

Abstract
Motivation: Experiments in animal models are often conducted to infer how humans will respond to stimuli by assuming that the same biological pathways will be affected in both organisms. The limitations of this assumption were tested in the IMPROVER Species Translation Challenge, where 52 stimuli were applied to both human and rat cells and perturbed pathways were identified. In the Inter-species Pathway Perturbation Prediction sub-challenge, multiple teams proposed methods to use rat transcription data from 26 stimuli to predict human gene set and pathway activity under the same perturbations. Submissions were evaluated using three performance metrics on data from the remaining 26 stimuli.
Results: We present two approaches, ranked second in this challenge, that do not rely on sequence-based orthology between rat and human genes to translate pathway perturbation state but instead identify transcriptional response orthologs across a set of training conditions. The translation from rat to human accomplished by these so-called direct methods is not dependent on the particular analysis method used to identify perturbed gene sets. In contrast, machine learning-based methods require performing a pathway analysis initially and then mapping the pathway activity between organisms. Unlike most machine learning approaches, direct methods can be used to predict the activation of a human pathway for a new (test) stimuli, even when that pathway was never activated by a training stimuli.
Availability: Gene expression data are available from ArrayExpress (accession E-MTAB-2091), while software implementations are available from http://bioinformaticsprb.med.wayne.edu?p=50 and http://goo.gl/hJny3h.
Contact: christoph.hafemeister@nyu.edu or atarca@med.wayne.edu.
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The fundamental assumption that underpins the use of animal models for the study of human disease is that there is conservation in the nature of the responses to injury and therapy. However, work reported by The Inflammation and Host Response to Injury, Large-Scale Collaborative Research Program (published in 2013) compared the transcriptional responses in peripheral blood with inflammatory injuries, such as burns, blunt force trauma, as well as to endotoxin in human patients and in mice, with startling and unexpected results (Seok et al., 2013). Humans had similar transcriptional responses to burns, trauma and endotoxemia. The murine model showed little correlation either to each other or to the human response (among genes that changed significantly in humans, the murine orthologs were close to random to their human counterparts with R2 values ranging between 0.0 and 0.1). These observations as well as earlier ones (Gruber et al., 2011; Quint et al., 2000) have elicited considerable interest and debate. Investigators have noted that candidate agents developed in animals to block the inflammatory response tested in >150 clinical trials have all failed when tested in humans (Seok et al., 2013). A major criticism has been that there is no systematic study of how well murine clinical models mimic human inflammatory diseases.
To address this question, an international crowdsourcing competition was convened in 2013 (IMPROVER Species Translation Challenge, IMPROVER STC) addressing the translatability of findings between rat and human model systems in four sub-challenges: (i) intra- and (ii) inter-species protein phosphorylation prediction, (iii) inter-species pathway perturbation prediction and (iv) species-specific network inference.
The work presented here is based on submissions to the third sub-challenge (SC3) by Team49 (A. L. Tarca, R. Romero) and Team133 (C. Hafemeister, R. Bonneau). For a set of test stimuli, SC3 asked participants to predict the perturbation state of gene sets representing pathways/biological processes in human cells given corresponding data in rat. The required submission was an ordering of the gene sets based on the enrichment in genes that are differentially expressed between stimuli treated and controls. Participants could tune their models on a set of training stimuli for which data for both rat and human were made available, as illustrated in Figure 1. Details of the experimental settings, generation, processing and quality control analysis of the dataset can be found in Poussin et al. (2014), and the raw data have been submitted to the ArrayExpress database and are available with the accession number E-MTAB-2091 (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2091/).
Fig. 1.

Overview of the Inter-species Pathway Perturbation Prediction Challenge. Participants were asked to predict the perturbation state of pathways in human cells (outlined in red) given corresponding data in rat. The organizers used a 25% cutoff on FDR to classify a pathway as significantly enriched in differentially expressed genes, based on the GSEA. Challengers were asked to give a confidence level that the GSEA FDR values on the test data (see red box) will be <0.25 for each stimuli
The gene sets were selected from the MSigDB database (Subramanian et al., 2005) and included biological pathways such as those available in KEGG (Ogata et al., 1999) and Reactome (Joshi-Tope et al., 2005), but also custom functionally related groups of genes (Supplementary Table S1). In the remainder of the article, we will use the term pathway to refer both biological pathways and gene sets for both simplicity reasons and to be consistent with the name of the sub-challenge: Inter-Species Pathway Perturbation Prediction.
Among the submissions to SC3, there were two main classes of methods: the first category of methods was based on machine learning applications in which the training was performed at the pathway level only (Fig. 2, panel C), i.e. the input was the pathway perturbation states in rat and human for a set of stimuli, and dependencies between them were exploited/modeled. Our methods represent the second category, which we call direct methods. These methods trained on the differential expression (DE) state of all individual genes across the different stimuli to identify a response homology between rat and human genes. Based on this homology, gene DE metrics in human are either borrowed (Team49) or estimated (Team133) from those in rat for the same stimuli. Gene DE metrics are then used to infer pathway perturbation (Fig. 2, panel A).
Fig. 2.
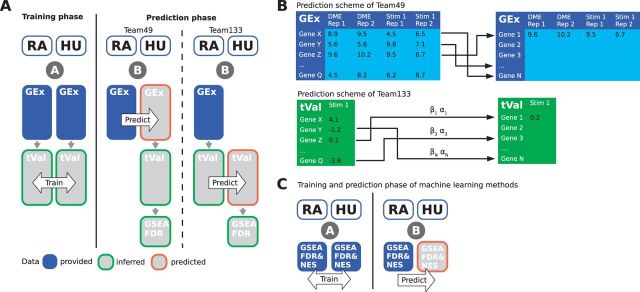
Illustration of the final methods of Team49 and Team133 used in SC3. (A) Both approaches rely on moderated t-tests, capturing the significance and direction of change of rat and human genes for the same stimuli in the training dataset, to find one rat gene as a predictor for each human gene (ortholog pairs). (B) Team49’s method finds the rat gene that is ranked similarly across all training stimuli when compared with a given human gene. The rat data in the test set are then used to impute the needed human expression data. Team133’s method fits a simple linear model with one coefficient and intercept per ortholog pair, and predicts t-values directly. (C) Machine learning methods bypass gene expression and DE tests altogether, and map rat pathway activity (NES or FDR values) to human pathway activity
This article describes in detail the approach of Team49 (A.L.T. and R.R.) and Team133 (C.H. and R.B.) who were tied for second place in this challenge. We describe commonalities and differences between these two direct methods and the machine learning-based approaches. The criteria used to discuss these methods include (i) prediction performance (overall and for the most challenging scenarios) using metrics estimated as in the official team ranking but also in alternative ways, (ii) applicability of the methods to instances when a given pathway was activated by few or no stimuli in the training set and (iii) the dependence of the rat to human pathway activity translation on the particular gene set analysis method used.
2 METHODS
In this section, we introduce the performance metrics used to rank predictions in the challenge, as well as the details of the methods developed by Team49 and Team133.
2.1 Performance metrics used in SC3
The three metrics used to rank the predictions in all three sub-challenges of the STC include the balanced accuracy (BAC), area under the precision-recall curve (AUPR) and the Pearson correlation coefficient. Although these metrics are described in detail elsewhere (Rhrissorrakrai et al., 2015), in this article we consider it relevant to discuss how these metrics were implemented. In the official scoring methodology, predictions for all pathways and test stimuli were pooled together, and then, the three performance metrics were computed. This implementation is justified considering that some individual pathways were activated in none or too few stimuli in the test set to allow computation of AUPR and Pearson correlation coefficient. An alternative implementation expected by some participating teams and feasible for BAC is to consider each pathway individually and average the performance over the different pathways. With such a mean BAC (mBAC) metric, finding the one stimulus (out of 26) for which a pathway A is truly activated (100% sensitivity) at the expense of a false positive (24/25 = 96% specificity) (scenario A) would be rewarded more than an alternative scenario (B) in which one predicts all pathways as inactive (0% sensitivity and 100% specificity). The pooled BAC metric as well as AUPR and Pearson correlation coefficient would assess these two scenarios as equivalent. The majority of human pathways was activated in less than two stimuli in the training dataset (Table 1), making it difficult to learn the relation between the activation status of these human pathways and rat gene expression and/or rat pathway activity. Although the mBAC metric results are reported in this article to aid explaining why the direct method of Team49 was designed in the way it was, and that the goal of improving pathway perturbation sensitivity for the most difficult scenarios can be achieved, the teams 49 and 133 fully recognize that the winner of this sub-challenge was Team50.
Table 1.
Distribution of pathways as a function of the number of stimuli that perturbed those pathways in the human test dataset
| Number of stimuli perturbing/activating a given pathway | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 12 | 13 |
| Pathways count | 70 | 52 | 45 | 22 | 22 | 7 | 5 | 7 | 4 | 7 | 3 | 1 | 1 |
Note: Of the 246 pathways, more than two-thirds were perturbed by two stimuli or less.
2.2 Differential expression analysis and gene set enrichment analysis used in SC3
The challenge organizers used existing methods to perform DE analysis on the measured gene expression levels, and to perform gene set enrichment analysis on the results. Raw expression values were processed and normalized altogether using GCRMA (Wu et al., 2004). The LIMMA package (Smyth, 2004) was used to compute pair-wise contrasts comparing gene expression levels for individual stimulus (—two to three replicates) and gene expression levels for DME control (four replicates) in both species. The output of this analysis was moderated t-values indicating the confidence and direction of the stimulus-specific DE. To identify enriched gene sets representative of specific pathways and biological processes perturbed by each stimulus, gene set enrichment analysis (GSEA) was performed (Subramanian et al., 2005). As the original GSEA method determines gene set significance by sample permutations, which was unfeasible here owing to a low number of samples, the challenge organizers used an alternative version of GSEA (pre-ranked GSEA) that is based on permutations of genes. The input in pre-ranked GSEA was the list ofgenes ranked by moderated t-scores, and the output was normalized enrichment scores (NES) and the associated false discovery rate (FDR) for each gene set and each stimuli. These data were made available to all SC3 participants, and our two methods, Team49 and Team133, used LIMMA and GSEA as described above.
2.3 Approach of Team49
The first submission of Team49 in this sub-challenge was based on a machine learning approach similar to the one we used previously (Tarca et al., 2013a) in the IMPROVER Diagnostic Signature Challenge as well as in the intra-species protein phosphorylation sub-challenge of STC (Dayarian et al., 2014). In this approach, the NES for one or few rat pathways were used as predictors in a linear discriminant analysis model that was fit to predict the activation status of one human pathway at a time. We considered that pathways that were not activated in four or more stimuli should be predicted as inactive in the test stimuli, as there was not enough information to train a model. This submission, referred to as Team49_alt1 was later replaced with the one obtained with a different method described below and referred to as Team49.
This second and final submission was based on the idea of finding an ‘orthologous’ rat gene for each human gene by learning from the training data. Then, for a given stimuli in the test set, the human expression data were borrowed from the available rat test expression data via the orthology mapping between human and rat genes. Human genes were subsequently ranked by moderated t-tests contrasting the stimuli-treated samples with the DME-treated samples. Finally, GSEA was applied on the ranked gene list to identify the pathways that were activated for the given stimuli by testing whether the genes of a given pathway were agglomerated toward the top or the bottom of the gene list. The procedure can be detailed as a series of steps:
1. Use the collection of gene sets and pathways provided by the organizers (c2.cp.v3.1.symbols.gmt file), and retain the 246 for which predictions were required. From each pathway, drop the genes not present on HG-U133 Plus2 array.
2. For each stimulus in the training set, compute the rank of each gene based on the moderated t-scores computed from data of stimuli- and DME-treated samples from the same batch. This analysis is done for both human and rat training data. Let us denote with and the rank that a given human gene h and a given rat gene r received, respectively, for stimuli i. The rank values are normalized so that 0 corresponds to the most downregulated gene, whereas 1.0 corresponds to the most upregulated gene. For each human gene h:
- a. Compute distance:
between the human gene h and all rat genes. The weight wi is defined as follows:(1) (2)
b. Choose as ortholog for the human gene h the rat gene that minimizes the D(h,r) distance defined above.
The weight w above gives more importance to the rank differences when the human gene is differentially expressed (rank far from 0.5) as opposed to when the human gene is not differentially expressed (rank closer to 0.5). This is because the ranks of genes that are not differentially expressed are more likely to be affected by noise than the genes ranked close to the top or to the bottom of the list.
3. GSEAP is applied using the human genes ranked based on moderated t-scores computed on expression data borrowed from the rat ortholog genes.
An illustration of the approach used by Team49 is given in Figure 2. The q-values for each pathway were converted into a final activation confidence value as follows:
Confidence = 1 if q-values < 0.25
Confidence = 0 if q-values > 0.45
Confidence is a linear function that decreases from 1 to 0 as q-values increases from 0.25 to 0.45.
This q-values-to-confidence levels conversion is different from the one used by the organizers to infer pathway activity (Confidence = 1 if q-values < 0.25, Confidence = 0 otherwise), by attempting to increase the sensitivity of the method at the expense of losing perhaps some specificity. Such a trade-off was expected to be meaningful in the context of a BAC being computed for each pathway separately and then averaged over all pathways (mBAC instead of pooled BAC).
2.4 Approach of Team133
The initial version of Team133’s method used linear regression models in t-value space to predict human t-values from rat ones, and used GSEA to identify perturbed pathways. As a first step, we used the LIMMA package (Smyth, 2004) to test every human and rat gene for DE across the 26 training stimuli when compared with their control treatment. From this analysis, we extracted the moderated t-values and called the resulting two matrices tr for rat and th for human. To level the differences in response magnitude that various stimuli can induce, we standardized tr so that the t-values for a given stimulus followed a distribution with mean 0 and SD 1. In our model, we represented the t-values of a human gene i as a linear fit of the t-values of some rat gene j, i.e. . The final values for the parameters were then the ones minimizing the sum of squares of the residuals of the linear regression model:
| (3) |
where s iterates over the stimuli.
Given the set of test stimuli for which we had only rat expression, we computed the tr matrix as described above. We then used the set of parameters for all human genes to predict th, the matrix of t-values. To obtain NES and associated FDR of differentially expressed pathways for every test stimulus, we applied GSEA pre-ranked by the corresponding t-values from th. Our final confidence scores indicating significantly perturbed pathways were then 1 − FDR.
The submission from the approach described above, referred to as Team133_alt1, was later replaced with another submission referred to as Team133. This second approach differed from the one described above in that it used similarities between a test stimulus response and the set of training stimuli responses. The motivating assumption was that stimuli that shared a high number of differentially expressed genes also perturbed the same pathways. Consequently, for a given test stimulus, we wanted to upweight similar stimuli during the training phase.
To upweight certain stimuli during parameter learning, we used a weighted regression and modified Equation 3 slightly to
| (4) |
We now minimized the weighted sum of squares of the residuals of the linear regression model, where the weights for a test stimulus test and a training stimulus were defined as the number of highly regulated genes that they have in common plus a pseudo count
| (5) |
Here top100 denotes the set of genes with the top 100 absolute t-values.
As opposed to the simplified approach described in the previous section, where there was only one single parameter estimation step, this weighted method required parameter estimation for every test stimulus because of the test stimulus-dependent weight.
3 RESULTS
3.1 Performance of the direct methods in SC3
The performance metrics, AUPR, BAC and Pearson correlation are presented in Figure 3 for all submissions to this sub-challenge. We also include some alternative submissions that were either submitted before the challenge deadline but replaced later with an updated submission (Team49_alt1 and Team133_alt1) or submitted after the challenge deadline (Team111_alt1) but still before the release of the gold standard data (the true activation status of the human pathways for the test stimuli). These alternative submissions included the following: (i) Team111_alt1, a version of Team111’s official submission including bug fixes; (ii) Team49_alt1, a machine learning-based approach of Team49 that was later replaced by the official Team49 submission; (c) Team133_alt1, the unweighted approach introduced in Section 2.4. As Team111’s challenge submission (Team111) contained a systematic error that led to a performance worse than random, we decided to not comment on the results. However, we have included the corrected method (Team111_alt1) below.
Fig. 3.
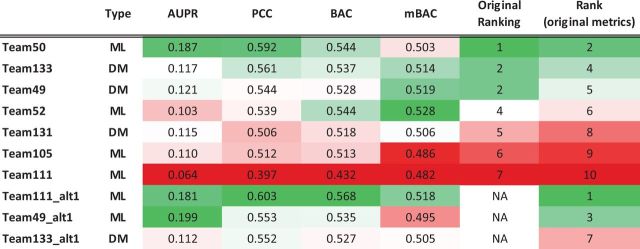
Table summarizing the ranking of different team submissions. Besides the original metrics AUPR, PCC and BAC, we have included mBAC, which computes BAC for each pathway and then computes the mean over pathways. Method type DM indicates direct methods, ML machine learning methods
As shown in Figure 3, according to the official ranking methodology, the machine learning-based approaches (Team111_alt1, Team50, Team49_alt1) consistently rank higher than the direct methods that we have proposed (Team133, Team49). Nevertheless, when considering the mBAC criteria (not used in the official ranking) alone, the direct methods perform better than most of their machine learning-based counterparts. The pooled BAC of the top three machine learning methods (Team111_alt1, Team50, Team49_alt1) was 0.55, on average, whereas our direct methods achieved a 0.53. The mBAC was 0.505, on average, for machine learning and 0.52 for direct methods. The team with the highest mBAC (Team52) shows the highest balance between sensitivity and specificity, whereas the highest ranking team based on the pooled BAC metric (Team111_alt1) shows a stronger bias toward specificity especially for pathways that are perturbed by only a few stimuli (see Supplementary Fig S1).
To evaluate how the different methods performed under the most difficult scenarios, we computed the BAC only for the human pathways in the test dataset for which no activation was observed in the training dataset. Most machine learning methods would have not been able to cope with such situations and hence were expected to predict these pathways as inactive for all stimuli in the test set. Of the 12 pathways that were neither perturbed in the rat nor the human training set, four were perturbed by at least one stimulus in the human test set. Results are given in the right panel of Table 2. Of all the methods, four did not predict any perturbations (Team50, Team105, Team111_alt1 and Team49_alt1) and had a sensitivity of 0, specificity of 1 (BAC = 0.5). Three methods made true-positive predictions: Team133 (sensitivity 0.29, specificity 0.99, BAC = 0.64), Team49 (sensitivity 0.29, specificity 0.96, BAC = 0.62), Team131 (sensitivity 0.14, specificity 0.83, BAC = 0.49). As in the official ranking, a pathway perturbation confidence threshold of 0.5 was used to label pathways as perturbed for a given stimuli.
Table 2.
Team performance on the pathways that were not perturbed in the training stimuli in human
| Method name | Type | 97 pathways not perturbed in the training stimuli in human |
12 pathways that were not perturbed in all training data |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TN | FN | TP | FP | Sens | Spec | BAC | mBAC | TN | FN | TP | FP | Sens | Spec | BAC | mBAC | ||
| Team49 | DM | 2322 | 72 | 15 | 113 | 0.17 | 0.95 | 0.56 | 0.56 | 293 | 5 | 2 | 12 | 0.29 | 0.96 | 0.62 | 0.56 |
| Team133 | DM | 2349 | 77 | 10 | 86 | 0.11 | 0.96 | 0.54 | 0.54 | 302 | 5 | 2 | 3 | 0.29 | 0.99 | 0.64 | 0.60 |
| Team52 | ML | 1806 | 58 | 29 | 629 | 0.33 | 0.74 | 0.54 | 0.53 | 295 | 7 | 0 | 10 | 0.00 | 0.97 | 0.48 | 0.48 |
| Team111_alt1 | ML | 2432 | 86 | 1 | 3 | 0.01 | 1.00 | 0.51 | 0.50 | 305 | 7 | 0 | 0 | 0.00 | 1.00 | 0.50 | 0.50 |
| Team50 | ML | 2435 | 87 | 0 | 0 | 0.00 | 1.00 | 0.50 | 0.50 | 305 | 7 | 0 | 0 | 0.00 | 1.00 | 0.50 | 0.50 |
| Team105 | ML | 2435 | 87 | 0 | 0 | 0.00 | 1.00 | 0.50 | 0.50 | 305 | 7 | 0 | 0 | 0.00 | 1.00 | 0.50 | 0.50 |
| Team49_alt1 | ML | 2435 | 87 | 0 | 0 | 0.00 | 1.00 | 0.50 | 0.50 | 305 | 7 | 0 | 0 | 0.00 | 1.00 | 0.50 | 0.50 |
| Team133_alt1 | DM | 2358 | 85 | 2 | 77 | 0.02 | 0.97 | 0.50 | 0.50 | 303 | 7 | 0 | 2 | 0.00 | 0.99 | 0.50 | 0.50 |
| Team111 | ML | 3 | 1 | 86 | 2432 | 0.99 | 0.00 | 0.49 | 0.50 | 0 | 0 | 7 | 305 | 1.00 | 0.00 | 0.50 | 0.50 |
| Team131 | DM | 2075 | 72 | 15 | 360 | 0.17 | 0.85 | 0.51 | 0.49 | 254 | 6 | 1 | 51 | 0.14 | 0.83 | 0.49 | 0.45 |
Note: True negatives (TN), false negatives (FN), true positives (TP) and false positives (FP) are summed over all pathways to obtain sensitivity, specificity and pooled BAC (BAC). Fifty-one pathways were omitted from the mBAC because they were not perturbed in any test stimuli in human and hence no sensitivity could be calculated for them. Method type DM indicates direct methods, ML machine learning methods.
Additionally, we performed the above analysis on all 97 pathways that are not perturbed in the training stimuli in human—another case where some machine learning approaches could have difficulties. Results are shown in the left panel of Table 2 and match the observations above—Team49 and Team133 had sensitivities of 0.17 and 0.11, and the highest BAC, while the machine learning methods predicted only zero positive results of all 87.
Finally, we computed specificity for all methods across the 70 pathways that are not perturbed in the human test set. The three machine learning-based methods (Team49_alt1, Team111_alt1 and Team50) and Team105 rank highest with a specificity > 0.98. The direct methods (Team49 and Team133) follow with a specificity of 0.95 (see Supplementary Table S2 for details).
3.2 Quality and overlap assessment of data-driven orthologs
Throughout this article, the quality of response ortholog pairs generated by the direct methods was assessed based on the quality of inter-species pathway perturbation prediction. However, such criteria are influenced by several factors such as the quality and identity of the gene sets and pathways considered, and by the sensitivity and specificity of the GSEA analysis used to determine pathway perturbation. In this section, however, we assess the quality of gene pairs directly by using (i) the correlation of their ranks between species across the same stimuli and (ii) their ability to predict DE status of each other.
Both approaches, Team49 and Team133_alt1, were designed to find one rat gene that best mimics the activity of a given human gene when cells are treated with stimuli from the training dataset. As an example, although Team49 chose for the human gene ALOX5AP the rat gene that had a similar rank across all stimuli (Fig. 4, top), Team133 searched for the rat gene with the highest linear correlation between its t-score and the human t-score (bottom) and, for this case, both teams found the rat gene Casp4 as the best match. Interestingly, the sequence-based rat ortholog from the HUGO Gene Nomenclature Committee (HGNC), Alox5ap, does not mimic the behavior of the human counterpart for the 26 stimuli. Additional examples of correlation plots between ranks of response ortholog genes are shown for both training and test stimuli in Supplementary Figures S2–S4. A significant association of gene pairs rank correlations between training and test sets was observed for all three sets of orthologs (Team49, Team133_alt1 and seqence-based from HGNC), as shown in Supplementary Figure S5. These data suggest that the ortholog pairs learned on the training data by the two direct methods do generalize to some extent on the test data.
Fig. 4.
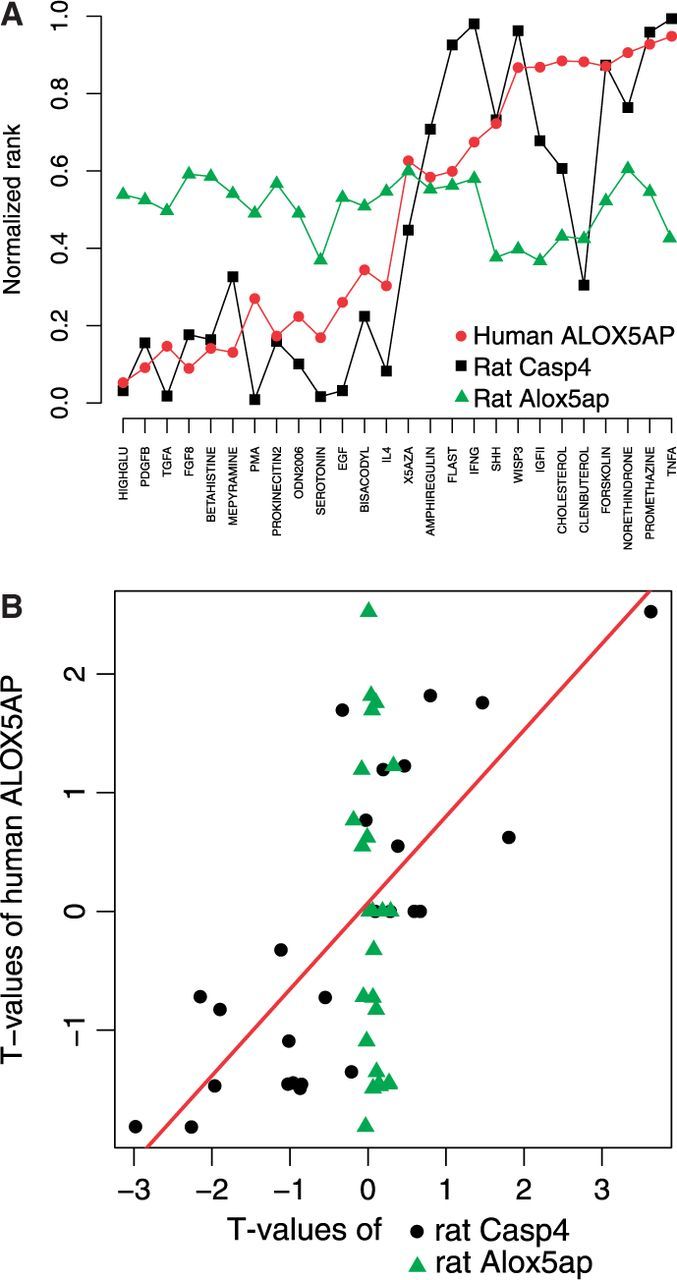
Data-driven orthologs. The ranks of human gene ALOX5AP, rat gene Casp4 and HGNC rat ortholog Alox5ap are standardized (0 most down-, 1 most upregulated among all genes) and shown across all stimuli (top). The moderated t-score for the human gene ALOX5AP is plotted against the standardized t-score for rat genes Casp4, Alox5ap (bottom)
To determine how well the rat response orthologs predict DE of individual human genes, we determined for each stimulus the DE status based on a moderate t-test P-value and noted the sign of the change. Then, for all gene pairs, we counted the following instances: (i) the rat and the human genes are DE in the same direction (true positive), (ii) the rat and the human genes are not DE (true negative), (iii) the rat gene is DE but the human gene is either not DE or changes in the opposite direction (false positive), (iv) only the human gene is DE (false negative). This allowed us to summarize performance metrics for the different ortholog sets separately for training and test data. Results are shown in Table 3. To be able to compare results with the sequence-based orthologs (HGNC), we used only gene pairs in this analysis, where the human gene had exactly one rat ortholog in HGNC. This requirement resulted in 11 540 gene pairs. We have also generated 1 million random ortholog mappings and show the results for the mapping with the highest sum of sensitivity and specificity. Table 3 results indicate that all tested ortholog sets were better in predicting the DE status of human genes than pairing genes at random. While Team133_alt1 had the highest training set performance, this mapping failed to generalize and exhibited lower test set performance. The best overall performance was observed for Team49 with a sensitivity of 0.19 on the test set stimuli at 0.89 specificity.
Table 3.
Ortholog pair performance in predicting DE in human
| Pairs | Training stimuli |
Test stimuli |
||||
|---|---|---|---|---|---|---|
| Sens | Spec | Prec | Sens | Spec | Prec | |
| Team49 | 0.25 | 0.91 | 0.30 | 0.19 | 0.89 | 0.16 |
| Team133_alt1 | 0.29 | 0.92 | 0.37 | 0.14 | 0.87 | 0.11 |
| HGNC | 0.08 | 0.91 | 0.12 | 0.12 | 0.90 | 0.12 |
| Random | 0.06 | 0.92 | 0.09 | 0.09 | 0.90 | 0.09 |
Note: Sens, sensitivity; Spec, specificity; Prec, precision.
To further assess the similarity of the two approaches, we studied the overlap of rat genes found as orthologs for human genes by the two direct methods. In this analysis, we focused on the Team133_alt1 method, as the official submission of Team133 used test stimulus-dependent orthologs and is not suitable for comparison with Team49’s method. A number of 410 human–rat response orthologs were found in common between the two teams (Supplementary Table S3) (. However, for the purpose of gene set analysis, this overlap is unnecessarily restrictive. If, for example, a human pathway has two genes hA and hB and one team found rat genes rD and rE as response orthologs, respectively, but the other team found rat genes rE and rD as response orthologs, respectively, the pathway enrichment analysis will be the same between teams. Therefore, in this additional overlap analysis, we simply count the rat genes selected in common by the two teams for a given human pathway. For every pathway i, we counted the number of common genes Ci between the two methods as well as the probability to observe Ci genes or more just by chance. Of the 246 pathways, 43 had an adjusted P-value < 0.05 using the FDR adjustment method (Benjamini and Hochberg, 1995). To see whether the overlap depends on the number of stimuli that a given pathway was perturbed by, we performed a Fisher’s exact test based on the contingency table: adjusted P-value < 0.05; active in at least one stimulus in human cells in either training or test set separately. No significant association (P > 0.35) was found between the number of active stimuli and the number of pathway with significant gene overlap between methods. These results suggest that the response orthologs found by the two teams show a level of similarity, and this was uniform across the pathways.
Finally, we have also performed a similar overlap analysis between the data-driven orthologs found by the direct methods and the list of orthologs from HGNC provided by the organizers of the challenge. We could not find a single gene set for which the overlap between HGNC orthologs and data-driven orthologs was significant and hence concluded that the sequence-based orthologs and expression profile-based orthologs are different.
4 DISCUSSION
The Systems Biology Verification IMPROVER Pathway perturbation prediction challenge verified methods and concepts of inter-species translation of pathway activity based on gene expression data. In this work, we focused on the solutions of Team49 and Team133, and put them in context of other submissions with the goal of investigating their advantages and limitations. Given the training data, both methods assigned one rat gene to each human gene based on a similar response pattern across the training stimuli. This orthology mapping is used to impute the missing human gene expression values (Team49) or to predict the t-values of DE tests in human from rat (Team133).
These two approaches are different in nature from another category of methods used by other teams in this challenge, namely, machine learning methods. These two categories of methods (direct methods versus machine learning methods) can be seen as a different sequence of two basic steps: gene set analysis and inference from rat data to human data. The machine learning-based methods require that a gene set analysis method is applied first and then inference of the pathway activation status from rat to human is learned. In contrast, direct methods that are the subject of this article perform the rat to human inference first and then apply the gene set analysis step. Therefore, one can see that mapping pathway activity from rat to human accomplished by the machine learning methods is dependent on the particular pathway analysis method used. This might be problematic because a given pathway may or may not appear to be activated by a given stimulus depending on the sensitivity and specificity of the gene set analysis method that was applied [see Tarca et al. (2013b) for a comparison of different methods]. In contrast, the rat-to-human inferences made by the direct methods are independent of the particular gene set analysis method that is involved, and they produce a correspondence between human and rat genes. This correspondence identifies rat genes thought to be under similar regulatory control in humans for the given type of cells and stimuli used in the experiments. The data-driven orthologs found by the direct methods predicted inter-species differential gene expression better than sequence-based orthologs and were significantly overlapping between the two methods; yet, they were divergent from sequence-based orthologs extracted from HGNC database.
Based on the three performance metrics (AUPR, Pearson correlation and BAC) computed by pooling predictions and gold standard over all pathways and stimuli, the top three teams would be Team111_alt1, Team50 and Team49_alt1, all machine learning methods. One could argue that this pooling of predictions favors methods that do not deal with the most difficult scenarios, i.e. with pathway activation inference for those pathways that were seldom or never activated in the training dataset, and simply predict these pathways as non-activated. An alternative would be to compute the performance metrics per pathway and then average over the 246 pathways; however, this is not feasible for AUPR and the correlation coefficient but is for BAC. Therefore, we compared the rankings of the machine learning methods with the ones of the direct methods based on the BAC alone computed as in the official challenge (pooled) and also per pathway and then averaged (mBAC). The pooled BAC was consistently higher for top three machine learning methods than for our direct methods. However, on average, our direct methods have a higher mBAC than the top three machine learning methods.
The inability of machine learning methods to predict the activation status of pathways that were never activated in the training dataset is reflected in the sensitivity of analysis restricted to those pathways. None of the top three methods, all machine learning based, produced a single true positive for these pathways. However, the direct method of Team133 achieved a sensitivity of 0.29 at specificity 0.99, (BAC = 0.64) and Team49 achieved a sensitivity of 0.29 at specificity 0.96 (BAC = 0.62). Machine learning methods define a stimuli effect on a given pathway into classes (activation versus non-activation), and hence require one or more instances of activations to work. An alternative machine learning approach around this issue is to transform the classification problem into a regression problem, as Team52 did, and use a continuous form of the pathway activity evidence (combination of NES scores and FDR values) in a regression model that can be fit regardless of the number of activation cases for a given pathway. Team52 had the highest mBAC statistics computed over all pathways, yet this method did not find any true positive for the zero activation pathways in the training dataset. A second alternative discussed in this issue by Team50 (Hormoz et al., 2014) would be to cluster pathways into groups and hence borrow activation information across pathways. However, from the outset, such an approach did not appear to be outperforming the methodology used by Team50 for their official submission in this challenge.
The task of using transcriptomics data in rat to infer pathway activity in human for a given stimuli proved to be challenging regardless of the approach that was used to tackle this problem. Compared with the task of predicting protein phosphorylation status in human based on gene expression and phosphorylation data in rat (sub-challenge SC2), the prediction of pathway activity (sub-challenge SC3) was much more difficult, with the best pooled BAC in SC2 being 0.77 compared with 0.56 in SC3. There may be several reasons for this, one of which being the fact that in the human training data only few outcomes are positives (6% of the pathway-stimulus combinations), which was not the case in SC2. A second reason for this apparent difficulty is that, by definition, the pathway activation status is dependent on tens or hundreds of member genes, and the call of whether a pathway is perturbed is dependent on the sensitivity and specificity of the gene set analysis method used—in this case pre-ranked GSEA.
More work would be needed to determine whether the use of more sensitive and more specific pathway analysis methods (Tarca et al., 2013b), such as MRGSE (Michaud et al., 2008), or the use of more relevant gene sets based on conserved co-regulated genes (Waltman et al., 2010) can lead to improved translation of pathway activity from rat to human.
Supplementary Material
ACKNOWLEDGEMENTS
The development of the methods and preparation of submissions described in this article were performed by A.T., C.H., R.B. and R.R. independently of P.M., K.R., E.B. and R.N. who contributed to the organization of the challenge. P.M. and K.R. were involved in the manuscript writing and post-challenge data analyses.
Funding: A.T. and R.R. were supported, in part, by the Perinatology Research Branch, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Department of Health and Human Services (NICHD/NIH) and, in part, with Federal funds from NICHD, NIH under Contract No. HHSN275201300006C. A.T. was also supported by the DSC best overall performer grant from Philip Morris International. R.B. and C.H. were supported by US National Science Foundation grants 0922738, 0929338, 1158273 and IOS- 1126971 and National Institutes of Health GM 32877-21/22, RC1-AI087266, RC4-AI092765, PN2-EY016586, IU54CA143907-01 and EY016586-06. The data and organization of the challenge were performed under a joint research collaboration between IBM and PMI and funded by PMI.
Conflict of interest: none declared.
REFERENCES
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995;57:289–300. [Google Scholar]
- Dayarian A, et al. Predicting protein phosphorylation from gene expression: Top methods from the IMPROVER Species Translation Challenge. Bioinformatics. 2015;31:462–470. doi: 10.1093/bioinformatics/btu490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber S, et al. Differential regulation of orthologous chitinase genes in mycoparasitic Trichoderma species. Appl. Environ. Microbiol. 2011;77:7217–7226. doi: 10.1128/AEM.06027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormoz S, et al. Inter-species inference of gene set enrichment in lung epithelial cells from large proteomic and transcriptomic data sets. Bioinformatics. 2014;31:492–500. doi: 10.1093/bioinformatics/btu569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Tope G, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud J, et al. Integrative analysis of RUNX1 downstream pathways and target genes. BMC Genomics. 2008;9:363. doi: 10.1186/1471-2164-9-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, et al. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poussin C, et al. The species translation challenge — a systems biology perspective on human and rat bronchial epithelial cells. Scientific Data. 2014;1:1–14. doi: 10.1038/sdata.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quint E, et al. Differential expression of orthologous Dlx genes in zebrafish and mice: implications for the evolution of the Dlx homeobox gene family. J. Exp. Zool. 2000;288:235–241. doi: 10.1002/1097-010x(20001015)288:3<235::aid-jez4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Rhrissorrakrai K, et al. Understanding the limits of animal models as predictors of human biology: lessons learned from the sbv improver species translation challenge. Bioinformatics. 2015;31:471–483. doi: 10.1093/bioinformatics/btu611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok J, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl Acad. Sci. USA. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarca A, et al. Methodological approach from the best overall team in the improver diagnostic signature challenge. Systems Biomed. 2013a;1:27–37. [Google Scholar]
- Tarca AL, et al. A comparison of gene set analysis methods in terms of sensitivity, prioritization and specificity. PLoS One. 2013b;8:e79217. doi: 10.1371/journal.pone.0079217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltman P, et al. Multi-species integrative biclustering. Genome Biol. 2010;11:R96. doi: 10.1186/gb-2010-11-9-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, et al. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 2004;99:909–917. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.