude (open circles in Fig. 7B). This indicates that alternans originates primarily from the Ca cycling system in this model. Moreover, the lower SERCA activity is the major cause of alternans at slow rates, based on the simulations above.
4. Discussion
TT disruption is one of the remodeling consequences of HF known to contribute to dyssynchronous Ca release and impair excitation-contraction coupling. In a recent study [14], Hong et al showed that loss of TTs also increased the propensity to arrhythmias in mouse hearts. However, the mechanistic link between TT disruption and arrhythmias is not well-understood [13]. In this study, we used computer modeling to investigate this issue and show that TT disruption plays an important role in promoting Ca alternans, a precursor which predisposes the heart to spatially discordant APD alternans and initiation of reentrant arrhythmias [46].
4.1. Mechanisms of TT disruption promoting Ca alternans
Our findings indicate that TT disruption per se does not cause Ca alternans, but promotes Ca alternans in combination with other changes. Two mechanisms of Ca alternans have been described [41, 47]. The first mechanism is SR load-dependent, which was rigorously analyzed in theoretical studies [28, 30] and supported by evidence from experiments [29, 32, 33]. This mechanism depends on a steep SR release-load relationship and reduced SERCA activity which work synergistically to promote Ca alternans, which is mathematically described as [30]:
| (1) |
where lk is the SR load of the previous paced beat and lk+1 is that of the present beat. g(lk) is the fractional SR Ca release and is the Ca taken up by the SR, with being the peak [Ca]i of the present beat. Eq.1 predicts that alternans occurs when
| (2) |
where g′ and h′ are the slopes of the two functions at the steady state, respectively. Therefore, alternans is promoted by a steep fractional SR release curve (large g′) and/or a shallow SR uptake function (small h′). A shallow uptake function indicates that the amount of Ca taken up by the SR is not very insensitive to the magnitude of cytosolic free [Ca]i. For example, in the two cases shown in Fig. 6, when SERCA activity was normal, Ca uptake following the large release beat was much greater than that following the small release beat (Fig. 6A). However, when SERCA activity was reduced as in HF, the Ca taken up by the SR following the large beat was actually less than that of the small release beat (in this case h′<0) (Fig. 6B), which is the mechanism promoting alternans based on Eq.2. Since this mechanism of alternans does not depend on refractoriness, alternans can occur at normal or slow heart rates, as indeed observed in experiments [29, 33, 35].
As shown in Fig. 2D, HF remodeling steepens the fractional SR release curve and TT disruption made it even steeper. Moreover, TT disruption results in a higher SR load (Fig. 2C). Therefore, in this mechanism of alternans, TT disruption promotes Ca alternans by steepening the fractional SR release curve and increasing SR load. However, as shown in Fig. 5A, alternans can occur without TT disruption, and TT disruption plays the role of a facilitator.
The second mechanism of Ca alternans is related to CRU refractoriness, and is also well-supported by experiments in which Ca alternans occurs without concomitant SR load alternans [35, 36, 39, 40]. Theoretical analysis (called 3R theory) [37, 41] shows that refractoriness alone is not sufficient, and the ability of CRUs to activate neighboring CRUs (Ca spark-induced secondary sparks) via diffusive coupling is also required. The mathematical description of this theory is
| (3) |
where Nk+1 is the number of Ca sparks in the present beat, Nk is the number of sparks in the previous beat, N0 is the total number of CRUs, and f is a nonlinear function describing the recruitment rate, which was derived as:
| (4) |
In Eqs. 3 and 4, α is the probability or percentage of sparks triggered by the random opening of the LCCs (the first R of the 3R theory); γ is the probability that a spark recruits a neighboring CRU to fire (the second R); and β is the probability that a CRU which on the previous beat remains refractory during the present beat (the third R). M is the number of neighbors of a CRU. α and γ are affected by many physiological factors (see Nivala and Qu [38]), such as LCC open probability, SR load, RyR leakiness, SERCA activity, NCX, and CRU spacing, and β is determined jointly by CRU refractoriness and PCL. Ca alternans occurs when β and γ are large, and α is in the intermediate range (gray region in Fig. 8A). It is intuitively clear that in order to have an alternating beat-to-beat pattern, the refractory period of CRUs must be engaged. Otherwise the same number of CRUs would fire from beat to beat. However, due to the randomness of sparks, just engaging the refractory period is not sufficient to result in alternans. Coupling between CRUs is needed to locally synchronize the CRUs to fire so that more CRUs fire on one beat than on the alternate beat, resulting in alternans. However, if all the CRUs are triggered by their LCCs (high α), there is no chance for recruitment to occur. Factors that decrease the number of sparks triggered by LCCs include: 1) reduced LCC conductance or open probability; 2) reduced LCC-RyR coupling fidelity (e.g., due to enlarged dyadic volume); and 3) TT disruption. In the first two cases, the beat-to-beat firing probability of a CRU is not 100% because of the reduced LCC conductance, open probability or reduced LCC-RyR coupling fidelity. That is, CRUs are not reliably triggered by their LCCs. The CRUs which are not activated by LCCs then provide the chance for spark-induced sparks to occur (this is why alternans requires α<1 in the 3R theory [37, 41]). In the third case, removal of LCC clusters provides the chance for spark-induced sparks to occur by creating orphaned RyR clusters. Thus, the role of TT disruption in promoting alternans is to allow recruitment to occur. As shown in Fig. 4A, TT disruption is required for alternans when LCC and LCC-RyR coupling are normal.
Figure 8. Mechanisms of TT disruption promoting Ca alternans.
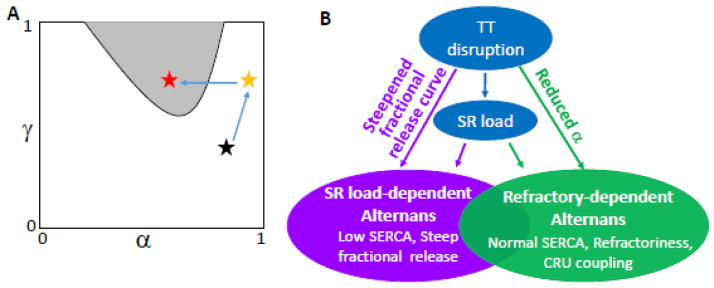
A. α-γ parameter space from Eqs. 3 and 4 demarcating the region of Ca alternans (gray shading) when refractoriness (β) is high, according to the 3R theory [37]. Assuming the black star as the normal position, elevation of [Ca]i and SR load due to elevation of [Na]i increases α and γ, bringing the system to the yellow star. TT disruption reduces LCC-triggered sparks and thus α, bringing the system into the alternans region (red star). B. Schematic plot summarizing the mechanisms by which TT disruption promotes Ca alternans.
In summary (Fig. 8B), TT disruption results in orphaned RyR clusters, which reduces the number of CRUs activated by LCC clusters and thereby increases the opportunity for recruitment. In the SR Ca load-dependent mechanism of alternans, recruitment causes a steeper SR fractional release curve which promotes alternans. In the RyR refractoriness-dependent mechanism of alternans, the recruitment allows synchronization of sparks to promote alternans. TT disruption may increase SR load which is also a factor promoting Ca alternans.
4.2. Implications to Ca alternans in HF
Electrophysiological remodeling in HF is complex and evolves over time as HF progresses [15–18]. As shown in animal studies [26, 48], the Ca transient can be increased in early stages of HF, but becomes prolonged with reduced amplitude in later stages of HF. It is likely that in the early stages, SERCA is still relatively normal and Ca is increased due to elevated [Na]i, while in the late stages, SERCA down-regulation and RyR leakiness depress the Ca transient. However, TT disruption may start early in the development of HF [6, 49]. Based on these observations, coupled with the mechanistic insights from our computational results, one can speculate that different mechanisms may be involved in Ca alternans at different stages of HF. In the early stage when SERCA activity is still relatively normal, Ca elevation due to [Na]i elevation increases both α and γ with or without increased RyR leakiness (from the black star to the yellow star in Fig. 8A) [38]. However, this does not cause alternans. TT disruption then reduces α to allow spark-induced sparks to occur, which results in alternans (from the yellow star to the red star in Fig. 8A). Therefore, TT disruption plays a very important role in promoting alternans in this stage. In the later stages of HF where SERCA is down-regulated, leaky RyR and TT disruption result in a steeper SR fractional release curve and shift the curve to a lower Ca load (Fig. 2D), promoting Ca alternans by the SR load-dependent mechanism. In this setting, TT disruption plays a less important role and increased RyR leakiness and SERCA down-regulation are the key factors. Note that in the early stage of HF, alternans requires the presence of refractoriness, and thus, tends to occur at fast heart rates, and may not be seen at normal heart rates. In the late stage of HF, however, refractoriness is not required, alternans can occur at either fast or slow heart rates, which may be responsible for pulsus alternans seen at normal heart rates in patients with severe end-stage HF.
4.3. Limitations
Several limitations should be mentioned. Although our cell model contains a spatially-distributed CRU network, the CRU model lacks asymmetrical details of geometry, RyR localization, and heterogeneous RyR cluster distribution [50–52]. In addition, in our simulations, the TTs were randomly disrupted, which may not be realistic. Spatial correlation or coarser patterns of TT disruption may cause more complex spatiotemporal behaviors of Ca alternans and other Ca cycling dynamics which need to be addressed in future studies. Refractoriness of Ca release is controversial, with time constants measured experimentally ranging from 100 to 650 ms [53–57]. Some studies have concluded that the refractoriness is mainly related to SR refilling and luminal Ca sensing, while others show that it is due to recovery of the RyR channels. The refractoriness in our model is caused by RyR recovery from inactivation and the time constant is in the slow range (Fig. 6). This accounts for alternans occurring at PCL=500 ms or less when alternans is refractoriness-dependent. A related issue is our use of the RyR model by Stern et al [58], in which spark termination is caused by cytosolic Ca-dependent inactivation of RyRs, for which there is no clear experimental evidence. To address this concern, we repeated some of the key simulations presented in this study using a completely different ventricular myocyte model developed by Restrepo et al [59] in which RyR opening and inactivation are regulated by luminal SR Ca via calsequestrin. We repeated the simulations shown in Fig. 3 and obtained similar results (see supplemental Fig. S1). We also used this model to simulate the effects of SERCA pump and confirmed that when SERCA was reduced to a sufficiently low level, alternans occurred at very slow heart rates (see supplemental Fig. S2), in agreement with our conclusions using the Stern et al model. In the future, other ventricular myocyte models or RyR models can be tested to further validate these conclusions. We expect that although quantitatively differences may exist, the qualitative effects of TT disruption on Ca alternans and the conditions for the two mechanisms of Ca alternans will hold true in general. Finally, in our model of HF (Fig. 7), we showed that alternans was primarily driven by the Ca cycling instability. At fast heart rates, however, APD restitution is engaged, and as shown in previous studies [30, 60, 61], the bidirectional coupling between voltage and Ca plays an important role in promoting alternans.
4.4. Conclusions
Despite these limitations, our present study provides mechanistic insights into: 1) the conditions for the two distinct mechanisms of Ca alternans in HF; 2) the roles of TT disruption in promoting Ca alternans; and 3) the mechanisms of alternans at different stages of HF development. Moreover, our study also shows that the spatial distribution of LCCs, not the magnitude of the whole-cell current per se, plays an important role in Ca alternans, i.e., alternans can be induced by altered spatial distribution of LCCs.
Supplementary Material
Highlights.
Effects of T-tubule disruption on calcium alternans are computationally simulated.
Two mechanisms of calcium alternans are demonstrated.
T-tubule disruption promotes calcium alternans under both mechanisms.
SERCA activity is a key parameter promoting the two mechanisms.
Different mechanisms responsible for alternans at different heart failure stages.
Acknowledgments
This work is supported by grants from National Institute of Health P01 HL078931, R56 HL118041, American Heart Association, Western States Affiliate, Beginning Grant-in-Aid 14BGIA18470026 (M.N.), and the Laubisch and Kawata Endowments.
Footnotes
Disclosure
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Soeller C, Cannell MB. Examination of the Transverse Tubular System in Living Cardiac Rat Myocytes by 2-Photon Microscopy and Digital Image Processing Techniques. Circ Res. 1999;84:266–75. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- 2.Brette F, Orchard C. Resurgence of cardiac t-tubule research. Physiology (Bethesda) 2007;22:167–73. doi: 10.1152/physiol.00005.2007. [DOI] [PubMed] [Google Scholar]
- 3.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, et al. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A. 2009;106:6854–9. doi: 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A. 2006;103:4305–10. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I, et al. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574:519–33. doi: 10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, et al. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–31. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swift F, Franzini-Armstrong C, Oyehaug L, Enger UH, Andersson KB, Christensen G, et al. Extreme sarcoplasmic reticulum volume loss and compensatory T-tubule remodeling after Serca2 knockout. Proc Natl Acad Sci U S A. 2012;109:3997–4001. doi: 10.1073/pnas.1120172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, et al. Remodeling of T-Tubules and Reduced Synchrony of Ca2+ Release in Myocytes From Chronically Ischemic Myocardium. Circ Res. 2008;102:338–46. doi: 10.1161/CIRCRESAHA.107.160085. [DOI] [PubMed] [Google Scholar]
- 9.Sachse FB, Torres NS, Savio-Galimberti E, Aiba T, Kass DA, Tomaselli GF, et al. Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ Res. 2012;110:588–97. doi: 10.1161/CIRCRESAHA.111.257428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GS, Steinbrecher JH, et al. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ Res. 2012;111:402–14. doi: 10.1161/CIRCRESAHA.112.274530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong T-T, Smyth JW, Chu KY, Vogan JM, Fong TS, Jensen BC, et al. BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm. 2012;9:812–20. doi: 10.1016/j.hrthm.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sacconi L, Ferrantini C, Lotti J, Coppini R, Yan P, Loew LM, et al. Action potential propagation in transverse-axial tubular system is impaired in heart failure. Proc Natl Acad Sci U S A. 2012;109:5815–9. doi: 10.1073/pnas.1120188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orchard CH, Bryant SM, James AF. Do t-tubules play a role in arrhythmogenesis in cardiac ventricular myocytes? The Journal of Physiology. 2013;591:4141–7. doi: 10.1113/jphysiol.2013.254540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong T, Yang H, Zhang S-S, Cho HC, Kalashnikova M, Sun B, et al. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med. 2014;20:624–32. doi: 10.1038/nm.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomaselli GF, Marbán E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–83. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 16.O’Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res. 1999;84:562–70. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- 17.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–67. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 18.Nattel S, Maguy A, Le Bouter S, Yeh Y-H. Arrhythmogenic Ion-Channel Remodeling in the Heart: Heart Failure, Myocardial Infarction, and Atrial Fibrillation. Physiol Rev. 2007;87:425–56. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- 19.Traube L. Ein fall von pulsus bigeminus nebst bemerkungen tiber die lebershwellungen bei klappenfehlern und uber acute leberatrophic. Ber Klin Wschr. 1872;9:185. [Google Scholar]
- 20.Rosenbaum DS. T wave alternans: a mechanism of arrhythmogenesis comes of age after 100 years. J Cardiovasc Electrophysiol. 2001;12:207–9. doi: 10.1046/j.1540-8167.2001.00207.x. [DOI] [PubMed] [Google Scholar]
- 21.Rosenbaum DS, Jackson LE, Smith JM, Garan H, Ruskin JN, Cohen RJ. Electrical alternans and vulnerability to ventricular arrhythmias. N Engl J Med. 1994;330:235–41. doi: 10.1056/NEJM199401273300402. [DOI] [PubMed] [Google Scholar]
- 22.Baravelli M, Salerno-Uriarte D, Guzzetti D, Rossi MC, Zoli L, Forzani T, et al. Predictive significance for sudden death of microvolt-level T wave alternans in New York Heart Association class II congestive heart failure patients: a prospective study. Int J Cardiol. 2005;105:53–7. doi: 10.1016/j.ijcard.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 23.Salerno-Uriarte JA, De Ferrari GM, Klersy C, Pedretti RF, Tritto M, Sallusti L, et al. Prognostic value of T-wave alternans in patients with heart failure due to nonischemic cardiomyopathy: results of the ALPHA Study. J Am Coll Cardiol. 2007;50:1896–904. doi: 10.1016/j.jacc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 24.Wilson LD, Jeyaraj D, Wan X, Hoeker GS, Said TH, Gittinger M, et al. Heart failure enhances susceptibility to arrhythmogenic cardiac alternans. Heart Rhythm. 2009;6:251–9. doi: 10.1016/j.hrthm.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wasserstrom JA, Sharma R, Kapur S, Kelly JE, Kadish AH, Balke CW, et al. Multiple Defects in Intracellular Calcium Cycling in Whole Failing Rat Heart. Circulation: Heart Failure. 2009;2:223–32. doi: 10.1161/CIRCHEARTFAILURE.108.811539. [DOI] [PubMed] [Google Scholar]
- 26.Kapur S, Aistrup GL, Sharma R, Kelly JE, Arora R, Zheng J, et al. Early development of intracellular calcium cycling defects in intact hearts of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2010;299:H1843–53. doi: 10.1152/ajpheart.00623.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aistrup GL, Gupta DK, Kelly JE, O’Toole MJ, Nahhas A, Chirayil N, et al. Inhibition of the late sodium current slows t-tubule disruption during the progression of hypertensive heart disease in the rat. Am J Physiol Heart Circ Physiol. 2013;305:H1068–79. doi: 10.1152/ajpheart.00401.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiferaw Y, Watanabe MA, Garfinkel A, Weiss JN, Karma A. Model of intracellular calcium cycling in ventricular myocytes. Biophys J. 2003;85:3666–86. doi: 10.1016/S0006-3495(03)74784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diaz ME, O’Neill SC, Eisner DA. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res. 2004;94:650–6. doi: 10.1161/01.RES.0000119923.64774.72. [DOI] [PubMed] [Google Scholar]
- 30.Qu Z, Shiferaw Y, Weiss JN. Nonlinear dynamics of cardiac excitation-contraction coupling: an iterated map study. Phys Rev E. 2007;75:011927. doi: 10.1103/PhysRevE.75.011927. [DOI] [PubMed] [Google Scholar]
- 31.Tao T, O’Neill SC, Diaz ME, Li YT, Eisner DA, Zhang H. Alternans of cardiac calcium cycling in a cluster of ryanodine receptors: a simulation study. Am J Physiol Heart Circ Physiol. 2008;295:H598–609. doi: 10.1152/ajpheart.01086.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie LH, Sato D, Garfinkel A, Qu Z, Weiss JN. Intracellular Ca alternans: coordinated regulation by sarcoplasmic reticulum release, uptake, and leak. Biophys J. 2008;95:3100–10. doi: 10.1529/biophysj.108.130955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Diaz ME, Eisner DA, O’Neill S. The effects of membrane potential, SR Ca2+ content and RyR responsiveness on systolic Ca2+ alternans in rat ventricular myocytes. J Physiol. 2009;587:1283–92. doi: 10.1113/jphysiol.2008.164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huertas MA, Smith GD, Gyorke S. Ca2+ alternans in a cardiac myocyte model that uses moment equations to represent heterogeneous junctional SR Ca2+ Biophys J. 2010;99:377–87. doi: 10.1016/j.bpj.2010.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL, Blatter LA. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol. 2000;524(Pt 3):795–806. doi: 10.1111/j.1469-7793.2000.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picht E, DeSantiago J, Blatter LA, Bers DM. Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ Res. 2006;99:740–8. doi: 10.1161/01.RES.0000244002.88813.91. [DOI] [PubMed] [Google Scholar]
- 37.Rovetti R, Cui X, Garfinkel A, Weiss JN, Qu Z. Spark-Induced Sparks As a Mechanism of Intracellular Calcium Alternans in Cardiac Myocytes. Circ Res. 2010;106:1582–91. doi: 10.1161/CIRCRESAHA.109.213975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nivala M, Qu Z. Calcium Alternans in a Couplon Network Model of Ventricular Myocytes: Role of Sarcoplasmic Reticulum Load. Am J Physiol Heart Circ Physiol. 2012;303:H341–52. doi: 10.1152/ajpheart.00302.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shkryl VM, Maxwell JT, Domeier TL, Blatter LA. Refractoriness of sarcoplasmic reticulum Ca release determines Ca alternans in atrial myocytes. American Journal of Physiology - Heart and Circulatory Physiology. 2012;302:H2310–H20. doi: 10.1152/ajpheart.00079.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Myles RC, De Jesus NM, Ohlendorf AKP, Bers DM, Ripplinger CM. Optical Mapping of Sarcoplasmic Reticulum Ca2+ in the Intact Heart: Ryanodine Receptor Refractoriness During Alternans and Fibrillation. Circ Res. 2014;114:1410–21. doi: 10.1161/CIRCRESAHA.114.302505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qu Z, Nivala M, Weiss JN. Calcium alternans in cardiac myocytes: Order from disorder. J Mol Cell Cardiol. 2013;58:100–9. doi: 10.1016/j.yjmcc.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nivala M, de Lange E, Rovetti R, Qu Z. Computational modeling and numerical methods for spatiotemporal calcium cycling in ventricular myocytes. Front Physiol. 2012;3:114. doi: 10.3389/fphys.2012.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shannon TR, Wang F, Bers DM. Regulation of Cardiac Sarcoplasmic Reticulum Ca Release by Luminal [Ca] and Altered Gating Assessed with a Mathematical Model. Biophys J. 2005;89:4096–110. doi: 10.1529/biophysj.105.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 45.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–4. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 46.Qu Z, Garfinkel A, Chen PS, Weiss JN. Mechanisms of discordant alternans and induction of reentry in simulated cardiac tissue. Circulation. 2000;102:1664–70. doi: 10.1161/01.cir.102.14.1664. [DOI] [PubMed] [Google Scholar]
- 47.Escobar AL, Valdivia HH. Cardiac Alternans and Ventricular Fibrillation: A Bad Case of Ryanodine Receptors Reneging on Their Duty. Circ Res. 2014;114:1369–71. doi: 10.1161/CIRCRESAHA.114.303823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shorofsky SR, Aggarwal R, Corretti M, Baffa JM, Strum JM, Al-Seikhan BA, et al. Cellular mechanisms of altered contractility in the hypertrophied heart: big hearts, big sparks. Circ Res. 1999;84:424–34. doi: 10.1161/01.res.84.4.424. [DOI] [PubMed] [Google Scholar]
- 49.Shah SJ, Aistrup GL, Gupta DK, O’Toole MJ, Nahhas AF, Schuster D, et al. Ultrastructural and cellular basis for the development of abnormal myocardial mechanics during the transition from hypertension to heart failure. Am J Physiol Heart Circ Physiol. 2014;306:H88–100. doi: 10.1152/ajpheart.00642.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hake J, Edwards AG, Yu Z, Kekenes-Huskey PM, Michailova AP, McCammon JA, et al. Modeling Cardiac Calcium Sparks in a Three-Dimensional Reconstruction of a Calcium Release Unit. J Physiol. 2012;590:4403–22. doi: 10.1113/jphysiol.2012.227926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cannell MB, Kong CH, Imtiaz MS, Laver DR. Control of sarcoplasmic reticulum Ca2+ release by stochastic RyR gating within a 3D model of the cardiac dyad and importance of induction decay for CICR termination. Biophys J. 2013;104:2149–59. doi: 10.1016/j.bpj.2013.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baddeley D, Jayasinghe ID, Lam L, Rossberger S, Cannell MB, Soeller C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc Natl Acad Sci U S A. 2009;106:22275–80. doi: 10.1073/pnas.0908971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol. 1996;270:C148–59. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- 54.DelPrincipe F, Egger M, Niggli E. Calcium signalling in cardiac muscle: refractoriness revealed by coherent activation. Nat Cell Biol. 1999;1:323–9. doi: 10.1038/14013. [DOI] [PubMed] [Google Scholar]
- 55.Sobie EA, Song LS, Lederer WJ. Local recovery of Ca2+ release in rat ventricular myocytes. J Physiol. 2005;565:441–7. doi: 10.1113/jphysiol.2005.086496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, et al. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. 2012;110:569–77. doi: 10.1161/CIRCRESAHA.111.260455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wasserstrom JA, Sharma R, O’Toole MJ, Zheng J, Kelly JE, Shryock J, et al. Ranolazine antagonizes the effects of increased late sodium current on intracellular calcium cycling in rat isolated intact heart. J Pharmacol Exp Ther. 2009;331:382–91. doi: 10.1124/jpet.109.156471. [DOI] [PubMed] [Google Scholar]
- 58.Stern MD, Pizarro G, Rios E. Local control model of excitation-contraction coupling in skeletal muscle. J Gen Physiol. 1997;110:415–40. doi: 10.1085/jgp.110.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Restrepo JG, Weiss JN, Karma A. Calsequestrin-mediated mechanism for cellular calcium transient alternans. Biophys J. 2008;95:3767–89. doi: 10.1529/biophysj.108.130419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shiferaw Y, Sato D, Karma A. Coupled dynamics of voltage and calcium in paced cardiac cells. Phys Rev E Stat Nonlin Soft Matter Phys. 2005;71:021903. doi: 10.1103/PhysRevE.71.021903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Groenendaal W, Ortega FA, Krogh-Madsen T, Christini DJ. Voltage and calcium dynamics both underlie cellular alternans in cardiac myocytes. Biophys J. 2014;106:2222–32. doi: 10.1016/j.bpj.2014.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.