

Abstract
The pharmaceutical industry has significantly contributed to improving human health. Drugs have been attributed to both increasing life expectancy and decreasing health care costs. Unfortunately, there has been a recent decline in the creativity and productivity of the pharmaceutical industry. This is a complex issue with many contributing factors resulting from the numerous mergers, increase in out-sourcing, and the heavy dependency on high-throughput screening (HTS). While a simple solution to such a complex problem is unrealistic and highly unlikely, the inclusion of metabolomics as a routine component of the drug discovery process may provide some solutions to these problems. Specifically, as the binding affinity of a chemical lead is evolved during the iterative structure-based drug design process, metabolomics can provide feedback on the selectivity and the in vivo mechanism of action. Similarly, metabolomics can be used to evaluate and validate HTS leads. In effect, metabolomics can be used to eliminate compounds with potential efficacy and side effect problems while prioritizing well-behaved leads with druglike characteristics.
Benefits of Drug Discovery
Extending Life Expectancy
The pharmaceutical industry has had a tremendous beneficial impact on human health. In fact, the increased use of pharmaceuticals in developed nations has significantly contributed to an increase in life expectancy (Figure 1a).1 Lichtenberg has estimated that 40% of the increase in life expectancy from 1982 to 2001 across 52 countries can be attributed to new drug launches.2 Specifically, greater than 80% of the gains in life expectancy for cancer patients have been attributed to new treatments that include medicines.3 A separate study indicates that 50–60% of life expectancy gains in cancer patients are directly attributed to pharmaceuticals.4 Similarly, the death rate from human immunodeficiency virus infection/acquired immunodeficiency syndrome (HIV/AIDS) decreased 85% after the approval of antiretroviral treatments in 19955 and, correspondingly, AIDS patients now have close to normal life expectancies.6 A comparable outcome has been observed with a 25% decline in coronary heart disease and mortality between 1997 and 2007, which is due, in part, to cholesterol-lowering drugs (Figure 1b).7,8 A recently completed 22-year follow-up study on the benefit of treating hypertension indicated that life expectancy increased one day for each month of treatment.9 In addition, the introduction of antibiotics has largely contributed to the eight year increase in life expectancy between 1944 and 1972 (Figure 1a).10 In fact, the major causes of death in 1900 were infectious diseases (pneumonia, tuberculosis, diarrhea, and enteritis),11 but due to the beneficial impact of antibiotics, by 2013 the leading causes of death can be attributed to an aging population (heart disease, cancer, stroke, and chronic lung disease).5 Clearly, there is a growing body of empirical evidence demonstrating the significant contribution of pharmaceuticals to increasing life expectancy.
Figure 1.

(a) A correlation between increasing U.S. life expectancy5 (solid black diamonds) and the decreasing U.S. mortality rate associated with infectious disease (solid red circles) between 1930 and 2006 (adapted from Armstong et al.10). (b) A correlation between the decrease in U.S. heart disease death rates5 and the decrease in U.S. LDL cholesterol levels (adapted from Kaufman et al.7). (c) Plot of the total U.S. health care cost per capita (solid black diamonds) and the associated expenditures on pharmaceuticals (solid red circles) between 2000 and 2011.12 (d) Plot of the decrease in new drugs approved by the FDA (solid black diamonds) between 1996 and 2013. Trend lines are provided to simply assist in visualizing the overall data trends and are not a model fit.
Reducing Health Care Costs
Health care costs in the U.S. have expanded at a rate significantly faster than inflation (Figure 1c).12 The costs of pharmaceuticals are a significant contributor to these rising health care expenditures, where pharmaceuticals expenditures account for approximately 10% of the total U.S. health care costs per year. Despite this inherent initial expense, pharmaceuticals have actually contributed to an overall decrease in the cost of health care.13 Simply, pharmaceutical use prevents serious health-related events that lowers the use of health care resources such as emergency room visits, hospital stays, medical personnel time (nurses, physicians, surgeons, etc.), surgery facilities, and diagnostic services (X-ray, magnetic resonance imaging (MRI), computerized tomography (CT) scans, etc.). In fact, a recent study of the Canadian health care system indicated that for each dollar spent on pharmaceuticals by a male patient, a decrease of 1.48 dollars was achieved in other health care resources.14 Similarly, the use of cholesterol lowering drugs in Scandinavia has led to a 34% reduction in hospital stays and a corresponding savings of $3872 per patient.15 Treating relapsing–remitting multiple sclerosis patients with cladribine (immunosuppressant) resulted in a similar decrease in hospital stays, emergency room visits, and missed work.16 Likewise, a randomized study across 28 countries analyzed the incremental cost-effectiveness ratio (ICER) of treating patients with clopidogrel, an antiplatelet agent.17 The treatment was determined to be cost-effective, with an ICER that ranged from $4833 to $6475. These studies, among others, have clearly indicated that the use of pharmaceuticals is a beneficial and cost-effective approach to lowering total health care costs.
Drug Discovery Challenges
Decline in New Drugs Approved by the U.S. Food and Drug Administration (FDA)
Despite the historical benefits of drug discovery to human health and well-being, there has been a disturbing decline over the last decades in the creativity and output of the pharmaceutical industry.18−21 Since 1996, there has been a precipitous drop in the number of new drugs approved by the FDA (Figure 1d). Also, an increasing number of these new drugs are simply repositioned22 or “me too”.23 Furthermore, there has been a steady decline in the efficiency of drug discovery as measured by the number of drugs per dollar of research and development (R&D) spending.18 What are the underlying problems with drug discovery causing this diminishing productivity? First and foremost, drug discovery is probably the most difficult endeavor embarked upon and, as a result, a high failure rate is commonly encountered. For example, an extremely high attrition rate occurs during the drug discovery process, where estimates indicate that it takes upward of 45 active research projects to generate one new drug application (NDA) to the FDA.19,24 Furthermore, the corresponding success rate of NDAs in clinical trials is only 11%.25 Further contributing to the situation is the high cost associated with the drug discovery process. The average cost for developing a new drug has been estimated to range from $800 million26 to $1.8 billion dollars.19 The high cost associated with drug discovery will severely limit the number of active research projects pursued by a company.
Potential Reasons for the Decline in New Drugs
Unfortunately, in response to these challenges, a number of serious and deleterious decisions have been made that have only aggravated the situation. The pharmaceutical industry has undergone an extensive round of mergers. Between 2000 and 2011, the pharmaceutical industry has lost nearly 300000 jobs according to the consulting firm Challenger, Gray & Christmas. Obviously, this drastic decline in highly trained personnel with extensive drug discovery experience has only contributed to the continuing decrease in the discovery of new drugs. Additionally, the pharmaceutical industry has significantly diminished drug development efforts in a number of therapeutic areas including Alzheimer’s and Parkinson’s disease,27 antibiotics,28 psychiatric disorders,29,30 vaccines,31 and even cardiovascular diseases.32 These therapeutic areas are being abandoned by some companies because of the low success rate or a low return on investment. Instead, the focus is on identifying the next “blockbuster” drug with profits in the billions of dollars per year.33 This approach effectively eliminates the possibility of discovering new therapies for important areas of human health. Conversely, the concentrated effort within a few research areas has inevitably led to the decrease in new drugs and the increase in the number of redundant drugs.34
Other ill-conceived managerial decisions have also contributed to the decline in the productivity and creativity of the pharmaceutical industry, which include outsourcing,35,36 the application of metrics,24,37 and restricting project timelines.38 In effect, drug discovery is being treated like any other manufacturing process where cost savings is driving business decisions, but assembly line science does not work because the process is highly interdisciplinary and unpredictable.39,40 Outsourcing simply leads to the isolation of critical components of the process, but breakthroughs and problem-solving require routine interactions between experts with diverse skill sets and experiences. Similarly, metrics emphasizes the bookkeeping of meaningless data to create the illusion of success, but the number of compounds synthesized, assays run, or structures solved is irrelevant if a drug is not the outcome. Finally, restricting research projects to a predefined timeline of only two to three years almost guarantees a pattern of repetitive failures. A priori predicting how long it will take any project to yield a positive outcome is simply foolish. Instead, an informed decision based on experience and results should be used to make a decision on whether to terminate or continue a project.
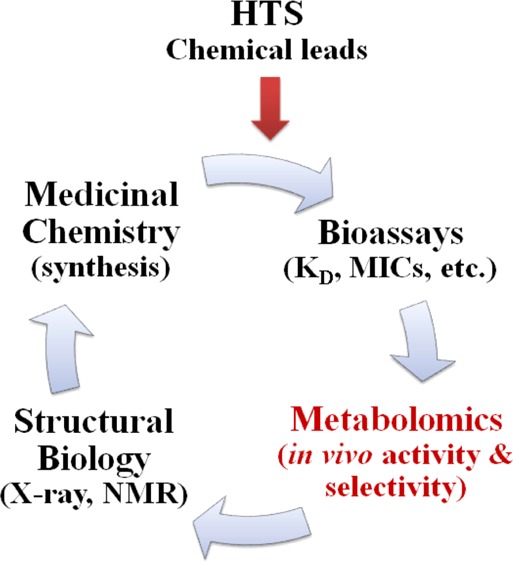
The focus on predefined timelines and assembly line science is a result of the integral role that HTS have been playing in the drug discovery process.41−44 This has occurred despite the fact that HTS has not contributed to an increase in the number of new drug discoveries.25,45−47 In general, HTS does not provide information on the in vivo mechanism of action, does not provide evidence of a direct binding interaction with the protein target, and has a tendency to identify a large number of hits with undesirable modes of action or false positives.48−54 Furthermore, the false leads that routinely emerge from HTS will contribute to delays in a drug discovery project and may eventually result in a project being terminated. In fact, poor HTS outcomes may also contribute to the observed failures of drug leads in the clinic.25,46 Of course, the HTS community has been aggressively responding to these issues by improving the quality and diversity of screening libraries,55−58 by improving the statistical analysis of HTS data sets,53,54 and by combining HTS with virtual screens.59 Unfortunately, a significant number of the problems encountered in the pharmaceutical industry are self-inflicted and will require a philosophical paradigm shift in how drug discovery is conducted. Clearly, these problems will not be solved with technology advancements like structure-based drug discovery,60 combinatorial chemistry,61 fragment-based screening,62 or metabolomics,63 but the adaption of metabolomics and other related approaches as an integral part of a drug discovery program has the potential of benefiting the drug discovery process.
Metabolomics
Metabolomics Overview
Metabolomics is a natural extension of genomics, transcriptomics, and proteomics, but instead of monitoring changes in the expression of genes or proteins, metabolomics monitors changes in the concentration of the low-molecular-weight (<1 kDa) compounds (metabolites) present in a cell, tissue, organ, organism, or biofluid (urine, plasma, cerebrospinal fluid, etc.). The metabolome is the entire collection of small molecules or metabolites within a biological sample that may include amino acids, carbohydrates, cofactors, fatty acids, nucleotides, and even xenobiotics that includes drugs and drug-associated metabolites. The number of metabolites for a particular biological system may range from thousands to hundreds of thousands of compounds. Importantly, changes in the metabolome or in a specific metabolite are a direct result of changes in the biological activity of an enzyme or protein. Correspondingly, metabolomics is commonly employed to define phenotypes.64 This is a critical difference between metabolomics and other “omics” techniques. The change in the expression level of a gene or protein does not necessarily correlate with a change in protein activity.65 Thus, metabolomics provides a direct measure of the state of the cell or biological system, where changes in the metabolome capture how the system responds to environmental or genetic stress. Specifically, a drug or an active chemical lead would be expected to perturb the metabolome of a cell or tissue upon treatment.
Targeted and untargeted metabolomics are two distinctly different approaches to investigating the metabolome. A targeted metabolomics approach follows changes to a specific metabolite or set of metabolites based on some prior information or hypothesis. Specifically, the identified or targeted metabolites are expected to respond to a drug treatment, disease state, genetic modification, or some other environmental stressor. Conversely, untargeted metabolomics is discovery based. The metabolites or metabolic pathways affected by these external stress factors are unknown. The goal of untargeted metabolomics is to monitor the entirety of the metabolome in order to identify the affected metabolites and pathways. In either case, the metabolome extracted from cells or biofluids are compared before and after the addition of the stress factor (drug treatment, genetic mutation, disease, etc.). A variety of analytical techniques are routinely used for metabolomics, but mass spectrometry (MS)64 and nuclear magnetic resonance (NMR) spectroscopy66 are the two most commonly employed methods. The application of NMR to metabolomics has been extensively reviewed, and only critical issues will be highlighted.66−77
Important Issues Related to Metabolomics
Metabolomics is a valuable tool of chemical biology and systems biology, and its application has been rapidly expanding (Figure 2). The ease of use and general utility has contributed to the continually increasing number of metabolomics studies. A standard protocol for analyzing bacterial cell cultures with metabolomics is illustrated in Figure 3.78 Unfortunately, the apparent simplicity of metabolomics also makes it easy to apply the technique incorrectly.79 This has resulted in numerous erroneous studies in the literature. Untargeted metabolomics consists of acquiring one-dimensional (1D) 1H NMR spectra for a set of cell lysates or biofluid samples that are then analyzed using standard multivariate statistical analysis techniques.80 The 1D 1H NMR spectra provides a fingerprint of the state of the metabolome, where principal component analysis (PCA), partial-least-squares (PLS), and orthogonal projection to latent structures discriminant analysis (OPLS-DA) determine if the metabolomes differentiate between the multiple classes (e.g., drug treated vs untreated cells). PLS and OPLS-DA S-plots and loading plots are then used to identify the spectral features (i.e., chemical shifts and associated metabolites) that primarily contribute to these observed class differentiations. Incorrect sample preparation and collection procedures may lead to biologically irrelevant changes to the metabolome.78 Inappropriate data processing and analysis protocols may bias the metabolomics data set, causing an erroneous interpretation.81,82 The lack of model validation may imply a class distinction that does not really exist.83 Similarly, class separations in a scores plot may be identified that are not statistically justified.84,85 Correspondingly, a current challenge in the field of metabolomics is optimizing and standardizing protocols.86,87
Figure 2.

Plot of manuscripts per year where the key word “metabolomics” was included in the topic field in the Web of Science database updated Jan 9, 2014.
Figure 3.

A flowchart of protocol used for the NMR analysis of bacterial metabolomes. Reprinted with permission from ref (78). Copyright 2013 Proteomass Scientific Society.
Unlike genomics or proteomics samples, metabolomics samples are not static and will change with time and from the handling and processing procedures.88 Verifying that the observed changes in a metabolomics sample are biologically relevant, as opposed to artifacts of sample preparation, is critical to a successful metabolomics study. Thus, multiple replicates of cell samples are grown, harvested, and lysed under identical conditions as is practically possible and as quickly as possible. Importantly, processing of samples should be completely randomized because, at a minimum, a time-bias will be imprinted on the samples if each class was processed sequentially. Similarly, each step of the process should be performed by the same individual. Again, variations in individual techniques may bias the outcome if each class was processed by different researchers. Despite these efforts, significant within class variability will occur because of the intrinsic nature of biological samples. Thus, it is also critical to prepare enough replicates per class in order to obtain statistical relevance to any observed class discrimination. Typically, a minimum of 6–10 replicates of cell lysates is required, but the sample size increases significantly for a clinical study.89 It is also critical to quantify the statistical significance of the cluster separations in the resulting scores plot.
Acquiring and processing the NMR spectra for each cell lysate can also induce unintended perturbations in the metabolomics data set. Randomizing the NMR data collection, utilizing automation, and obtaining efficient solvent suppression minimizes these concerns. Specifically, high-quality solvent suppression is required to avoid baseline correcting the NMR spectrum. A minimal and uniform approach to processing the NMR spectrum (Fourier transformation and phase correction) is preferred because baseline correction, applying a window function, zero filling, and other processing steps will modify the data in a biologically irrelevant manner. Proper preprocessing of the metabolomics data set, which includes removing noise,90 normalization, scaling, and aligning (or binning), is also necessary for a reliable PCA, PLS, or OPLS-DA model and meaningful interpretation of the metabolomics data. Removing noise regions eliminates the possibility that serendipitous covariant noise peaks may contribute to irrelevant class separation. Normalization corrects for the variability in total metabolite concentration or NMR sensitivity due to experimental differences in the total number of cells harvested, in the efficiency in extracting the metabolomes, and potentially due to changes in instrument performance. Similarly, scaling accounts for large differences in metabolite concentrations within a given sample (dynamic range issue) and prevents intense peaks from dominating the multivariate statistical analysis. Binning or aligning the spectra corrects for small variations in peak position and peak shape due to differences in sample conditions (pH, ionic strength, concentration, etc.) and instrument variability between replicates. In practice, multiple program packages are used for each step of the NMR data processing and analysis including separate software for processing of the 1D 1H NMR spectra, preprocessing the metabolomics data set, and the multivariate statistical analysis. This situation clearly contributes to the lack of a standard set of protocols and the significant variations between individual metabolomics studies. To address this issue, we have developed an open-source platform for the complete handling of NMR metabolomics data and the generation of validated multivariate statistical models (MVAPACK, http://bionmr.unl.edu/mvapack.php).91 We have also developed software to analyze the statistical significance of cluster separations in PCA, PLS, and OPLS-DA scores plots (PCA/PLS-DA utilities, http://bionmr.unl.edu/pca-utils.php).84,85
Applying Metabolomics to Drug Discovery
The first step of the drug discovery process is to identify initial chemical leads with reasonable affinity (typically a KD of 10 μM or less) and novel structures (patentable) that have druglike characteristics (Lipinski’s Rule of Five)92 and are synthetically achievable and malleable. This is primarily accomplished by using a high-throughput screen of a chemical library composed of hundreds of thousands to millions of compounds.41 The chemical leads are then evolved to high-affinity ligands (KD ≤ nM) through an iterative process involving structure-based drug design, traditional medicinal chemistry techniques, and multiple activity assays. This process may include cell-based assays to measure cell viability when the desired activity of a chemical lead is cell death (i.e., infectious disease and cancer). While this process is generally very efficient and high affinity ligands typically emerge, there is a fundamental difference between a true drug lead and a tight-binding ligand. It is well-known that an increase in affinity can be achieved by increasing the size and hydrophobicity of a compound, which are generally detrimental to druglike characteristics.48−52 In fact, the drift of corporate libraries toward these undesirable traits has been well-documented,93 which inevitably led to the development of Lipinski’s Rule of Five.92 Furthermore, compounds with poor physiochemical properties also tend to provide misleading biological activity. For this class of compounds, the inhibition of a protein target likely occurs through undesirable mechanisms such as micelle formation or induced protein aggregation or precipitation.48−52 Correspondingly, the process of increasing binding affinity may actually change the mode of action of a lead candidate in an undesirable direction.
Besides improving efficacy, increasing the binding affinity of chemical leads is also intended to improve selectivity. Reasonably, the more a particularly compound has been optimized to bind to the exact sequence and structural characteristics of a specific active site, the less likely it will bind to other proteins. Of course, a secondary assay using a panel of functional homologues with high sequence and structure similarity to the therapeutic target is routinely used to evaluate compound selectivity. Unfortunately, these protein panels are rarely exhaustive. Despite these efforts, the lack of efficacy and issues with toxicity, which include off-target activity, are the primary reasons potential drugs fail in the clinic.94 Correspondingly, evaluating issues related to bioavailability (absorption, distribution), in vivo activity, in vivo selectivity, compound stability (metabolism, excretion), and toxicity are an integral, but extremely challenging component of the drug discovery process. This critical information is a primary outcome of clinical trials, but of course this is a costly and highly inefficient means of identifying problematic drug leads. Ideally, serious problems would be identified and eliminated prior to initiating a clinical trial. Metabolomics can assist in this endeavor.
In essence, as the affinity of a chemical lead is enhanced it is also critical to validate and maintain a desirable in vivo mode of action. Is the compound being taking up by the cell? Is it inhibiting the intended protein target? Are there any off-target effects or toxicity issues? Because the metabolome captures the state of the cell and is a direct measure of protein activity, any observed changes in the metabolome as a result of a drug treatment would provide information on the drug’s activity and selectivity. In this manner, untreated wild-type cells represent a negative control and a knockout mutant cell line, where the protein target has been genetically inactivated to provide a positive control. The difference between these two samples represents the activity of the protein target as measured by the observed changes in the metabolome. This difference is observed with a PCA, PLS, or OPLS-DA scores plot generated from a set of 1D 1H NMR spectra. Simply, each 1D 1H NMR spectrum is reduced to a single point in the principal component (PC)-space of a scores plot. Similar spectra, and correspondingly similar metabolomes, will cluster together in a scores plot. Conversely, NMR spectra obtained from distinctly different metabolomes will form separate clusters in a scores plot.
The in vivo activity of a chemical lead can then be ascertained by treating both the wild-type and mutant cells with the lead compound and interpreting the resulting clustering patterns in a PCA, PLS, or OPLS-DA scores plot (Figure 4). A chemical lead that is inactive in vivo would have no impact on the metabolome of either the wild-type or mutant cells. The separation in the scores plot would simply reflect the difference in the activity of the target protein. So, the treated and untreated wild-type cells will cluster together and separate from the treated and untreated mutant cells in the scores plot (Figure 4A). A chemical lead with the desired in vivo activity and selectivity, only inhibiting the identified protein target, would perturb the metabolome of the wild-type cells in a manner similar to the mutant cells. Thus, wild-type cells treated with the chemical lead would cluster together with both the treated and untreated mutant cells in the scores plot (Figure 4B). The untreated wild-type cells would still form a separate cluster because the protein target is still active. The metabolome of the mutant cells is not affected by treatment with the chemical lead because the protein target is already inactivated and the chemical lead does not inhibit a secondary protein. Conversely, if the chemical lead was not selective, then the treated wild-type and mutant cells would form a third cluster separate from the untreated wild-type and mutant clusters (Figure 4C). Changes in the metabolome from the treated cells would result from inactivating multiple proteins, but the untreated mutant cells would only have the target protein inactivated. Finally, if the chemical lead actually inhibits the wrong protein target in vivo, then four separate clusters would be observed in a scores plot (Figure 4D). Effectively, a different set of active or inactive proteins is present in the four different cell cultures. The treated wild-type cells would have the wrong protein inactivated, whereas the treated mutant cells would have both the original target protein and the wrong protein inactivated.
Figure 4.

Illustration of hypothetical PCA scores plot for the following scenarios (A) inactive compound, (B) active and selective inhibitor, (C) active, nonselective inhibition of target and secondary protein, and (D) active, nonselective preferential inhibition of secondary protein. Labels correspond to: wild-type cells (open circles), drug-treated wild-type cells (black solid circles), knockout mutant cells where the protein target of the drug is inactive (open triangles), and drug-treated mutant cells (black solid triangles). Abbreviations correspond to mutant cells (mut) and wild-type cells (wt). Adapted from Forgue et al. (2006).95
Importantly, the results of the metabolomics study would be used to prioritize chemical leads and focus effort. Compounds that maintained the desired in vivo activity and selectivity in the metabolomics experiments while providing a route to improve binding affinity would be identified as chemical leads and proceed to more advanced animal and absorption, distribution, metabolism, and excretion (ADME) studies. Conversely, compounds that lack in vivo activity in the metabolomics study are likely to experience bioavailability issues and compounds that are nonselective in the metabolomics study are likely to encounter toxicity issues. These compounds should be excluded if these problems cannot be easily corrected. Compounds that unexpectedly target the wrong protein in the metabolomics study should simply be abandoned. Importantly, the cluster analysis can be supplemented with an in-depth and exhaustive analysis of metabolite concentration changes (Figure 3). Changes to a specific set of metabolite should be correlated with each of the clusters observed in the scores plot. S-Plots from PLS and OPLS-DA are routinely used to identify the metabolites primarily contributing to the observed class differentiation in the scores plot. Similarly, loadings plots provide information on the relative contribution to the class differentiation which can be used to infer metabolite concentration changes. In addition, 13C-labeled metabolites combined with two-dimensional (2D) 1H–13C heteronuclear single quantum coherence (HSQC) experiments can be used to identify and quantify metabolite concentration changes. Finally, this information can be combined together to generate a metabolic network to capture the global changes to the metabolome.
Examples of Using Metabolomics in Drug Discovery
Employing metabolomics as part of a drug discovery project is very appealing because of its relative simplicity, universal application, and high information content. For instance, while obtaining a knockout mutant may not always be practical (i.e., the therapeutic target may be essential), there are other equally valid approaches to obtaining a positive control. The activity of the target protein can be manipulated using RNA interference (RNAi), a known drug, monoclonal antibody, or any other means of modulating the activity of a protein. The technique is not restricted to metabolomic extracts from cell lysates. The approach can be applied to tissues, organs, organisms, or a variety of biofluids. Similarly, metabolomics is also extremely valuable even if the in vivo target of the chemical leads is unknown. The successful application of metabolomics to a drug discovery project is simply dependent on identifying the appropriate set of classes to compare (e.g., wild-type vs mutant, healthy vs disease, drug treated vs untreated, etc.). The following are a few illustrated examples of using metabolomics to assist drug discovery.
8-Azaxanthine (AZA) is an inhibitor of urate oxidase and was shown to inhibit new hyphal growth in Aspergillus nidulans.95 The corresponding metabolomics study demonstrated that the drug was a selective in vivo inhibitor (Figure 5a).95 The metabolome of an A. nidulansuaZ14 mutant that eliminated urate oxidase was shown to be distinct from the metabolome from wild-type mycelia. Treating both the wild-type and uaZ14 mutant mycelia with AZA resulted in a PCA scores plot with a clustering pattern consistent with a selective and active inhibitor and a potential antifungal (Figure 4B). d-Cycloserine (DCS) is a second-line treatment for tuberculosis that has been used for over 50 years despite the lack of an in vivo mechanism of action. DCS has been shown to inhibit both alanine racemase (Alr) and d-alanine-d-alanine ligase (Ddl) in vitro. A metabolomics study was conducted to determine if alanine racemase was the lethal target of DCS using Mycobacterium smegmatis mc2155 as a model system.96 The results indicated that DCS is a promiscuous inhibitor and that Alr is not the lethal target of DCS (Figure 5b). Treating both the wild-type mc2155 cells and an alr null mutant (TAM23) with DCS resulted in a PCA scores plot with a clustering pattern consistent with a nonselective inhibitor (Figure 4C). In fact, DCS is well known to have serious side effects that include seizures and mental disorders, presumably due to off-target activity (agonist against N-methyl-d-aspartate (NMDA) receptors). This is clearly consistent with the metabolomics results and indicative of potential problems with nonselective inhibitors: the likelihood of side effects or toxicity issues. Again, this demonstrates the value of incorporating metabolomics in a drug discovery effort in order to identify and eliminate such problems prior to clinical trials. These types of comparative metabolomics analysis can also be used to identify a potential in vivo mode of action to identify novel inhibitors. The Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) screened a large chemical library against M. tuberculosis using cell-based assays. TAACF only reported MIC50 values, where the mechanism of action was unknown. A metabolomics study was conducted to infer an in vivo mode of action for three highly active compounds identified by TAACF.97 Instead of comparing the metabolomes between wild-type and mutant cell lines as before, this study compared the activity of a collection of known antibiotics with defined cellular targets against the TAACF unknowns. Simply, multiple drugs inhibiting the same cellular target would be expected to induce a similar change in the metabolome. This is exactly what was observed (Figure 5c). Drugs that target cell wall synthesis, mycolic acid biosynthesis, or transcription, translation, or DNA supercoiling formed separate and distinct clusters in the OPLS-DA scores plot. The TAACF unknowns clustered together with the cell wall inhibitors implying a similar mechanism of action. A subsequent literature search verified that the TAACF unknowns have been previously shown to disrupt bacterial membranes. Again, this demonstrates the value of incorporating metabolomics while evolving chemical leads. The metabolomics analysis can verify that the in vivo mechanism of action is maintained as the binding affinity is improved.
Figure 5.

(a) The PCA scores plot comparing A. nidulansuaZ14 mutant (green X’s), wild-type with AZA (solid red squares), uaZ14 mutant with AZA (solid blue circles), and wild-type cells (solid black diamonds). The clustering pattern is comparable to the hypothetical PCA scores plot depicted in Figure 4B, which is consistent with an active and selective inhibitor. Abbreviations correspond to mutant cells (mut), wild-type cells (wt), and 8-azaxanthine (AZA). Reprinted with permission from ref (95). Copyright 2006 American Chemical Society. (b) PCA scores plot comparing Mycobacterium smegmatis mc2155 (solid blue squares), TAM23 (solid black circles), GPM14 (solid red diamonds), GPM16 (solid green upright triangles), TAM23 pTAMU3 (solid gold inverted triangles), mc2155 with DCS (solid purple squares), and TAM23 with DCS (solid red circles), GPM14 with DCS (solid purple diamonds), GPM16 with DCS (solid cyan upright triangles), and TAM23 pTAMU3 with DCS (solid orange inverted triangles). The clustering pattern is comparable to the hypothetical PCA scores plot depicted in Figure 4C, which is consistent with an active and nonselective inhibitor. Abbreviations correspond to mutant cells (mut), wild-type cells (wt) and d-cycloserine (DCS). Reprinted with permission from ref (96). Copyright 2007 American Chemical Society. (c) 2D OPLS-DA scores plot demonstrating the clustering pattern for 12 antibiotics with known biological targets and three compounds of unknown in vivo activity: untreated M. smegmatis cells (solid black squares), chloramphenicol (solid cyan diamonds), ciprofloxacin (solid gold diamonds), gentamicin (solid pink diamonds), kanamycin (solid purple diamonds), rifampicin (solid red diamonds), streptomycin (solid yellow diamonds), ethambutol (solid light-green inverted triangles), ethionamide (solid cyan inverted triangles), isoniazid (solid pink inverted triangles), ampicillin (solid red upright triangles), d-cycloserine (solid upright purple triangles), vancomycin (solid upright orange triangles), amiodorone (solid purple circles), chlorprothixene (solid light-green circles), and clofazimine (solid red circles) treated M. smegmatis cells. The ellipses correspond to the 95% confidence limits from a normal distribution for each cluster. The untreated M. smegmatis cells (solid black squares) was designated the control class, and the remainder of the cells were designated as treated. The OPLS-DA used one predictive component and six orthogonal components to yield a R2X of 0.715, R2Y of 0.803, Q2 of 0.671, and a CV-ANOVA p-value of 1.54 × 10–34. Reprinted with permission from ref (97). Copyright 2012 American Chemical Society.
Conclusions
Metabolomics is a valuable and versatile technique that is making important contributions to systems biology, personalized medicine, biomarker discovery, toxicology, and disease diagnostics. Similarly, the inclusion of metabolomics as a routine component of the drug discovery process may improve the efficiency and success rate of drug discovery. Given the current challenges facing the pharmaceutical industry and the high reliance on high-throughput screens for lead identification, metabolomics fills an important need. Metabolomics is useful for verifying a desirable in vivo mechanism of action for HTS chemical leads and for further validating and verifying the mode of action is maintained as the binding affinity is iteratively increased. In this manner, metabolomics can be used to identify truly novel leads with a unique cellular target and bioactivity. Metabolomics is also useful for determining if a chemical lead is a selective in vivo inhibitor. This is a unique asset of metabolomics that provides a simple mechanism to remove chemical leads with potential side effects or toxicity issues. For a drug discovery effort to effectively benefit from adapting metabolomics, the technology needs to be properly applied. Incorrect sample preparation and collection procedures, inappropriate data processing and analysis protocols, incorrect application of statistical techniques, and the lack of model validation are all common occurrences that result in erroneous outcomes. Nevertheless, these issues are aggressively being addressed by the field, which has led to the development and adaptation of standards by The Metabolomics Standards Initiative (MSI, http://msi-workgroups.sourceforge.net/)86 and the Coordination of Standards in Metabolomics (COSMOS, http://cosmos-fp7.eu/).87 NMR metabolomics is also limited to systems where a drug treatment modulates the metabolome in a direct response to the drug’s biological activity. For instance, drugs designed to prevent plaque formation in Alzheimer’s patients by physically inhibiting β-amyloid aggregation may not perturb the metabolome because the drug does not affect protein activities. Furthermore, the metabolomes from both a healthy control and the disease state needs to be readily accessible from a cell culture, tissue, organ, or biofluid for a comparative analysis. This is relatively straightforward to achieve for drugs targeting any disease with well-established cell lines (various cancers, neuronal cells, pathogenic bacteria, viruses, etc.). The expanding collection of disease-specific human stem cell lines also significantly increases the number of diseases amenable to analysis by metabolomics. Animal models may also be used, but this would dramatically increase the cost while decreasing throughput. Alternatively, a knock-down mutant cell line can be generated for a specific therapeutic target, where the comparison would simply be between wild-type and mutant cell lines. In essence, the availability of a cell-based or target-specific HTS assay implies that it is also possible to conduct a metabolomics study.
Acknowledgments
I would like to thank Bradley Worley for his contributions to the manuscript. The projects described were supported by National Institutes of Health Grants P20 RR-17675, P30 GM103335, R01 CA163649-01A1, and R01 AI087668-01A1. Research was performed in facilities renovated with support from the NIH under grant RR015468-01.
Glossary
Abbreviations Used
- 1D
one-dimensional
- 2D
two-dimensional
- ADME
absorption, distribution, metabolism, and excretion
- Alr
alanine racemase
- AZA
8-azaxanthine
- COSMOS
Coordination of Standards in Metabolomics
- CT
computerized tomography
- DCS
d-cycloserine
- Ddl
d-alanine-d-alanine ligase
- FDA
Food and Drug Administration
- GPM14
M. smegmatis Alr overproducing mutant
- GPM16
M. smegmatis DCS resistant mutant
- HIV/AIDS
human immunodeficiency virus infection/acquired immunodeficiency syndrome
- HSQC
heteronuclear single quantum coherence
- HTS
high-throughput screening
- ICER
incremental cost-effectiveness ratio
- mc2155
M. smegmatis wild-type
- MRI
magnetic resonance imaging
- MS
mass spectrometry
- MSI
Metabolomics Standards Initiative
- mut
mutant
- NDA
new drug application
- NMDA
N-methyl-d-aspartate
- NMR
nuclear magnetic resonance
- OPLS-DA
orthogonal projection to latent structures discriminant analysis
- PC
principal component
- PCA
principal component analysis
- PLS
partial-least-squares
- R&D
research and development
- RNAi
RNA interference
- TAACF
Tuberculosis Antimicrobial Acquisition and Coordinating Facility
- TAM23
M. smegmatisalr null mutant
- TAM23 pTAMU3
M. smegmatis TAM23 complemented with wild-type alr gene
- wt
wild-type
Biography
Robert Powers is a Professor in the Department of Chemistry at the University of Nebraska—Lincoln (UNL). Dr. Powers earned his Ph.D. with Professor David Gorenstein at Purdue University at West Lafayette, IN, and was then an intramural research training award (IRTA) postdoctoral fellow at the National Institutes of Health with Drs. G. Marius Clore and Angela Gronenborn in the Laboratory of Chemical Physics. He started his career in the pharmaceutical industry with American Cyanamid in 1992, which was acquired by American Home Products, followed by a merger with Genetics Institute to form Wyeth. Dr. Powers was an Associate Director before joining UNL in 2003. Dr. Powers’ research specializes in the development of techniques using NMR, metabolomics, structural biology, and bioinformatics to aid in drug discovery.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Liu L.; Cline R. R.; Schondelmeyer S. W.; Schommer J. C. Pharmaceutical expenditures as a correlate of population health in industrialized nations. Ann. Pharmacother. 2008, 42, 368–374. [DOI] [PubMed] [Google Scholar]
- Lichtenberg F. R. The impact of new drug launches on longevity: evidence from longitudinal, disease-level data from 52 countries, 1982–2001. Int. J. Health Care Finance Econ. 2005, 5, 47–73. [DOI] [PubMed] [Google Scholar]
- Lakdawalla D. N.; Sun E. C.; Jena A. B.; Reyes C. M.; Goldman D. P.; Philipson T. J. An economic evaluation of the war on cancer. J. Health Econ. 2010, 29, 333–346. [DOI] [PubMed] [Google Scholar]
- Lichtenberg F. R.The Expanding Pharmaceutical Arsenal in the War on Cancer; National Bureau of Economic Research: Cambridge, MA, 2004; Vol. I, Feb 2004. [Google Scholar]
- Hoyert D. L.; Xu J. Deaths: Preliminary Data for 2011. Natl. Vital Stat. Rep. 2012, 61, 1–52. [PubMed] [Google Scholar]
- Dieffenbach C. W.; Fauci A. S. Thirty years of HIV and AIDS: future challenges and opportunities. Ann. Intern. Med. 2011, 154, 766–771. [DOI] [PubMed] [Google Scholar]
- Kaufman H. W.; Blatt A. J.; Huang X.; Odeh M. A.; Superko H. R. Blood cholesterol trends 2001–2011 in the United States: analysis of 105 million patient records. PLoS One 2013, 8, e63416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford E. S.; Ajani U. A.; Croft J. B.; Critchley J. A.; Labarthe D. R.; Kottke T. E.; Giles W. H.; Capewell S. Explaining the decrease in U.S. deaths from coronary disease, 1980–2000. N. Engl. J. Med. 2007, 356, 2388–2398. [DOI] [PubMed] [Google Scholar]
- Kostis J. B.; Cabrera J.; Cheng J. Q.; Cosgrove N. M.; Deng Y.; Pressel S. L.; Davis B. R. Association between chlorthalidone treatment of systolic hypertension and long-term survival. JAMA, J. Am. Med. Assoc. 2011, 306, 2588–2593. [DOI] [PubMed] [Google Scholar]
- Armstrong G. L.; Conn L. A.; Pinner R. W. Trends in infectious disease mortality in the United States during the 20th century. JAMA, J. Am. Med. Assoc. 1999, 281, 61–66. [DOI] [PubMed] [Google Scholar]
- CDC on Infectious Diseases in the United States: 1900–99. Population Dev. Rev. 1999, 25, 635–640. [Google Scholar]
- Health Care Cost and Utilization Report: 2011; Health Care Cost Institute, Inc.: Washington, DC, 2012; pp 1–18
- Levy R. Costs and benefits of pharmaceuticals: the value equation for older Americans. Care Manage. J. 2002, 3, 135–142. [DOI] [PubMed] [Google Scholar]
- Cremieux P.-Y.; Ouellette P.; Petit P. Do drugs reduce utilisation of other healthcare resources?. Pharmacoeconomics 2007, 25, 209–221. [DOI] [PubMed] [Google Scholar]
- Pedersen T. R.; Kjekshus J.; Berg K.; Olsson A. G.; Wilhelmsen L.; Wedel H.; Pyorala K.; Miettinen T.; Haghfelt T.; Faergeman O.; Thorgeirsson G.; Jonsson B.; Schwartz J. S. Cholesterol lowering and the use of healthcare resources. Results of the Scandinavian Simvastatin Survival Study. Circulation 1996, 93, 1796–1802. [DOI] [PubMed] [Google Scholar]
- Ali S.; Paracha N.; Cook S.; Giovannoni G.; Comi G.; Rammohan K.; Rieckmann P.; Sarensen P. S.; Vermersch P.; Greenberg S.; Scott D. A.; Joyeux A. Reduction in healthcare and societal resource utilization associated with cladribine tablets in patients with relapsing-remitting multiple sclerosis: analysis of economic data from the CLARITY study. Clin. Drug Invest. 2012, 32, 15–27. [DOI] [PubMed] [Google Scholar]
- Weintraub W. S.; Mahoney E. M.; Lamy A.; Culler S.; Yuan Y.; Caro J.; Gabriel S.; Yusuf S. Long-term cost-effectiveness of clopidogrel given for up to one year in patients with acute coronary syndromes without ST-segment elevation. J. Am. Coll. Cardiol. 2005, 45, 838–845. [DOI] [PubMed] [Google Scholar]
- Scannell J. W.; Blanckley A.; Boldon H.; Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nature Rev. Drug Discovery 2012, 11, 191–200. [DOI] [PubMed] [Google Scholar]
- Paul S. M.; Mytelka D. S.; Dunwiddie C. T.; Persinger C. C.; Munos B. H.; Lindborg S. R.; Schacht A. L. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nature Rev. Drug Discovery 2010, 9, 203–214. [DOI] [PubMed] [Google Scholar]
- Sams-Dodd F. Target-based drug discovery: is something wrong?. Drug Discovery Today 2005, 10, 139–147. [DOI] [PubMed] [Google Scholar]
- Cuatrecasas P. Drug discovery in jeopardy. J. Clin. Invest. 2006, 116, 2837–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburn T. T.; Thor K. B. Drug repositioning: identifying and developing new uses for existing drugs. Nature Rev. Drug Discovery 2004, 3, 673–683. [DOI] [PubMed] [Google Scholar]
- Garattini S. Are me-too drugs justified?. J. Nephrol. 1997, 10, 283–94. [PubMed] [Google Scholar]
- Ullman F.; Boutellier R. A case study of lean drug discovery: from project driven research to innovation studios and process factories. Drug Discovery Today 2008, 13, 543–550. [DOI] [PubMed] [Google Scholar]
- Kola I.; Landis J. Opinion: Can the pharmaceutical industry reduce attrition rates?. Nature Rev. Drug Discovery 2004, 3, 711–716. [DOI] [PubMed] [Google Scholar]
- Dickson M.; Gagnon J. P. Key factors in the rising cost of new drug discovery and development. Nature Rev. Drug Discovery 2004, 3, 417–429. [DOI] [PubMed] [Google Scholar]
- Miller G. Is Pharma running out of brainy ideas?. Science 2010, 329, 502–504. [DOI] [PubMed] [Google Scholar]
- Livermore D. Can better prescribing turn the tide of resistance?. Nature Rev. Microbiol. 2004, 2, 73–78. [DOI] [PubMed] [Google Scholar]
- Hendrie C. A. The funding crisis in psychopharmacology: an historical perspective. J. Psychopharmacol. 2010, 24, 439–440. [DOI] [PubMed] [Google Scholar]
- Nierenberg A. A. The perfect storm: CNS drug development in trouble. CNS Spectrums 2010, 15, 282–283. [DOI] [PubMed] [Google Scholar]
- Offit P. A. Why Are Pharmaceutical Companies Gradually Abandoning Vaccines?. Health Affairs 2005, 24, 622–630. [DOI] [PubMed] [Google Scholar]
- Garber A. M. Perspective: an uncertain future for cardiovascular drug development?. N. Engl. J. Med. 2009, 360, 1169–1171. [DOI] [PubMed] [Google Scholar]
- Booth B.; Zemmel R. Opinion: Quest for the best. Nature Rev. Drug Discovery 2003, 2, 838–841. [DOI] [PubMed] [Google Scholar]
- Wermuth C. G. Similarity in drugs: reflections on analogue design. Drug Discovery Today 2006, 11, 348–354. [DOI] [PubMed] [Google Scholar]
- Seidelmann O. Outsourcing of chemical research: promising field for service providers. Chim. Oggi 2004, 18–21. [Google Scholar]
- Cavalla D. The extended pharmaceutical enterprise. Drug Discovery Today 2003, 8, 267–274. [DOI] [PubMed] [Google Scholar]
- Ullman F.; Boutellier R. Drug discovery: are productivity metrics inhibiting motivation and creativity?. Drug Discovery Today 2008, 13, 997–1001. [DOI] [PubMed] [Google Scholar]
- Mackay M.; Street S. D. A.; McCall J. M. Risk reduction in drug discovery and development. Curr. Top. Med. Chem. (Sharjah, United Arab Emirates) 2005, 5, 1087–1090. [DOI] [PubMed] [Google Scholar]
- Handen J. S. The industrialization of drug discovery. Drug Discovery Today 2002, 7, 83–85. [DOI] [PubMed] [Google Scholar]
- Schmid E. F.; Smith D. A. Is pharmaceutical R&D just a game of chance or can strategy make a difference?. Drug Discovery Today 2004, 9, 18–26. [DOI] [PubMed] [Google Scholar]
- Mayr L. M.; Fuerst P. The future of high-throughput screening. J. Biomol. Screening 2008, 13, 443–448. [DOI] [PubMed] [Google Scholar]
- Szymanski P.; Markowicz M.; Mikiciuk-Olasik E. Adaptation of high-throughput screening in drug discovery—toxicological screening tests. Int. J. Mol. Sci. 2012, 13, 427–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr L. M.; Bojanic D. Novel trends in high-throughput screening. Curr. Opin. Pharmacol. 2009, 9, 580–588. [DOI] [PubMed] [Google Scholar]
- Martis E. A.; Radhakrishnan R.; Badve R. R. High-Throughput Screening: The Hits and Leads of Drug Discovery—An Overview. J. Appl. Pharm. Sci. 2011, 1, 2–10. [Google Scholar]
- Lahana R. How many leads from HTS?. Drug Discovery Today 1999, 4, 447–448. [DOI] [PubMed] [Google Scholar]
- Macarron R.; Banks M. N.; Bojanic D.; Burns D. J.; Cirovic D. A.; Garyantes T.; Green D. V. S.; Hertzberg R. P.; Janzen W. P.; Paslay J. W.; Schopfer U.; Sittampalam G. S. Impact of high-throughput screening in biomedical research. Nature Rev. Drug Discovery 2011, 10, 188–195. [DOI] [PubMed] [Google Scholar]
- Dove A. Screening for content-the evolution of high throughput. Nature Biotechnol. 2003, 21, 859–864. [DOI] [PubMed] [Google Scholar]
- Kubinyi H. Opinion: Drug research: myths, hype and reality. Nature Rev. Drug Discovery 2003, 2, 665–668. [DOI] [PubMed] [Google Scholar]
- Seidler J.; McGovern S. L.; Doman T. N.; Shoichet B. K. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 2003, 46, 4477–4486. [DOI] [PubMed] [Google Scholar]
- Rishton G. M. Reactive compounds and in vitro false positives in HTS. Drug Discovery Today 1997, 2, 382–384. [Google Scholar]
- McGovern S. L.; Caselli E.; Grigorieff N.; Shoichet B. K. A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J. Med. Chem. 2002, 45, 1712–1722. [DOI] [PubMed] [Google Scholar]
- McGovern S. L.; Helfand B. T.; Feng B.; Shoichet B. K. A Specific Mechanism of Nonspecific Inhibition. J. Med. Chem. 2003, 46, 4265–4272. [DOI] [PubMed] [Google Scholar]
- Malo N.; Hanley J. A.; Cerquozzi S.; Pelletier J.; Nadon R. Statistical practice in high-throughput screening data analysis. Nature Biotechnol. 2006, 24, 167–175. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. Interpreting Steep Dose–Response Curves in Early Inhibitor Discovery. J. Med. Chem. 2006, 49, 7274–7277. [DOI] [PubMed] [Google Scholar]
- Snowden M. A.; Green D. V. S. The impact of diversity-based, high-throughput screening on drug discovery: “chance favours the prepared mind”. Curr. Opin. Drug Discovery Dev. 2008, 11, 553–558. [PubMed] [Google Scholar]
- Djuric S. W.; Akritopoulou-Zanze I.; Cox P. B.; Galasinski S. Compound collection enhancement and paradigms for high-throughput screening—an update. Annu. Rep. Med. Chem. 2010, 45, 409–428. [Google Scholar]
- Petrone P. M.; Wassermann A. M.; Lounkine E.; Kutchukian P.; Simms B.; Jenkins J.; Selzer P.; Glick M. Biodiversity of small molecules—a new perspective in screening set selection. Drug Discovery Today 2013, 18, 674–680. [DOI] [PubMed] [Google Scholar]
- Harper G.; Pickett S. D.; Green D. V. S. Design of a compound screening collection for use in high throughput screening. Comb. Chem. High Throughput Screening 2004, 7, 63–71. [DOI] [PubMed] [Google Scholar]
- Bajorath J. Integration of virtual and high-throughput screening. Nature Rev. Drug Discovery 2002, 1, 882–894. [DOI] [PubMed] [Google Scholar]
- Anderson A. C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797. [DOI] [PubMed] [Google Scholar]
- Ramstrom O.; Lehn J.-M. Drug discovery by dynamic combinatorial libraries. Nature Rev. Drug Discovery 2002, 1, 26–36. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; McDowell R. S.; O’Brien T. Fragment-Based Drug Discovery. J. Med. Chem. 2004, 47, 3463–3482. [DOI] [PubMed] [Google Scholar]
- Nicholson J. K.; Connelly J.; Lindon J. C.; Holmes E. Innovation: Metabonomics: a platform for studying drug toxicity and gene function. Nature Rev. Drug Discovery 2002, 1, 153–161. [DOI] [PubMed] [Google Scholar]
- Fiehn O. Metabolomics—the link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [PubMed] [Google Scholar]
- Guo Y.; Xiao P.; Lei S.; Deng F.; Xiao G. G.; Liu Y.; Chen X.; Li L.; Wu S.; Chen Y.; Hui J.; Tan L.; Xie J.; Zhu X.; Liang S.; Deng H. How is mRNA expression predictive for protein expression? A correlation study on human circulating monocytes. Acta Biochim. Biophys. Sin. 2008, 40, 426–436. [DOI] [PubMed] [Google Scholar]
- Nicholson J. K.; Lindon J. C.; Holmes E. “Metabonomics”: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Powers R. Using NMR-based metabolomics to study the regulation of biofilm formation. Future Med. Chem. 2012, 4, 1273–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebregiworgis T.; Powers R. Application of NMR Metabolomics to Search for Human Disease Biomarkers. Comb. Chem. High Throughput Screening 2012, 15, 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers R. NMR metabolomics and drug discovery. Magn. Reson. Chem. 2009, 47, S2–S11. [DOI] [PubMed] [Google Scholar]
- Bollard M. E.; Stanley E. G.; Lindon J. C.; Nicholson J. K.; Holmes E. NMR-based metabonomic approaches for evaluating physiological influences on biofluid composition. NMR Biomed. 2005, 18, 143–162. [DOI] [PubMed] [Google Scholar]
- Coen M.; Holmes E.; Lindon J. C.; Nicholson J. K. NMR-Based Metabolic Profiling and Metabonomic Approaches to Problems in Molecular Toxicology. Chem. Res. Toxicol. 2008, 21, 9–27. [DOI] [PubMed] [Google Scholar]
- Dunn W. B.; Broadhurst D. I.; Atherton H. J.; Goodacre R.; Griffin J. L. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40, 387–426. [DOI] [PubMed] [Google Scholar]
- Griffin J. L. Metabonomics: NMR spectroscopy and pattern recognition analysis of body fluids and tissues for characterisation of xenobiotic toxicity and disease diagnosis. Curr. Opin. Chem. Biol. 2003, 7, 648–654. [DOI] [PubMed] [Google Scholar]
- Reo N. V. NMR-based metabolomics. Drug Chem. Toxicol. 2002, 25, 375–382. [DOI] [PubMed] [Google Scholar]
- Serkova N. J.; Niemann C. U. Pattern recognition and biomarker validation using quantitative 1H-NMR-based metabolomics. Expert Rev. Mol. Diagn. 2006, 6, 717–731. [DOI] [PubMed] [Google Scholar]
- Wishart D. S. Quantitative metabolomics using NMR. TrAC, Trends Anal. Chem. 2008, 27, 228–237. [Google Scholar]
- Lei S.; Powers R. NMR Metabolomics Analysis of Parkinson’s Disease. Curr. Metabolomics 2013, 1, 191–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Halouska S.; Gaupp R.; Lei S.; Snell E.; Fenton R. J.; Barletta R. G.; Somerville G. A.; Powers R. Revisiting Protocols for the NMR Analysis of Bacterial Metabolomes. J. Integr. OMICS 2013, 2, 120–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadhurst D. I.; Kell D. B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar]
- Worley B.; Powers R. Multivariate analysis in metabolomics. Curr. Metabolomics 2013, 1, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg R. A.; Hoefsloot H. C.; Westerhuis J. A.; Smilde A. K.; van der Werf M. J. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics 2006, 7, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig A.; Cloareo O.; Holmes E.; Nicholson J. K.; Lindon J. C. Scaling and normalization effects in NMR spectroscopic metabonomic data sets. Anal. Chem. 2006, 78, 2262–2267. [DOI] [PubMed] [Google Scholar]
- Kjeldahl K.; Bro R. Some common misunderstandings in chemometrics. J. Chemom. 2010, 24, 558–564. [Google Scholar]
- Worley B.; Halouska S.; Powers R. Utilities for quantifying separation in PCA/PLS-DA scores plots. Anal. Biochem. 2013, 433, 102–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth M. T.; Halouska S.; Shortridge M. D.; Zhang B.; Powers R. Analysis of Metabolomic PCA Data using Tree Diagrams. Anal. Biochem. 2010, 399, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansone S.-A.; Fan T.; Goodacre R.; Griffin J. L.; Hardy N. W.; Kaddurah-Daouk R.; Kristal B. S.; Lindon J.; Mendes P.; Morrison N.; Nikolau B.; Robertson D.; Sumner L. W.; Taylor C.; van d. W. M.; van O. B.; Fiehn O. The Metabolomics Standards Initiative. Nature Biotechnol. 2007, 25, 846–848. [DOI] [PubMed] [Google Scholar]
- Salek R. M.; Haug K.; Steinbeck C. Dissemination of metabolomics results: role of MetaboLights and COSMOS. Gigascience 2013, 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canelas A.; Ras C.; ten Pierick A.; van Dam J.; Heijnen J.; van Gulik W. Leakage-free rapid quenching technique for yeast metabolomics. Metabolomics 2008, 4, 226–239. [Google Scholar]
- Blaise B. J. Data-Driven Sample Size Determination for Metabolic Phenotyping Studies. Anal. Chem. 2013, 85, 8943–8950. [DOI] [PubMed] [Google Scholar]
- Halouska S.; Powers R. Negative impact of noise on the principal component analysis of NMR data. J. Magn. Reson. 2006, 178, 88–95. [DOI] [PubMed] [Google Scholar]
- Worley B.; Powers R.. MVAPACK: A Complete Data Handling Package for NMR Metabolomics ACS Chem. Biol. 2014, accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technol. 2004, 1, 337–341. [DOI] [PubMed] [Google Scholar]
- Gribbon P.; Sewing A. High-throughput drug discovery: what can we expect from HTS?. Drug Discovery Today 2005, 10, 17–22. [DOI] [PubMed] [Google Scholar]
- Schuster D.; Laggner C.; Langer T. Why drugs fail—a study on side effects in new chemical entities. Curr. Pharm. Des. 2005, 11, 3545–3559. [DOI] [PubMed] [Google Scholar]
- Forgue P.; Halouska S.; Werth M.; Xu K.; Harris S.; Powers R. NMR Metabolic Profiling of Aspergillus nidulans to Monitor Drug and Protein Activity. J. Proteome Res. 2006, 5, 1916–1923. [DOI] [PubMed] [Google Scholar]
- Halouska S.; Chacon O.; Fenton R. J.; Zinniel D. K.; Barletta R. G.; Powers R. Use of NMR Metabolomics To Analyze the Targets of d-Cycloserine in Mycobacteria: Role of d-Alanine Racemase. J. Proteome Res. 2007, 6, 4608–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halouska S.; Fenton R. J.; Barletta R. G.; Powers R. Predicting the in Vivo Mechanism of Action for Drug Leads Using NMR Metabolomics. ACS Chem. Biol. 2012, 7, 166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]