

ABSTRACT
Mutational analyses have indicated that the carboxyl-terminal domain (CTD) of hepadnavirus core protein and its state of phosphorylation are critical for multiple steps in viral replication. Also, CTD interacts with host proteins in a phosphorylation state-dependent manner. To ascertain the role of CTD in viral replication without perturbing its sequence and the role of CTD-host interactions, CTD of the human hepatitis B virus (HBV) or duck hepatitis B virus (DHBV) core protein, either the wild type (WT) or with alanine or glutamic acid/aspartic acid substitutions at the phosphorylation sites, was expressed in cells replicating DHBV with the WT core protein. A dramatic decrease in phosphorylation of the DHBV core protein (DHBc) was observed when the WT and most HBV core protein CTD (HCTD) variants were coexpressed in trans, which was accompanied by a profound reduction of viral core DNA and, in particular, the double-stranded DNA. One HCTD variant that failed to change DHBc phosphorylation also had no effect on DHBV core DNA. All WT and variant HCTDs and DHBc CTDs (DCTDs) decreased the DHBV covalently closed circular (CCC) DNA. Identification of CTD-host interactions indicated that CDK2 binding by CTD may mediate its inhibitory effect on DHBc phosphorylation and reverse transcription via competition with DHBc for the host kinase, whereas importin α binding by CTD may contribute to inhibition of CCC DNA production by competitively blocking the nuclear import of viral nucleocapsids. These results suggest the possibility of blocking multiple steps of viral replication, especially CCC DNA formation, via inhibition of CTD functions.
IMPORTANCE Mutational analyses have suggested that the carboxyl-terminal domain (CTD) of hepadnavirus core protein is critical for viral replication. However, results from mutational analyses are open to alternative interpretations. Also, how CTD affects virus replication remains unclear. In this study, we took an alternative approach to mutagenesis by overexpressing CTD alone in cells replicating the virus with the wild-type core protein to determine the roles of CTD in viral replication. Our results revealed that CTD can inhibit multiple stages of viral replication, and its effects may be mediated at least in part through specific host interactions. They suggest that CTD, or its mimics, may have therapeutic potential. Furthermore, our experimental approach should be broadly applicable as a complement to mutagenesis for studying protein functions and interactions while at the same time providing a means to identify the relevant interacting factors.
INTRODUCTION
Hepadnaviruses are a family of small hepatotropic DNA viruses, including the global human pathogen hepatitis B virus (HBV) and closely related animal viruses, such as the duck hepatitis B virus (DHBV) (1). Hepadnaviruses contain a small (ca. 3-kb), partially double-stranded (DS), relaxed circular (RC) DNA genome enclosed within an icosahedral capsid that is, in turn, formed by multiple copies (240 or 180) of the viral capsid or core protein (2, 3). All hepadnaviruses replicate their genomic DNA via an RNA intermediate, termed the pregenomic RNA (pgRNA), by reverse transcription (4). Upon entering the host cells, the virion RC DNA is released into the nucleus for conversion into a covalently closed circular (CCC) DNA, which then serves as the viral transcriptional template for the synthesis of all viral RNAs, including pgRNA, by the host RNA polymerase II. After being packaged together with the viral reverse transcriptase (RT) protein into assembling immature nucleocapsid (NC) (5, 6), the pgRNA is converted, by the multifunctional RT, first to a single-stranded (SS) DNA and then to the characteristic RC DNA (4, 7). The mature (i.e., RC DNA-containing) NCs are then encapsulated by the viral envelope proteins and secreted extracellularly as virions, or they can deliver their RC DNA content to the nucleus to be converted to more CCC DNA via an intracellular amplification pathway (8–10).
The viral core protein consists of two separate domains: the N-terminal domain (NTD) that is sufficient to form the capsid shell and the C-terminal domain (CTD) that is dispensable for capsid assembly but nevertheless essential for viral replication (11–13) (Fig. 1A and C). The CTD is highly basic, is rich in arginine, and contains multiple sites of serine/threonine (S/T) phosphorylation (13–16). The HBV core protein (HBc) contains three major S phosphorylation sites (S155, S162, and S170) (15), and three additional S/T phosphorylation sites (T160, S168, and S176) have recently been identified (17) (Fig. 1B). Besides these six known phosphorylation sites, another potential CTD phosphorylation site (S178) (Fig. 1B) is also conserved among most HBV isolates. Similarly, the duck hepatitis B virus (DHBV) core protein (DHBc) contains six known S/T phosphorylation sites at its CTD (S230, S232, T239, S245, S257, and S259) (14, 16) (Fig. 1E). Mutational analyses indicate that the phosphorylation state of HBc CTD (HCTD) is important for RNA packaging and DNA synthesis (17–19). The phosphorylation state of DHBc CTD (DCTD) has only a minor effect on viral RNA packaging but is essential for viral DNA synthesis such that phosphorylation at the S/T sites is required for minus-strand DNA synthesis and dephosphorylation is needed for the synthesis and accumulation of mature RC DNA (13, 16, 20–22). Several kinases have been reported to phosphorylate the core protein in vitro, including protein kinase C (23, 24) and the serine-arginine protein kinases 1 and 2 (SRPK1 and SRPK2) (25), but it remains uncertain what, if any, specific sites of the core protein or CTD are phosphorylated by these kinases in vivo. We have recently reported that the host cyclin-dependent kinase 2 (CDK2) can phosphorylate the functionally critical S/T-P sites in HCTD and DCTD in vitro and in vivo and CDK2 or a CDK2-like kinase is incorporated into NCs (26), accounting for the major part of the so-called endogenous kinase activity reported decades ago (27). The putative cellular phosphatase(s) that mediates dephosphorylation of mature NCs remains to be identified.
FIG 1.
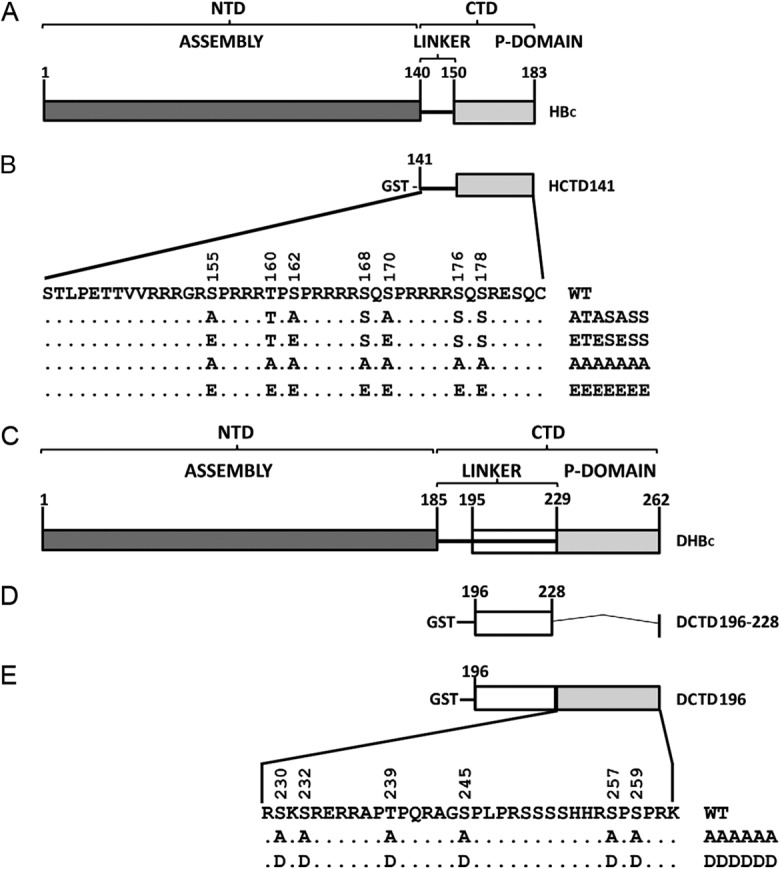
Schematic diagram of HBc and DHBc domains and GST-HCTD and -DCTD fusion constructs. (A) HBc contains an N-terminal domain (NTD) from amino acids 1 to 140 and a C-terminal domain (CTD) that includes a flexible linker region from amino acids 141 to 149 and a phosphor (P) domain from 150 to 183. (B) The WT GST-HCTD fusion protein, HCTD141, contains the CTD from residue 141 to the end of HBc. Alanine (A) or glutamic acid (E) substitutions at the three major phosphorylation sites (S155, S162, and S170) or all seven potential phosphorylation sites (T160, S168, S176, and S178 in addition to the aforementioned three major sites) in the WT GST-HCTD fusion protein were named HCTD141-ATASASS, HCTD141-ETESESS, HCTD141-AAAAAAA, and HCTD141-EEEEEEE, respectively. (C) DHBc contains an NTD from amino acids 1 to 185 and a CTD from 186 to 262, including a flexible linker region from amino acids 186 to 228 and the phosphor (P) domain from 229 to 262. (D) The linker region from amino acids 196 to 228 was fused with GST and named DCTD196–228. (E) The WT GST-DCTD fusion protein, DCTD196, contains the linker region and the phosphor domain, in which six phosphorylation sites (S230, S232, T239, S245, S257, and S259) are localized. All six phosphorylation sites in DCTD196 were substituted for A or aspartic acid (D), and the resultant mutants were named DCTD196-AAAAAA and DCTD196-DDDDDD, respectively.
Hepadnavirus CCC DNA is the molecular basis of viral persistence, but the exact mechanism for CCC DNA formation is still obscure. It is generally accepted that RC DNA within the mature NC must be released, which is probably facilitated by preferential destabilization of the mature NCs (28), and imported into the nucleus to be converted to CCC DNA (10, 29). It is still unclear how the mature NC disassembles and delivers RC DNA to the nucleus, but several studies have proposed that NCs can be directed to the nuclear pore by nuclear localization signals (NLSs) located on the CTD of the viral core protein, following the classical nuclear targeting pathway (24, 30, 31), in which soluble transport receptors importin α/importin β bind to the NLS of the core protein and mediate attachment of the complex to the nuclear pore complexes (NPCs). At the nuclear pore, with the assistance of components of NPC, mature NCs may selectively disintegrate and deliver their RC DNA content to the nucleus (30, 31). The multiple NLSs of HCTD overlap the phosphorylation sites (15, 24, 32–35), while the single NLS reported for DHBc was localized upstream of the DCTD phosphorylation sites in the so-called linker region (36) (Fig. 1). It has been suggested that the phosphorylation state of CTD could affect its NLS function and thus potentially RC DNA nuclear import and CCC DNA formation (15, 24, 30, 36).
The roles of CTD phosphorylation of the hepadnavirus core protein in viral replication, as outlined above, have been analyzed by extensive mutational analyses of the phosphorylation sites. However, it remains possible that the sequence changes per se, rather than the CTD phosphorylation state, were responsible for the various effects reported. Furthermore, it is still unclear how the CTD phosphorylation state affects virus replication. One possibility is that the core protein exerts its multiple roles by interacting, dynamically, with distinct viral or host factors at different stages of viral replication, in a CTD phosphorylation state-dependent fashion (37). Efforts to manipulate directly the host kinases or other CTD-interacting host factors as a means of affecting CTD phosphorylation state and functions have been complicated by the pleiotropic and toxic effects of these manipulations on cell biology due to the multiple roles of these host factors in the cells.
To further ascertain the role of CTD in viral replication, without perturbing its sequence, and the role that CTD-host interactions may play at different stages of viral replication, we took an alternative approach to mutagenesis by overexpressing HCTD or DCTD, either wild type (WT) or with A or E/D substitutions at the phosphorylation sites, in cells replicating DHBV with the WT core protein (Fig. 1). We have recently shown that both HCTD and DCTD, when expressed without the NTD, serve as appropriate substrates for phosphorylation by cellular kinases in vivo and in vitro, mimicking the CTD in the context of full-length HBc or DHBc (26, 37). Furthermore, the isolated HCTD and DCTD can interact with host factors such as B23 and I2PP2A in a phosphorylation-regulated manner, as in the context of the full-length proteins (37). Thus, we reasoned that the isolated CTD might be able to affect viral replication, in trans, by competing for the cellular kinases or other host factors with the WT, full-length DHBc. The effects of HCTD/DCTD on various steps of DHBV replication, including core protein phosphorylation, capsid assembly, pgRNA packaging, reverse transcription, and CCC DNA synthesis were analyzed. Our results revealed that CTD could indeed regulate hepadnavirus reverse transcription and CCC DNA formation, and its effects may be mediated at least in part through specific host interactions.
MATERIALS AND METHODS
Plasmids and antibodies.
pCMV-DHBV-Env− directs the expression of the WT DHBV pgRNA under the control of the human cytomegalovirus promoter and is defective in the expression of all DHBV surface proteins (10, 38, 39). HCTD coding sequences from 141 to 183 (WT and phosphorylation site mutants) were generated by PCR amplification and subcloned to pEBG (39), downstream of the glutathione S-transferase (GST)-coding sequences, for expression of the GST-HCTD fusion proteins. The WT sequence is designated HCTD141, and the phosphorylation site mutants, S/T to A and S/T to E at the three SP sites (S155, S162, and S170), are designated HCTD141-ATASASS and HCTD141-ETESESS, respectively (Fig. 1B). These three GST-HCTD fusion protein expression constructs have been described previously (37). In addition, HCTD141-AAAAAAA and HCTD141-EEEEEEE were generated in this study with all seven potential phosphorylation sites (T160, S168, S176, and S178, in addition to the aforementioned three sites) substituted by A or E (Fig. 1B). The GST-DCTD fusion protein expression constructs have also been described previously (37). Briefly, DCTD coding sequences from 196 to 262, the WT sequence, or sequences with S/T-to-A or S/T-to-D substitutions at all six phosphorylation sites (S230, S232, T239, S245, S257, S259) were subcloned to pEBG for expression of DCTD196, DCTD196-AAAAAA, or DCTD196-DDDDDD, respectively (Fig. 1E), and DCTD coding sequences from 196 to 228 (the linker region) were subcloned for expression of DCTD196–228 (Fig. 1D).
A rabbit polyclonal antibody against DHBc was generously provided by William Mason (40). Monoclonal mouse anti-GST (Thermo Scientific), mouse anti-importin α (Sigma), and polyclonal rabbit anti-CDK2 (Santa Cruz) were commercially obtained. Goat anti-rabbit IgG horseradish peroxidase (HRP)-conjugated antibody (Southern Biotech) and goat anti-mouse HRP-conjugated antibody (Invitrogen) were used as secondary antibodies.
Cell cultures and transient transfections.
Cells from the human embryonic kidney cell line HEK293 were cultured in Dulbecco's modified Eagle's medium (DMEM)-F12 supplemented with 10% fetal bovine serum (FBS) (HyClone), as previously described (10). HEK293 cells were seeded in 60-mm or 100-mm dishes and transfected at 70% confluence with mixtures of pCMV-DHBV-Env– and the indicated GST-HCTD/DCTD expression constructs or with GST-HCTD/DCTD expression constructs alone, using a CalPhos mammalian transfection kit (Clontech) (41).
Detection of DHBc and GST-HCTD/DCTD fusion protein expression.
Five days posttransfection, HEK293 cells were harvested, and the core protein expression levels were analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and Western blotting as described previously (26, 39), with minor modifications. Briefly, HEK293 cells were lysed in NP-40 lysis buffer (10) (50 mM Tris-HCl [pH 8.0], 1 mM EDTA, 1% NP-40, and 1× protease inhibitor cocktail [Roche]), and the cell lysates were resolved on a 12.5% SDS-PAGE gel. Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (PerkinElmer) and detected by incubation with the rabbit polyclonal antibody against DHBc (40) (1:5,000 dilution), followed by the goat anti-rabbit secondary antibody (1:20,000 dilution) and Western Lightning enhanced chemiluminescence reagent (PerkinElmer), and the signals were visualized with a Chemi-Doc MP imaging system (Bio-Rad) and quantified using Image Lab software version 4.1 (Bio-Rad). Samples were also probed with the mouse monoclonal antibody against GST (1:1,000 dilution), followed by the goat anti-mouse secondary antibody (1:20,000 dilution).
Analysis of viral DNA.
DHBV core and protein-free (PF) DNAs were isolated 5 days posttransfection, as previously described (10), with minor modifications. Briefly, for isolation of core DNA, HEK293 cells were lysed in NP-40 lysis buffer as described above. After removal of the nuclear pellet by brief centrifugation, the supernatant (cytoplasmic lysate) was incubated with micrococcal nuclease (MNase) (Roche) (150 units/ml) and CaCl2 (5 mM) at 37°C for 90 min, and proteinase K (Invitrogen) was then used to digest viral DNA-protein complexes after NCs were precipitated with polyethylene glycol and disrupted by SDS. Viral core DNA was then purified by phenol-chloroform extraction and ethanol precipitation. PF DNA was isolated using Hirt extraction (10, 42). Briefly, HEK293 cells were lysed in SDS lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 150 mM NaCl, and 1% SDS). After being incubated for 5 min at room temperature, the cell lysates were mixed with 2.5 M KCl and incubated at 4°C overnight with gentle rotation. The lysate was then spun at 14,000 × g for 20 min, and the supernatant was extracted three times with phenol and once with chloroform. The DNA was then recovered by ethanol precipitation.
Purified core or PF DNA was analyzed by agarose gel electrophoresis and Southern blotting, as previously described (43–45). Briefly, viral DNA was separated on a 1% agarose gel. The gel was then subjected to denaturalization in a solution containing 0.5 M NaOH and 1.5 M NaCl, followed by neutralization in a buffer containing 1.5 M NaCl and 1 M Tris-HCl (pH 7.5). The DNA was then transferred onto a positively charged nylon membrane (GE) and detected with a 32P-radiolabeled DHBV DNA probe prepared by using the Random Priming labeling kit (Roche). Hybridization signals were visualized by phosphorimaging (Typhoon, GE) and quantified using the Quantity One software, version 4.5.0 (Bio-Rad).
Analysis of viral RNA packaging.
DHBV RNA packaging was analyzed 5 days posttransfection, as previously described (21). Briefly, intact NCs from MNase-digested cell lysate were analyzed by native agarose gel electrophoresis, followed by detection using a 32P-labeled DHBV plus-strand-specific riboprobe against the 5′ end of the pgRNA. Hybridization signals were visualized and quantified as described above. Under the conditions used for RNA packaging assay, plus-strand-specific riboprobes (complementary to viral plus-strand DNA or pgRNA) do not detect the plus-strand DNA (or minus-strand DNA) (46), likely due to the fact that the plus-strand DNA is always hybridized to the minus-strand DNA in the viral NC and is thus unavailable to hybridize to the riboprobes. The same membrane was subsequently probed with an anti-DHBV core polyclonal antibody (40) to detect the core protein, and the signals were visualized using a Chemi-Doc MP imaging system (Bio-Rad) and quantified using Image Lab software version 4.1 (Bio-Rad).
GST pulldown assay.
Three days posttransfection, GST fusion proteins and potentially bound host proteins were purified from HEK293 cells and analyzed as described previously (37). Briefly, HEK293 cells were lysed in pulldown lysis buffer containing 50 mM Tris (pH 8.0), 150 mM NaCl, 1 mM EDTA, 1% NP-40, and protease inhibitors (complete protease inhibitor cocktail; Roche) and phosphatase inhibitors (10 mM sodium fluoride, 50 mM β-glycerophosphate, 10 mM sodium pyrophosphate, and 2 mM sodium orthovanadate). Cell debris and nuclear pellet were removed by centrifugation at 11,000 × g for 20 min at 4°C, and the cleared supernatant was treated with RNase A (67 μg/ml; Sigma) for 1 h at room temperature with gentle agitation. The supernatant was removed from the precipitates and applied to the glutathione (GSH) resin (Sigma) overnight at 4°C with rotating. The resin was collected by two-minute centrifugation at 800 × g, and unbound proteins were removed. The resin was washed extensively in the pulldown lysis buffer, except that 1% NP-40 was replaced with 0.1% NP-40. Bound proteins were eluted in 20 mM GSH (Sigma) in 200 mM Tris (pH 8.0) with the protease inhibitor cocktail (Roche) and analyzed by SDS-PAGE and detected by protein staining and Western blotting.
RESULTS
HCTD but not DCTD strongly inhibited DHBc phosphorylation.
To ascertain if the isolated HCTD or DCTD could affect viral replication, HEK293 cells were cotransfected with pCMV-DHBV-Env–, which expresses a surface protein-defective DHBV genome that allows enhanced amplification of CCC DNA (38), together with the indicated GST-HCTD (WT or mutant; Fig. 1B) or GST-DCTD (WT or mutant; Fig. 1D and E) expression plasmid. As we have shown before, HCTD and DCTD can bind specific host factors differently depending on the state of CTD phosphorylation (37). HCTD and DCTD mutants mimicking either the nonphosphorylated state (HCTD141-ATASASS and -AAAAAAA and DCTD196-AAAAAA) or the phosphorylated state (HCTD141-ETESESS and -EEEEEEE and DCTD196-DDDDDD) (Fig. 1) were included along with the WT HCTD and DCTD fusion proteins. HCTD141-ATASASS and -ETESESS had the three major HCTD phosphorylation sites (all within the SP motif) substituted, but the other non-SP sites were unchanged. HCTD141-AAAAAAA and -EEEEEEE and DCTD196-AAAAAA and -DDDDDD had all seven potential phosphorylation sites in HCTD and all six phosphorylation sites in DCTD substituted, respectively. An additional DCTD construct was included containing only the linker region (196 to 228), as we have shown recently that this region alone, without the downstream phosphorylated region in DCTD, could mediate CTD binding to specific host factors (37). HEK293 cells were chosen, as they are known to support high-level DHBV and HBV replication, including CCC DNA formation (10, 39, 47). Furthermore, they can be transfected at high efficiency to allow overexpression of the HCTD or DCTD fusion proteins so as to enhance the likelihood of their successful competition for host factors with the full-length DHBc protein. DHBV, rather than HBV, was used as the target virus for regulation by HCTD and DCTD in trans, as DHBV is known to form CCC DNA much more efficiently than HBV in cell cultures, particularly when the expression of the viral envelope proteins was eliminated (10). Although hyperamplification of DHBV CCC DNA was shown to be cytotoxic in primary duck hepatocytes (not in chicken hepatoma cell line LMH) (48), no apparent cytotoxicity was observed under transient-transfection conditions in established cell lines (10, 49).
We first examined if the expression pattern of DHBc was affected by the HCTD or DCTD fusion proteins via SDS-PAGE and Western blotting. As reported previously (21, 50), DHBc clearly exhibited heterogeneity in mobility on SDS-PAGE gels, with at least two main distinct bands detected (Fig. 2A, C, and D, top), representing hyperphosphorylated (Hyper-P; top band) and hypophosphorylated (Hypo-P; bottom band) DHBc (14, 21, 50). Compared with the pattern of DHBc when GST was coexpressed, which showed more hyperphosphorylated DHBc than hypophosphorylated DHBc (Fig. 2A [lanes 1 to 5], C [lane 1], D [lanes 1 to 7]), the DHBc phosphorylation pattern was clearly changed when HCTD141 (WT), HCTD141-ATASASS, HCTD141-ETESESS, and HCTD141-EEEEEEE were coexpressed, with more hypophosphorylated DHBc than hyperphosphorylated DHBc (Fig. 2A [lanes 6 to 8 and 10] and B). These results clearly demonstrated that these HCTD variants decreased DHBc phosphorylation, leading to more hypophosphorylated DHBc than the hyperphosphorylated form and a 4- to 5-fold decrease in the ratio of hyperphosphorylated DHBc to hypophosphorylated DHBc (Fig. 2B). In sharp contrast, the variant HCTD141-AAAAAAA, which mimics the nonphosphorylated state at all seven HCTD sites, had no appreciable effect on DHBc phosphorylation (Fig. 2A [lane 9] and B). Expression of HCTD-AAAAAAA was poor compared with that of the other HCTD variants (Fig. 2A), but increasing the expression of this variant, to levels above those of the other variants when they inhibited DHBc phosphorylation, still failed to change DHBc phosphorylation (Fig. 2C, lanes 11 to 13 compared with the GST control in lane 1 and the other HCTD variants in lanes 2, 5, 8, and 14). For reasons unknown at this time, the HCTD constructs increased the expression of DHBc (with a given DHBV plasmid amount transfected), as most clearly shown in Fig. 2C. We also determined whether DCTD could have a similar effect as HCTD on DHBc phosphorylation. As shown in Fig. 2D and E, coexpression of DCTD196-AAAAAA and DCTD196-DDDDDD had no effect on DHBc phosphorylation (Fig. 2D [lanes 9 and 10] and E), and coexpression of DCTD196 and DCTD196–228 had only a slight inhibitory effect on DHBc phosphorylation (Fig. 2D [lanes 8 and 11] and E). These results thus indicated that HCTD had a strong inhibitory effect on DHBc phosphorylation, whereas DCTD had little to no effect.
FIG 2.

Effect of HCTD and DCTD on DHBc phosphorylation. HEK293 cells were cultured in 60-mm dishes and transfected with pCMV-DHBV-Env– and the indicated GST-HCTD (A and C) or GST-DCTD (D) expression plasmids, as detailed below. In all transfections, the total DNA amount per transfection was kept constant (10 μg) by adding the GST expression plasmid as a filler when needed. The expression of DHBc and GST-HCTD/DCTD fusion proteins was analyzed by SDS-PAGE and Western blotting with a DHBc-specific (panels A, C, and D, top) or GST-specific (panels A, C, and D, bottom) antibody, respectively. In panel A, lanes 1 to 5, 0.0625 μg, 0.125 μg, 0.25 μg, 0.5 μg, or 1 μg of pCMV-DHBV-Env– was transfected into HEK293 cells, together with the control GST expression plasmid. For lanes 6 to 10, 0.5 μg (lanes 6 to 8 and 10; thick horizontal line) or 0.125 μg (lane 9; thin horizontal line) of pCMV-DHBV-Env–,together with 1 μg of the indicated GST-HCTD expression plasmid, was cotransfected into HEK293 cells. In panel C, 1 μg of pCMV-DHBV-Env– was cotransfected into HEK293 cells together with the GST expression plasmid alone (lane 1) or with increasing amounts (1 μg for lanes 2, 5, 8, 11, 14; 4 μg for lanes 3, 6, 9, 12, 15; 9 μg for lanes 4, 7, 10, 13, 16) of the indicated GST-HCTD expression plasmid. In panel D, lanes 1 to 7, 0.0625 μg, 0.125 μg, 0.25 μg, 0.5 μg, 1 μg, 2 μg, or 4 μg of pCMV-DHBV-Env– was transfected into HEK293 cells, together with the GST expression plasmid. For lanes 8 to 11, 0.125 μg of pCMV-DHBV-Env– and 9.875 μg of the indicated GST-DCTD expression plasmid were cotransfected into HEK293 cells. (B and E) The relative levels of hyperphosphorylated DHBc (Hyper-P; the top band of DHBc in panels A and D) and hypophosphorylated DHBc (Hypo-P; the bottom band of DHBc in panels A and D) were quantified and the ratios of Hyper-P to Hypo-P (Hyper-P/Hypo-P) DHBc from GST-HCTD (A) or -DCTD (D) cotransfections were normalized to that of the control (GST) cotransfection that is set to 100 (lane 4 in panel A and lane 2 in panel D). The arrowheads denote the GST-DCTD fusion proteins that were detected by the antibody against DHBc. Data are presented as means and the standard errors of the means (SEM) from four (B) or three (E) independent experiments.
As will be described below, we noticed that DHBV CCC DNA formation could be saturated when viral core DNA levels reached a certain limit, which would lead to underestimation of any effect of HCTD or DCTD on CCC DNA formation. To keep viral core DNA levels under this limit, the amount of pCMV-DHBV-Env– was deliberately decreased when cotransfected with HCTD141-AAAAAAA (Fig. 2A, lane 9) or all of the DCTD variants (Fig. 2D, lanes 8 to 11), compared with cotransfection with the other HCTD variants (Fig. 2A, lanes 6 to 8 and 10). This was done because HCTD141-AAAAAAA and the DCTD variants did not decrease DHBV core DNA, but the other HCTD variants did. To facilitate comparison of the HCTD or DCTD cotransfections to the appropriate control GST cotransfection, the control GST cotransfection was done with a range of amounts of the pCMV-DHBV-Env– plasmid. Therefore, the HCTD or DCTD cotransfections were compared to the appropriate GST control transfection showing a similar level of DHBc expression (or capsid assembly, pgRNA packaging, core DNA synthesis) (see the figure legends for details), when determining the effects of the HCTD and DCTD fusion proteins on capsid assembly (or pgRNA packaging, core DNA synthesis, and CCC DNA formation). Thus, the effect on a particular step in the viral life cycle was always normalized to the preceding step. As shown in Fig. 2 and as will be described later in the legends to Fig. 3 and 4, DHBc phosphorylation, capsid assembly, pgRNA packaging, and core DNA synthesis efficiency were not affected by the amount of pCMV-DHBV-Env– transfected (or DHBc expression levels). This result suggested that these steps of the viral life cycle, in contrast to CCC DNA formation (Fig. 5 and 6), were not saturated under our experimental conditions.
FIG 3.
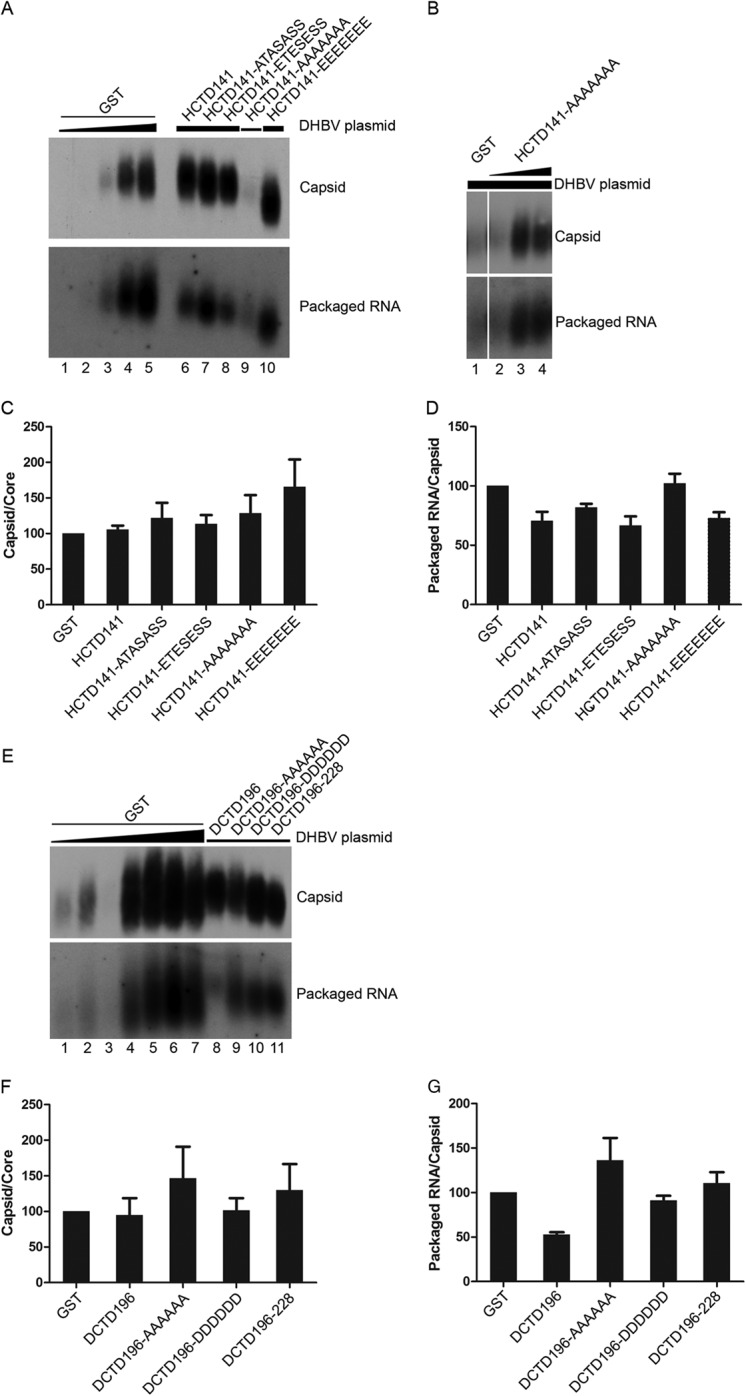
Effect of HCTD and DCTD on DHBV capsid assembly and pgRNA packaging. HEK293 cells were cotransfected as described in the legend to Fig. 2. Cytoplasmic capsids were analyzed by native agarose gel electrophoresis and transferred to a nitrocellulose membrane. A radiolabeled, plus-strand-specific DHBV riboprobe was used to detect the packaged pgRNA (A, B, and E, bottom), and the capsid protein on the same membrane (A, B, and E, top) was detected by an anti-DHBc antibody. Transfections shown in panels A and E were the same as those shown in Fig. 2A and D, respectively. Lanes 1 to 4 in panel B shows the same transfections as in lane 1 and lanes 11 to 13 in Fig. 2C. (C and F) The relative levels of assembled capsids from the GST-HCTD (A) or GST-DCTD (E) cotransfections were normalized to those of the total core protein detected by SDS-PAGE (shown in Fig. 2) (capsid/core) to estimate the assembly capability of DHBc. The capsid/core ratios from GST-HCTD (A) or GST-DCTD (E) were compared to that of the control (GST) transfection that is set to 100 (lane 4 in panel A and lane 2 in panel E). (D and G) The relative levels of packaged pgRNA were normalized to those of assembled capsids (packaged pgRNA/capsid) in the same manner. The packaged pgRNA/capsid ratios from the GST-HCTD (A) or GST-DCTD (E) cotransfections were compared to that of the control (GST) transfection that is set to 100 (lane 4 in panel A and lane 2 in panel E) to estimate DHBV RNA packaging capability. Data are presented as means and SEM from three independent experiments.
FIG 4.

Effect of HCTD and DCTD on DNA synthesis. (A, B, and E) HEK293 cells were cotransfected as described in the legend to Fig. 3A, B, and E, except that in panel A, the HCTD141-AAAAAAA cotransfection (with strong core DNA signal) was loaded in lane 10 after HCTD141-EEEEEEE in lane 9. Replicative viral DNA intermediates were isolated from cytoplasmic NCs and analyzed by Southern blotting using a radiolabeled DHBV DNA probe. (C and F) The relative levels of total core DNA from the GST-HCTD (A) and GST-DCTD (E) cotransfections were normalized to those of the packaged pgRNA (shown in Fig. 3A and E, respectively), and the core DNA/packaged RNA ratios were compared with that of the control (GST) cotransfection, set to 100 (lane 4 in panel A and lane 2 in panel E). (D) The relative levels of RC DNA from the GST-HCTD cotransfections were normalized to those of SS DNA. The RC DNA/SS DNA ratios were compared to that of the control (GST) cotransfection, set to 100 (lane 4 in panel A). Data are presented as means and SEM from three independent experiments. RC, relaxed circular DNA; SS, full-length single-stranded DNA; M, DNA marker.
FIG 5.
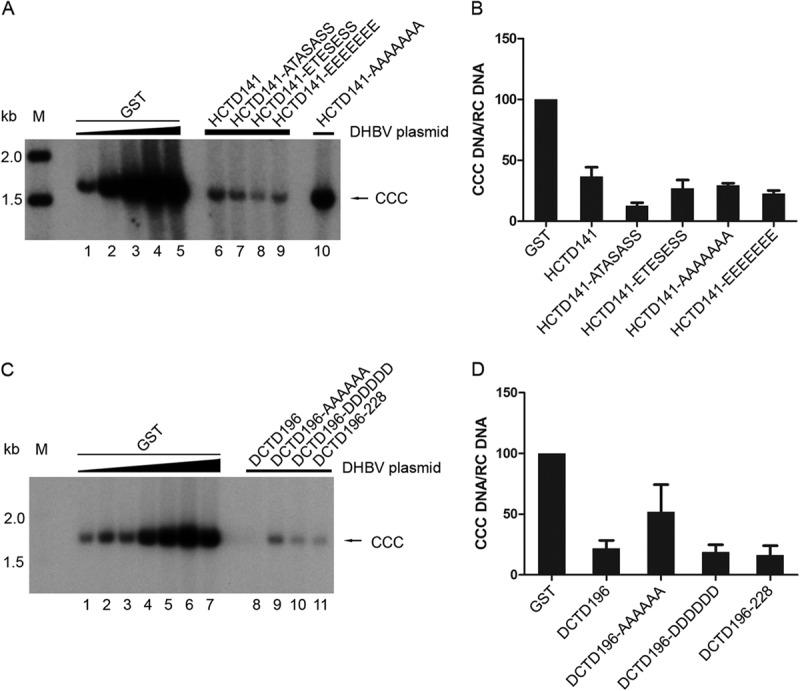
Effect of HCTD and DCTD on CCC DNA synthesis. (A and C) HEK293 cells were cotransfected as described in the legend to Fig. 4A and E. CCC DNA was extracted using Hirt extraction and analyzed by Southern blotting using a radiolabeled DHBV DNA probe. (B and D) The relative levels of CCC DNA from the GST-HCTD (A) and GST-DCTD (C) cotransfections were normalized to those of RC DNA (shown in Fig. 4A and E, respectively), and the CCC DNA/RC DNA ratios were compared with that of the control (GST) cotransfection having similar RC DNA levels, set to 100 (lane 3 in panel A for the HCTD141-AAAAAAA cotransfection, lane 1 in panel A for the other GST-HCTD cotransfections, lane 3 in panel C for DCTD196 cotransfections, and lane 4 in panel C for the other GST-DCTD cotransfections). Data are presented as means and SEM from three independent experiments. CCC, covalently closed circular DNA; M, DNA marker.
FIG 6.

Titration assay to estimate the efficiency of CCC DNA formation from increasing amounts of RC DNA. (A, B, D, E) Lanes 1 to 6, HEK293 cells in 60-mm dishes were transfected with pCMV-DHBV-Env– (0.03125, 0.0625, 0.125, 0.25, and 0.5 μg for lanes 1 to 5 in panels A and B; 0.0625, 0.125, 0.25, 0.5, 1, and 2 μg for lanes 1 to 6 in panels D and E). The GST expression plasmid was cotransfected to keep the total DNA amount per transfection constant (10 μg). RC DNA (A, D) and CCC DNA (B, E) were isolated and analyzed as described in the legends to Fig. 4 and 5, respectively. (C, F) The ratios of CCC DNA to RC DNA (CCC DNA/RC DNA) were estimated as described in the legend to Fig. 5 and normalized to that of lane 1, set to 1. Graph C is based on the results shown in panels A and B, and graph F is based on panels D and E. The average copy numbers of RC DNA, CCC DNA, and RC plus CCC DNA per cell are indicated at the bottom of the graphs. S1 to S6, 486, 162, 54, 18, 6, and 2 pg of the 3-kb DHBV DNA was loaded as quantification standards. M, DNA marker; RC, relaxed circular DNA; SS, full-length single-stranded DNA; CCC, covalently closed circular DNA.
HCTD and DCTD had little effect on DHBV capsid assembly and pgRNA packaging.
To address whether the coexpressed GST-HCTD/DCTD would have any effect on DHBV capsid assembly and pgRNA packaging, we estimated the levels of DHBV assembled capsids and pgRNA packaged in capsids by the native agarose gel assay. Given the effect on DHBc protein expression levels by the HCTDs as described above, the efficiencies of DHBV capsid assembly and pgRNA packaging under the different conditions were determined by normalizing the level of assembled capsids to that of total DHBc protein or the level of packaged pgRNA to that of assembled capsids, respectively. As shown in Fig. 3, compared with the GST-alone control samples, coexpression of neither HCTD nor DCTD led to any significant changes in the level of assembled capsids (Fig. 3A, B, and E, top) after normalization to total DHBc protein levels (Fig. 2). Similarly, coexpression of none of the HCTD or DCTD variants had any major effect (less than 2-fold at most) on the normalized levels of DHBV pgRNA packaging (Fig. 3A, B, and E, bottom). Among the GST-alone coexpression controls, the levels of capsid assembly (Fig. 3A [lanes 1 to 5] and E [lanes 1 to 7]) were proportional to the total DHBc levels (Fig. 2A [lanes 1 to 5] and D [lanes 1 to 7]), and levels of pgRNA packaging were proportional to those of capsid assembly (Fig. 3A [lanes 1 to 5] and E [lanes 1 to 7]). This indicated that DHBc capsid assembly and pgRNA packaging did not reach saturation under these conditions. Thus, these results indicated that HCTD and DCTD had little to no effect on DHBc assembly or pgRNA packaging.
Effect of HCTD and DCTD on DHBV DNA synthesis.
To determine whether the coexpressed GST-HCTD/DCTD would have any effect on DHBV DNA synthesis, the levels of DHBV core DNA were analyzed by Southern blotting. Core DNA synthesis efficiency was estimated by normalizing levels of total core DNA to RNA packaging levels. When HCTD141, HCTD141-ATASASS, HCTD141-ETESESS, and HCTD141-EEEEEEE were coexpressed, DHBV core DNA synthesis was dramatically inhibited (7- to 12-fold reduction) (Fig. 4A [lanes 6 to 9] and C). When the expression of these HCTD variants was increased by transfecting more expression plasmids, as shown in Fig. 2C, they decreased DHBV core DNA in a dose-dependent manner. At the largest amount of HCTDs transfected, almost no core DNA was detectable (data not shown). These were the same HCTD variants that also inhibited DHBc phosphorylation (Fig. 2). Notably, HCTD141-AAAAAAA, the one HCTD variant that did not affect DHBc phosphorylation (Fig. 2), also had little effect on core DNA synthesis (Fig. 4A [lane 10] and C), even when its expression was increased (Fig. 4B and C). The apparent increase in DHBV DNA synthesis by HCTD141-AAAAAAA (Fig. 4B, lanes 2 to 4), relative to the GST control, was caused by the increase in DHBc protein expression (Fig. 2C, lanes 11 to 13). On the other hand, none of the DCTD variants had any major effect on DHBV core DNA synthesis, with only DCTD196 (WT) showing a modest (2-fold) inhibitory effect (Fig. 4E and F), correlating with its modest to no inhibition on DHBc phosphorylation (Fig. 2). Among the GST-alone coexpression controls, the levels of core DNA (Fig. 4A [lanes 1 to 5] and E [lanes 1 to 7]) were proportional to the RNA packaging levels (Fig. 3A [lanes 1 to 5] and E [lanes 1 to 7]), indicating that core DNA synthesis did not reach saturation under these conditions.
A close inspection of the core DNA pattern also revealed that 4 of the 5 HCTD variants, again with the exception of HCTD141-AAAAAAA, were able to preferentially reduce the levels of mature RC DNA (i.e., plus-strand DNA synthesis), beyond their inhibitory effects on total core DNA synthesis (Fig. 4A and D). When the mature RC DNA levels were normalized to those of the SS DNA, coexpression of HCTD141, HCTD141-ATASASS, HCTD141-ETESESS, and HCTD141-EEEEEEE led to a further 4- to 5-fold reduction of RC DNA (Fig. 4A [lanes 6 to 9] and D). Again, HCTD141-AAAAAAA, the one HCTD variant which had little effect on total core DNA synthesis, also had no effect on RC DNA (Fig. 4A [lane 10] and D).
HCTD and DCTD strongly reduced the DHBV CCC DNA levels.
Next, we examined whether DHBV CCC DNA formation might be affected by the coexpression of the GST-HCTD/DCTD fusion proteins. CCC DNA formation efficiency was estimated by normalizing levels of CCC DNA to those of RC DNA. As alluded to above, titration assay showed that the efficiency of CCC DNA formation from RC DNA decreased when the level of RC DNA was increased to certain levels (as is described in the legend to Fig. 6). Thus, the normalized levels of CCC DNA from each HCTD/DCTD cotransfection were compared to that from the indicated GST control cotransfection that showed similar levels of RC DNA (see Fig. 4A and E). As shown in Fig. 5, coexpression of all the HCTD variants, including HCTD141-AAAAAAA, which had no effect on DHBc phosphorylation or core DNA synthesis, led to a significant decrease of CCC DNA (Fig. 5A [lanes 6 to 9 compared with lane 1 as the GST control; lane 10 compared with lane 3] and B). Similarly, the level of CCC DNA decreased significantly when DCTD196, DCTD196-DDDDDD, and DCTD196–228 were coexpressed (Fig. 5C [lane 8 compared with lane 3; lane 10 and 11 compared with lane 4] and D). Coexpression of DCTD196-AAAAAA also led to a decrease of DHBV CCC DNA, though the extent of the decrease was not as great as that achieved with the other DCTD fusion proteins (Fig. 5C [lane 9 compared with lane 4] and D). These results indicate that HCTD and DCTD could inhibit the CCC DNA production from RC DNA. This effect of the CTD fusion proteins on CCC DNA was independent of their effects on DHBc phosphorylation or core DNA synthesis, because HCTD141-AAAAAAA and most DCTD variants didn't change the phosphorylation of DHBc or core DNA synthesis (Fig. 2 and 4) but did suppress CCC DNA formation (Fig. 5).
Relative efficiency of CCC DNA formation was decreased when RC DNA was increased.
As mentioned above, when DHBV core DNA was increased, by increasing the amount of DHBV DNA transfected, the ratio of CCC DNA to core RC DNA was decreased, indicating a decrease in the relative efficiency of CCC DNA formation from RC DNA. This was clearly shown in Fig. 6, and the same result could also be seen by comparing the CCC DNA levels shown in Fig. 5 to core RC DNA levels shown in Fig. 4 for the GST control lanes (Fig. 4A and 5A [lanes 1 to 5] and 4E and 5C [lanes 1 to 7]). This result indicated that the CCC DNA formation reached saturation; when core RC DNA was increased to a certain level, CCC DNA formation could not be increased proportionally. Absolute quantification showed that the range of the average copy number per cell of RC DNA was from 1 to 282 when the amount of pCMV-DHBV-Env– transfected was increased, while the average copy number per cell of CCC DNA was estimated to be from 2 to 443 (Fig. 6C and F). Before the total RC DNA synthesized (estimated by adding the core RC DNA and CCC DNA that was derived from the core RC DNA) reached ca. 60 copies per cell, the copy number of CCC DNA per cell increased proportionally with the increase in core RC DNA (Fig. 6A to C). However, once the total RC DNA reached above ca. 70 copies per cell, the increase in CCC DNA no longer kept pace with the RC DNA increase (Fig. 6D to F). Nevertheless, it was notable that HEK293 cells were able to make over 400 copies of CCC DNA per cell (Fig. 6F), which was as efficient as what was reported in primary duck hepatocytes (49).
HCTD and DCTD interactions with CDK2 and importin α.
To determine whether interactions with specific host factors might be involved in the effects of HCTD and DCTD on DHBc phosphorylation, core DNA synthesis, and CCC DNA formation, we performed GST pulldown experiments using the same HCTD/DCTD fusion proteins and HEK293 cells that were used for the functional analyses described above and analyzed the host proteins that might be associated with the CTD fusion proteins by SDS-PAGE and Western blotting. As shown in Fig. 7A, two host proteins, CDK2 and importin α, were copurified with GST-HCTD/DCTD fusion proteins. To correct for nonspecific binding of CDK2 or importin α to GST alone or the affinity resin and the variation in the expression levels of the fusion proteins, we estimated the binding capabilities of GST-HCTD/DCTD to CDK2 or importin α by normalizing the level of CDK2 or importin α to the amount of GST or GST-HCTD/DCTD in the pulldown. HCTD141, HCTD141-ATASASS, HCTD141-ETESESS, and HCTD141-EEEEEEE had higher binding capacities to CDK2 than HCTD141-AAAAAAA and all the DCTD fusion proteins (Fig. 7B). In contrast, none of the HCTD variants, except HCTD141-ATASASS, showed specific binding to importin α, but all DCTD fusion proteins could bind importin α specifically (Fig. 7C). These results suggested that HCTD and DCTD variants could interact with the host CDK2 and importin α, which was influenced by the sequence (HCTD versus DCTD) and the phosphorylation state of CTD (HCTD141-ATASASS versus all the other HCTD variants).
FIG 7.
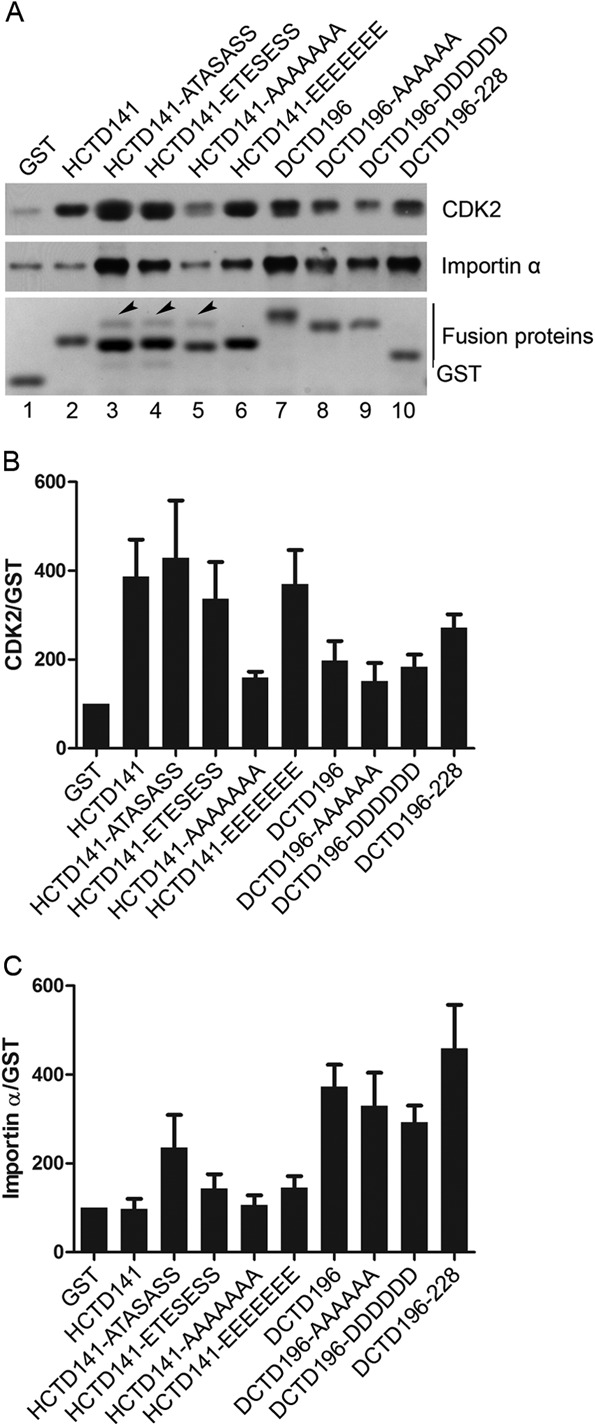
Interactions between CDK2 or importin α and HCTD or DCTD detected by GST pulldown assay. HEK293 cells in 100-mm dishes were transfected with 20 μg of the indicated GST-HCTD or GST-DCTD fusion constructs. Three days posttransfection, cells were harvested and GST pulldown assay was performed. (A) Copurified proteins were resolved by SDS-PAGE and detected by either Coomassie blue staining (bottom) or Western blotting using the CDK2-specific (top) or importin α-specific (middle) antibody. Arrowheads indicate the unidentified protein that was copurified with the GST-HCTD fusion proteins. (B and C) The relative levels of CDK2 and importin α associated with the HCTD or DCTD variants were normalized to those of the GST fusion proteins in the pulldown. The ratios of CDK2 or importin α to the GST fusion proteins were compared to that of the control (GST), set to 100. Data are presented as means and SEM from three independent experiments.
DISCUSSION
In this study, we analyzed the effects of the hepadnavirus core protein CTD and the CTD-host interactions on multiple steps in viral replication by coexpression of GST-CTD fusion proteins in trans in DHBV replicating cells. Our results showed that WT HCTD and all of its phosphomimetic and/or nonphosphomimetic mutants, except HCTD141-AAAAAAA, changed the phosphorylation state of DHBc dramatically and led to a profound reduction of viral core DNA and, in particular, the plus-strand DNA. Intriguingly, WT HCTD and all mutant HCTDs tested, including HCTD141-AAAAAAA, also strongly decreased the level of DHBV CCC DNA. In contrast, the WT and mutant DCTDs had a much smaller effect on DHBc phosphorylation or core DNA. Nevertheless, the DCTDs, like the HCTDs, also significantly decreased DHBV CCC DNA. GST pulldown assays revealed that the isolated HCTD and DCTD could interact with the cellular CDK2 and importin α, with different binding capacities, which could, at least partially, explain their inhibitory effects on DHBc phosphorylation/core DNA synthesis and CCC DNA formation, respectively.
One HCTD mutant, HCTD141-AAAAAAA, mimicking the nonphosphorylated CTD, and all the DCTDs tested had no or only a slight effect on DHBc phosphorylation and also showed low CDK2 binding capabilities. In contrast, the WT and all the other HCTD variants reduced DHBc phosphorylation significantly and also showed strong CDK2 binding. Therefore, there was a strong correlation between CDK2 binding and inhibition of DHBc phosphorylation among all the HCTD and DCTD variants tested, suggesting that CDK2 binding by the isolated CTD may mediate its inhibitory effect on DHBc phosphorylation via competition with the CTD in the full-length DHBc for the host kinase (Fig. 8). These results also support our previous findings indicating that CDK2 is a major cellular kinase responsible for DHBc phosphorylation in vivo (26).
FIG 8.

Model for HCTD and DCTD effects on DHBc phosphorylation, core DNA synthesis, and CCC DNA formation through interactions with different host factors. Shown schematically is a simplified diagram of the DHBV life cycle, with its major steps highlighted. The interactions between the HCTD and DCTD variants with the host CDK2, importin α, a putative cellular phosphatase (PPase), or unknown factors (indicated by the question marks) are proposed to sequester these factors so that they are unavailable for interactions with the CTD on the full-length core protein, thus inhibiting viral replication at the various steps. The gray letters in parentheses denote that those fusion proteins fail to interact with the relevant host factors. The capsid is depicted as hexagons, and capsid phosphorylation is denoted by the letter “P” inside the circles. The dashed line of the mature NCs (i.e., containing RC DNA) denotes the fact that they are less stable than the immature NCs (i.e., containing pgRNA or SS DNA). RC, relaxed circular DNA; SS, single-stranded DNA; CCC, covalently closed circular DNA. See the text for details.
It remains to be understood why HCTD141-AAAAAAA had a weaker CDK2 binding capacity than all the other HCTD variants. It is possible that the S-to-A substitutions removed the CTD phosphorylation sites, which were required for CDK2 binding to CTD. However, the fact that HCTD141-EEEEEEE, in which the phosphorylation sites were also removed, showed CDK2 binding capacity similar to that of the WT HCTD and the other HCTD mutants seems to rule out this possibility. Another possibility is that the S-to-A substitutions at all seven (potential) phosphorylation sites in CTD led to a change in its net charge, which interfered with CDK2 binding. The fact that HCTD141-ATASASS, which only had S-to-A substitutions in the three SP motifs, showed stronger CDK2 binding than HCTD141-AAAAAAA suggested that phosphorylation (or negative charges) at the four non-SP sites in CTD could be important for CDK2 binding even though CDK2, as a classical proline-directed kinase, preferentially phosphorylates the SP motifs as the substrates (26). The weaker CDK2 binding by DCTD compared to HCTD is another intriguing observation that warrants further investigation. These results suggest that HBV and DHBV may interact with their respective host kinases differently. It also remains possible that DCTD may have evolved to interact with the homologous duck CDK2 better than the heterologous human CDK2 tested here, although DHBc is phosphorylated and DHBV replicates well in HEK293 cells.
None of the HCTD or DCTD variants showed any significant effect on DHBV RNA packaging. This is consistent with the previous suggestion, obtained through mutagenesis of the CTD phosphorylation sites in the context of the full-length DHBc, that CTD phosphorylation does not play a major role in DHBV pgRNA packaging (18, 20–22). On the other hand, previous mutagenesis studies in the context of the full-length DHBc indicated that CTD phosphorylation in DHBV is required for the synthesis of the minus-strand DNA, while its dephosphorylation facilitates the maturation of the plus-strand DNA and the stabilization of the mature NCs (16, 21). Here, we have shown that all the HCTD variants that decreased DHBc phosphorylation also decreased DHBV core DNA, whereas the one HCTD variant and all the DCTD variants that failed to inhibit DHBc phosphorylation also couldn't reduce DHBV core DNA. This nearly perfect correlation between the effects on DHBc phosphorylation and DHBV core DNA synthesis among all the HCTD and DCTD variants tested here thus confirmed that DHBc phosphorylation is indeed required for core DNA synthesis. Our results provide further evidence that the host CDK2, by binding and phosphorylating CTD, is an important, bona fide host factor critical for viral replication. It is not yet clear why the DCTD did not efficiently inhibit DHBc phosphorylation. One possibility is that the affinity of the isolated DCTD for human CDK2 is not as high as that of the same CTD in the context of the full-length DHBc.
The strong effect of the HCTD variants on DHBV plus-strand DNA synthesis suggested they might also have sequestered the putative cellular phosphatase(s) that mediates CTD dephosphorylation thought to be critical for plus-strand elongation and stabilization of mature NCs (Fig. 8) (21). Although the net effect of these HCTD variants on DHBc phosphorylation was a decrease in phosphorylation, it remains possible that the dynamics of CTD phosphorylation and dephosphorylation, not just the net outcome of these dynamic events, are important for NC maturation and were disturbed by the HCTD variants expressed in trans in our experiments. Alternatively, these results could suggest that plus-strand DNA synthesis and NC maturation requires additional host factors, unrelated to CTD (de)phosphorylation, that were effectively sequestered by the HCTD variants. Interestingly, whatever putative phosphatase(s) or other host factors important for plus-strand DNA synthesis may be, they seemed to share the CTD interaction properties with CDK2, in that they could be sequestered effectively by all HCTD variants tested, except HCTD141-AAAAAAA, but not by any of the DCTD variants.
In contrast to the effects on DHBc phosphorylation and core DNA synthesis, which varied depending on the HCTD state of phosphorylation and between the HCTD and DCTD fusion proteins, all HCTD and DCTD variants expressed in trans strongly decreased DHBV CCC DNA, independent of their effects on DHBc phosphorylation or core DNA synthesis. As delivery of RC DNA in mature NCs to the nucleus is an important step during the conversion of RC DNA to CCC DNA, and CTD is thought to play an important role in directing the nuclear import of NCs, the HCTDs and DCTDs likely suppressed CCC DNA synthesis by blocking the nuclear import of NCs via competition with the CTD on the mature NCs for key factors in the nuclear import pathway. Indeed, we could show that the DCTDs could interact with importin α (Fig. 8). The observation that the DCTD linker region alone was able to bind importin α and inhibit CCC DNA formation as efficiently as the full CTD containing also the downstream phosphor domain suggests that this region, rather than the phosphor domain, harbors the binding site for importin α. Interestingly, we have previously shown that this same region in DCTD also harbors the binding site for two other host proteins (37), suggesting that this linker region is a focal point for host interactions. One HCTD variant, HCTD141-ATASASS, which showed a stronger inhibitory effect on CCC DNA than all the other HCTD variants, also showed increased importin α binding compared to that of all other HCTD variants, further implicating this nuclear import factor in CCC DNA formation. However, the fact that the other HCTD variants that could not bind to importin α still decreased CCC DNA suggests that other host factors involved in CCC DNA formation might also be targeted by HCTDs (Fig. 8).
The differential binding of importin α by DCTD versus HCTD implies that DHBV and HBV may employ different pathways to mediate nuclear import of RC DNA for CCC DNA formation. This is consistent with the observation that DHBV CCC DNA formation is more efficient than HBV, at least in commonly used cell cultures (10, 51). Our observation that CCC DNA formation could not be increased further beyond certain levels, even when core RC DNA continued to increase, suggests that a factor(s) in at least one step of CCC DNA formation from RC DNA was limiting and could be saturated.
The present study illustrates the role of CTD and the CTD-host interactions in the multiple steps of hepadnaviruses replication and the role of the dynamic CTD phosphorylation state on host interaction and viral replication. The experimental approach to effecting changes in protein phosphorylation state or interactions by overexpression in trans of part of the target protein (analogous to dominant negative inhibition), without mutagenesis of the target protein sequence itself, should also be broadly applicable to test vigorously conclusions generated from mutagenesis studies while at the same time providing a means to identify the putative factors involved. The fact that we were able to effect strong changes in DHBc phosphorylation, core DNA synthesis, and CCC DNA formation, all in the absence of any overt cytotoxicity, contrasts sharply with the inability to achieve similar effects by targeting the host factors involved with chemical inhibitors (26, 37). Other than the obvious issue of off-target effects of chemical inhibitors, these results may also suggest that the overexpressed CTDs might have preferentially sequestered a subpopulation of the relevant host factors (CDK2, importin, etc.) that are hijacked by the virus during its replication while leaving the rest of these factors largely unaffected to carry out their normal cellular functions. Thus, our results further suggest the exciting possibility that CTD, or CTD mimics, could have therapeutic potential by inhibiting multiple steps of viral replication, especially CCC DNA synthesis, which cannot be targeted directly by current antiviral therapy.
ACKNOWLEDGMENTS
The excellent technical assistance of Laurie Luckenbaugh is gratefully acknowledged. We thank William Mason for the anti-DHBV core antibody.
This work was supported by Public Health Service grant R01 AI074982 (to J.H.) from the National Institutes of Health and a scholarship from the China Scholarship Council (to K.L.).
REFERENCES
- 1.Seeger C, Zoulim F, Mason WS. 2007. Hepadnaviruses, p 2977–3030 InKnipe DM, Howley PM (ed), Fields virology. Lippincott, Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Wynne SA, Crowther RA, Leslie AG. 1999. The crystal structure of the human hepatitis B virus capsid. Mol Cell 3:771–780. doi: 10.1016/S1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 3.Conway JF, Cheng N, Zlotnick A, Wingfield PT, Stahl SJ, Steven AC. 1997. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 386:91–94. doi: 10.1038/386091a0. [DOI] [PubMed] [Google Scholar]
- 4.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. doi: 10.1016/0092-8674(82)90157-X. [DOI] [PubMed] [Google Scholar]
- 5.Hirsch RC, Lavine JE, Chang LJ, Varmus HE, Ganem D. 1990. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as well as for reverse transcription. Nature 344:552–555. doi: 10.1038/344552a0. [DOI] [PubMed] [Google Scholar]
- 6.Bartenschlager R, Schaller H. 1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J 11:3413–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones SA, Hu J. 2013. Hepatitis B virus reverse transcriptase: diverse functions as classical and emerging targets for antiviral intervention. Emerg Microbes Infect 2:e56. doi: 10.1038/emi.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tuttleman JS, Pourcel C, Summers J. 1986. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47:451–460. doi: 10.1016/0092-8674(86)90602-1. [DOI] [PubMed] [Google Scholar]
- 9.Wu TT, Coates L, Aldrich CE, Summers J, Mason WS. 1990. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 175:255–261. doi: 10.1016/0042-6822(90)90206-7. [DOI] [PubMed] [Google Scholar]
- 10.Gao W, Hu J. 2007. Formation of hepatitis B virus covalently closed circular DNA: removal of genome-linked protein. J Virol 81:6164–6174. doi: 10.1128/JVI.02721-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu M, Summers J. 1991. A domain of the hepadnavirus capsid protein is specifically required for DNA maturation and virus assembly. J Virol 65:2511–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nassal M. 1992. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J Virol 66:4107–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlicht HJ, Bartenschlager R, Schaller H. 1989. The duck hepatitis B virus core protein contains a highly phosphorylated C terminus that is essential for replication but not for RNA packaging. J Virol 63:2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu M, Summers J. 1994. Phosphorylation of the duck hepatitis B virus capsid protein associated with conformational changes in the C terminus. J Virol 68:2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao W, Ou JH. 1995. Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J Virol 69:1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perlman DH, Berg EA, O'Connor PB, Costello CE, Hu J. 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci U S A 102:9020–9025. doi: 10.1073/pnas.0502138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung J, Hwang SG, Chwae YJ, Park S, Shin HJ, Kim K. 2014. Phosphoacceptors threonine 162 and serines 170 and 178 within the carboxyl-terminal RRRS/T motif of the hepatitis B virus core protein make multiple contributions to hepatitis B virus replication. J Virol 88:8754–8767. doi: 10.1128/JVI.01343-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan YT, Li J, Liao W, Ou J. 1999. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259:342–348. doi: 10.1006/viro.1999.9798. [DOI] [PubMed] [Google Scholar]
- 19.Lewellyn EB, Loeb DD. 2011. Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PLoS One 6:e17202. doi: 10.1371/journal.pone.0017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gazina EV, Fielding JE, Lin B, Anderson DA. 2000. Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J Virol 74:4721–4728. doi: 10.1128/JVI.74.10.4721-4728.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basagoudanavar SH, Perlman DH, Hu J. 2007. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J Virol 81:1641–1649. doi: 10.1128/JVI.01671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu M, Summers J. 1994. Multiple functions of capsid protein phosphorylation in duck hepatitis B virus replication. J Virol 68:4341–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kann M, Gerlich WH. 1994. Effect of core protein phosphorylation by protein kinase C on encapsidation of RNA within core particles of hepatitis B virus. J Virol 68:7993–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kann M, Sodeik B, Vlachou A, Gerlich WH, Helenius A. 1999. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J Cell Biol 145:45–55. doi: 10.1083/jcb.145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daub H, Blencke S, Habenberger P, Kurtenbach A, Dennenmoser J, Wissing J, Ullrich A, Cotten M. 2002. Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J Virol 76:8124–8137. doi: 10.1128/JVI.76.16.8124-8137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ludgate L, Ning X, Nguyen DH, Adams C, Mentzer L, Hu J. 2012. Cyclin-dependent kinase 2 phosphorylates S/T-P sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J Virol 86:12237–12250. doi: 10.1128/JVI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albin C, Robinson W. 1980. Protein kinase activity in hepatitis B virus. J Virol 34:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui X, Ludgate L, Ning X, Hu J. 2013. Maturation-associated destabilization of hepatitis B virus nucleocapsid. J Virol 87:11494–11503. doi: 10.1128/JVI.01912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rabe B, Vlachou A, Pante N, Helenius A, Kann M. 2003. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci U S A 100:9849–9854. doi: 10.1073/pnas.1730940100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitz A, Schwarz A, Foss M, Zhou L, Rabe B, Hoellenriegel J, Stoeber M, Pante N, Kann M. 2010. Nucleoporin 153 arrests the nuclear import of hepatitis B virus capsids in the nuclear basket. PLoS Pathog 6:e1000741. doi: 10.1371/journal.ppat.1000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eckhardt SG, Milich DR, McLachlan A. 1991. Hepatitis B virus core antigen has two nuclear localization sequences in the arginine-rich carboxyl terminus. J Virol 65:575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh CT, Chu CM, Liaw YF. 1995. A single serine mutation on the nuclear localization signal of hepatitis B virus core protein abolishes the inhibition of nuclear transport by surface proteins. Biochem Biophys Res Commun 213:1068–1074. doi: 10.1006/bbrc.1995.2236. [DOI] [PubMed] [Google Scholar]
- 34.Yeh CT, Liaw YF, Ou JH. 1990. The arginine-rich domain of hepatitis B virus precore and core proteins contains a signal for nuclear transport. J Virol 64:6141–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li HC, Huang EY, Su PY, Wu SY, Yang CC, Lin YS, Chang WC, Shih C. 2010. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog 6:e1001162. doi: 10.1371/journal.ppat.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mabit H, Breiner KM, Knaust A, Zachmann-Brand B, Schaller H. 2001. Signals for bidirectional nucleocytoplasmic transport in the duck hepatitis B virus capsid protein. J Virol 75:1968–1977. doi: 10.1128/JVI.75.4.1968-1977.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ludgate L, Adams C, Hu J. 2011. Phosphorylation state-dependent interactions of hepadnavirus core protein with host factors. PLoS One 6:e29566. doi: 10.1371/journal.pone.0029566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Summers J, Smith PM, Horwich AL. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J Virol 64:2819–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Grammatikakis N, Hu J. 2002. Role of p50/CDC37 in hepadnavirus assembly and replication. J Biol Chem 277:24361–24367. doi: 10.1074/jbc.M202198200. [DOI] [PubMed] [Google Scholar]
- 40.Jilbert AR, Wu TT, England JM, Hall PM, Carp NZ, O'Connell AP, Mason WS. 1992. Rapid resolution of duck hepatitis B virus infections occurs after massive hepatocellular involvement. J Virol 66:1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen DH, Gummuluru S, Hu J. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J Virol 81:4465–4472. doi: 10.1128/JVI.02510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 43.Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J Virol 77:1885–1893. doi: 10.1128/JVI.77.3.1885-1893.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78:13122–13131. doi: 10.1128/JVI.78.23.13122-13131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perlman D, Hu J. 2003. Duck hepatitis B virus virion secretion requires a double-stranded DNA genome. J Virol 77:2287–2294. doi: 10.1128/JVI.77.3.2287-2294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, Hu J. 2011. Secretion of genome-free hepatitis B virus—single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog 7:e1002255. doi: 10.1371/journal.ppat.1002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scaglioni PP, Melegari M, Wands JR. 1997. Posttranscriptional regulation of hepatitis B virus replication by the precore protein. J Virol 71:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lenhoff RJ, Summers J. 1994. Construction of avian hepadnavirus variants with enhanced replication and cytopathicity in primary hepatocytes. J Virol 68:5706–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Summers J, Smith PM, Huang MJ, Yu MS. 1991. Morphogenetic and regulatory effects of mutations in the envelope proteins of an avian hepadnavirus. J Virol 65:1310–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pugh J, Zweidler A, Summers J. 1989. Characterization of the major duck hepatitis B virus core particle protein. J Virol 63:1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kock J, Rosler C, Zhang JJ, Blum HE, Nassal M, Thoma C. 2010. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog 6:e1001082. doi: 10.1371/journal.ppat.1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]