

Abstract
One of the neuropathological hallmarks of Alzheimer’s disease (AD) is the accumulation of beta-amyloid peptides (Aβ) in senile plaques. Aβ-induced oxidative stress is believed to be responsible for degeneration and apoptosis of neurons and consequent cognitive and memory deficits. Here, we investigated the possible neuroprotective effect of the heat shock protein 90 (Hsp90) inhibitor geldanamycin (GA) against amyloid pathogenesis in adult male Wistar rats. GA or vehicle was injected into the lateral cerebral ventricles of rats 24 h before injection of Aβ (1–42) in CA1 area of hippocampus. The learning and memory of the rats were assessed 7 days after injection of Aβ using passive avoidance (PA) task. As potential contributing factors in Aβ-induced memory decline, we evaluated apoptotic markers and also used terminal-transferase UTP nick end labeling (TUNEL) technique to detect apoptosis in the hippocampus of Aβ-injected rats. Our behavioral data suggest that GA pretreatment can significantly suppress memory deficits in Aβ-injected rats. There was also not only a marked increase in Hsp70 level but also upregulated 70 kDa ribosomal protein S6 kinase (p70S6K) in the hippocampus of GA-treated groups with a reduction in apoptotic factors including caspase-3, poly (ADP-ribose) polymerase, Bax/Bcl-2 ratio, and TUNEL-positive cells as well. Thus, we conclude that GA exerts its protective effects against Aβ (1–42) toxicity and memory deficits, at least in part, by upregulating of Hsp70 and P70S6K.
Keywords: Alzheimer’s disease, Hsp90, Geldanamycin, p70S6K, Learning, Memory
Introduction
Reflecting an aging population, Alzheimer's disease (AD) is among the first ten leading causes of death and the first most prevalent form of dementia occurring after age 65 (Reitz et al. 2011). Insoluble fibrillar plaques of amyloid β-proteins (Aβ) and neurofibrillary deposits of hyperphosphorylated tau proteins accumulate over the course of decades in the brains of patients with AD (Querfurth and LaFerla 2010). During the natural course of AD, an early degeneration of the internal temporal regions, notably of the hippocampus, is accompanied with declining memory and inhibition of hippocampal long-term potentiation (LTP) (Chen et al. 2000). Various underlying mechanisms have been discussed for the pathophysiology of memory and cognition loss in AD, among which neuronal apoptosis and synapse loss in hippocampus with associated atrophy are the common hallmarks (Mormino et al. 2009; Reixet et al. 2007). With regard to separate modes of Aβ and hyperphosphorylated tau-mediated toxicity, a fundamental tenet is Aβ-induced oxidative stress that results in the activation of apoptotic signaling and leads to neuronal death (Cai et al. 2011).
Geldanamycin (GA), a naturally occurring benzoquinone ansamycin, specifically binds to and inhibits the molecular chaperone heat shock protein 90 (Hsp90) that leads to activation of heat shock factor 1 (HSF-1) and, subsequently, induction of expression of stress proteins (Zou et al. 1998; Lu et al. 2002). A common feature of neurodegenerative disease is disturbance in protein turnover and a resulting intracellular accumulation of misfolded proteins, which underscores the major role of molecular chaperones and enhancement of protein quality control as a therapeutic approach (Selkoe 2004). Several experiments using administration of GA in vitro have indicated its neuroprotective activity via preventing α-synuclein aggregation and neurotoxicity and protecting dopaminergic neuron death (McLean et al. 2004; Shen et al. 2005). In the terms of AD studies, GA has been shown to promote the binding of tau protein to microtubules, leading to a significant reduction in tau aggregation (Dou et al. 2003), although its action on Aβ-mediated apoptosis and oxidative stress has not been yet studied.
Hsps are ubiquitous and highly conserved proteins, mainly responsible for the proper folding of nascent polypeptides and providing their quality control until degradation (Wickner et al. 1999). Expression of Hsps is tightly regulated by HSF-1, which closely interacts with and is inhibited by Hsp90. Upon dissociation of Hsp90, expressions of Hsp70 and Hsp40, as well as other chaperones, are induced. The most studied function of Hsps is the promotion of the formation of less toxic protein aggregates and/or by targeting toxic, misfolded proteins for degradation through the ubiquitin-proteasome system. In this manner, Hsp70 protein plays a role in suppressing apoptotic cascades by directly mediating the conformation of molecules involved in signal transduction, beyond chaperone activity in protein folding and stabilization (Lu et al. 2002; Sabirzhanov et al. 2012; Gallo 2006). Conversely, Hsp90 has been suggested to contribute to upregulation of proapoptotic factors (Solier et al. 2012). Also, Hsp70 acts as a sensor of cellular redox changes and is involved in occupying the pocket of the chaperone protein (Miyata et al. 2012). Besides the Hsps, 70 kDa ribosomal protein S6 kinase (p70S6K), a key factor in mammalian target of rapamycin (mTOR) signaling pathway, is a translational regulator that has been connected to GA following Hsp90 inactivation (Ohji et al. 2006). Levels of p70S6K in the cortex correlate with Aβ toxicity and neuronal apoptosis (Lafay-Chebassier et al. 2005). Deletion of p70S6K also has a significant impact on normal expression of behavioral learning and hippocampal synaptic plasticity (Antionet et al. 2008).
In this study, we investigated the protective impact of GA-mediated inhibition of Hsp90 and p70S6K modification on the Aβ-induced learning and long-term memory decline in adult male Wistar rats, using passive avoidance (PA) task. At the molecular level, we aimed to determine GA effect on the cellular level of apoptotic markers, Hsps and p70S6K, which are supposed to play significant roles in mediating the GA protection against Aβ toxicity.
Materials and methods
Materials
Antibodies directed against caspase-3, Bax, Bcl-2, Poly (ADP-ribose) polymerase (PARP), Hsp70, Hsp90, p70S6kinase (p70S6K), p-p70S6K, and β-actin were obtained from Cell Signaling Technology (Beverly, MA, USA). GA was purchased from Selleckchem (Houston, TX, USA). Terminal-transferase UTP nick end labeling (TUNEL) assay was done using In Situ Cell Death Detection kit (Roche, Switzerland). Electrochemiluminescence (ECL) kit was provided from Amersham Biosciences, USA. All the other reagents, unless otherwise stated, were from Sigma-Aldrich (St. Louis, MO, USA).
Animals
Ninety adult male Wistar rats weighing 220–260 g (12–14 weeks old) were procured from Pasteur Institute, Tehran, Iran. Animals were housed four per cage and maintained at the room temperature and under a 12 h light/dark cycle with access to water and food ad libitum. Rats adapted to laboratory conditions for more than a week prior to test. All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 80-23, revised 1996) and were approved by the research and ethics committee of the Shahid Beheshti University of Medical Sciences.
Experimental design
The rats were divided into six groups: Control group, which only received carrier [DMSO (3 μl/rat) in lateral ventricle and PBS (3 μl/side) in CA1 regions bilaterally]; Aβ-injected group, which received bilateral intra-CA1 injection of Aβ [30 ng/3 μl phosphate buffer saline (PBS) per side], without receiving any treatment; two Hsp90 inhibitor groups, which received intracerebroventricular (i.c.v.) infusion of GA [either (1 μg/kg)/3 μl 2 % DMSO or (2 μg/kg)/3 μl 2 % DMSO] with PBS injection (3 μl/side) in CA1; and two treatment groups which received i.c.v. administration of GA [either (1 μg/kg)/3 μl 2 % DMSO or (2 μg/kg)/3 μl 2 % DMSO], 24 h prior to intra-hippocampal Aβ (30 ng/3 μl PBS per side) injection. The aforementioned groups were studied by two experimental protocols: behavioral experiments (n = 8 rats/group) and molecular studies (n = 3 rats/group).
Surgery and microinjection
Rats were anesthetized with intraperitoneal (i.p) injection of ketamine hydrochloride (100 mg/kg) and xylazine (10 mg/kg), and were placed in a stereotaxic frame. Microinjections were performed by 30-gauge injector cannula that was connected to a short piece of polyethylene tube and a 10 μl Hamilton syringe. For i.c.v. administration of GA, 30-gauge injector cannula was aimed at the lateral ventricle (stereotaxic coordinates: incisor bar −3.3 mm, 0.5 mm posterior to the bregma, 1.5 mm lateral to the sagittal suture and 4 mm down from top of the skull), according to the atlas of Paxinos and Watson of the rat brain (Paxinos and Watson 2007). For intra-hippocampal administration, stainless steel guide cannulae (23 gauge), 6 mm in length, were implanted in the CA1 area of the hippocampus (stereotaxic coordinates: incisor bar −3.3 mm, 3.8 mm posterior to the bregma, ±3.2 mm lateral to the sagittal suture, and 2.8 mm down from top of the skull) bilaterally (Paxinos and Watson 2007). Cannulae were secured with jewelers’ screws and dental acrylic cement. Two stainless steel stylets were used to close the guide cannulae. Animals received a total volume of 3 μl (1 μg and 2 μg/kg) GA into the right ventricle and 3 μl Aβ (10 ng/μl) microinjection into both sides of the CA1 area of hippocampus. All microinjections were performed slowly over 2 min, and injection needles were left in their places for an additional 60 s, in order to prevent the possible leakage of the drugs and achieve a proper diffusion of the drug.
Preparation of Aβ1–42 and fibril formation
Aβ1–42 was dissolved in PBS then aliquoted and stored at −20 °C. The 200 ng/μl concentrations of Aβ prepared in PBS (0.1 M) and were incubated at 37 °C for 5 days to permit formation of fibrils. This solution was further diluted with PBS for injection (10 ng/μl).
Behavioral testing
Step-through passive avoidance apparatus
This test was carried out as described previously (Davoodi et al. 2011). The passive avoidance apparatus (modified shuttle box) consisted of a lighted chamber (20 cm × 40 cm × 20 cm) made from transparent plastic that was connected by a rectangular opaque guillotine door (8 cm × 8 cm) to the dark compartment of the same dimensions with black opaque walls and ceiling. The floor of both chambers was made of stainless steel rods (3 mm diameter) spaced 1 cm apart. The floor of the dark chamber could be electrified. The shock was delivered to the animal’s feet via a shock generator.
Training trial
Seven days after surgery, behavioral tests were started. All experimental groups were given two trials to habituate them to the apparatus. For these trials, rats were placed in the lighted compartment of the apparatus facing away from the door and 10 s later, the guillotine door was raised. Upon entering the dark compartment, the door was closed and 30 s later animals were taken from the dark compartment into their home cage. The habituation trial was repeated after 30 min and followed after the same interval by the first acquisition (learning) trial. During the adaptation trials, latency to enter the dark compartment was measured to ensure that all animals entered the dark compartment under 20 s. In the training session, each rat was placed in the light chamber and after the animal had spontaneously entered the dark compartment, the guillotine door was pulled down and a 50-Hz square wave, 1-mA constant current shock was applied for 1.5 s. The entrance latency to the dark compartment, step-through latency (STL), was recorded when the animal placed all four paws in the dark compartment. The animal was removed from the dark compartment about 30 s after receiving the shock and returned to its home cage, and the trial was repeated 2 min later. This procedure was repeated until the rat remained in the illuminated compartment for 120 consecutive seconds.
Retention test
The retention test was performed 24 h after PA acquisition trials. The rat was placed in the lighted chamber as in the training procedure, but no foot shock was administered if they entered the dark chamber. After 10 s, the guillotine door was raised and STL and time in dark compartment (TDC) were recorded up to 300 s. If the animal did not enter the dark compartment within 300 s, the retention test was terminated. Behavioral tests were performed at 8:00 to 11:00 a.m.
Western blot analysis
Seven days following Aβ injection, rats was decapitated and their brains were removed. Then, the hippocampi were isolated and homogenized in lysis buffer containing Tris (1 M, pH 7.4), sodium deoxycholate, NaCl, EDTA, sodium dodecyl sulfate (SDS), Triton X-100 (1 %), and complete protease inhibitor cocktail. The supernatant of the extract was collected and the protein concentrations were determined according to Bradford’s method (Bradford 1976). Equal amounts of total proteins (60 μg) were loaded and subjected to electrophoresis on a 12 % SDS-PAGE gel and transferred to polyvinylidene fluoride membranes. Blots were blocked by blocking solution [2 % non-fat dry milk in Tris-buffered saline containing Tween 80 (TBST, 0.1 % Tween 80, 50 mM Tris–HCl pH 7.4, 150 mM NaCl)] and incubated with primary antibodies at 4 °C overnight. After washing with TBST, membranes were incubated with a horseradish peroxidase-conjugated secondary antibody for 90 min at the room temperature. Immunoreactive proteins were detected by ECL kit. To normalize protein content, blots were stripped in stripping buffer containing 100 mM 2-mercaptoethanol, 2 % (w/v) SDS, 62.5 mM Tris–HCl (pH 6.7) and then probed with anti β-actin antibody. Densitometric analysis of the protein bands was done by Image J software.
Measurement of glutathione level
The concentration of glutathione (GSH) was determined in whole cell lysate using dithionitrobenzoic acid method at 412 nm (Ellman 1959).
Immunostaining
All animals were anesthetized and were transcardially perfused with PBS (pH 7.4), followed by 4 % paraformaldehyde (PFA) in 0.1 M pH 7.4 phosphate buffer (PB) (n = 4 rats/group). The brains were rapidly removed, post-fixed for 24 h in 4 % PFA–PB, and embedded with paraffin. Then, 5 μm coronal sections of hippocampal formation were prepared and TUNEL assay was performed using the In Situ Cell Death Detection Kit. Tissue sections were deparaffinized in xylene, rehydrated, and immersed in 3 % hydrogen peroxide, in order to block the endogenous peroxidase activity. After rinsing with PBS, sections were treated with proteinase K solution at 37 °C for 30 min. The sections were then incubated for 60 min at 37 °C with 50 μl of TUNEL reaction mixture, and then incubated for 30 min at 37 °C with 50 μl of converter-POD. Following washing with PBS, they were incubated again for 10 min at 15–25 °C with 50 μl of diaminobenzidine (DAB) substrate solution. Washing with PBS was done at the final stage. Counter staining was achieved with 0.5 % methyl green. Tissue was incubated in DNase solution for 10 min at 15–25 °C and sections were then dehydrated with their cover slip for analysis under light microscopy. To obtain the mean percentage of apoptotic cells to normal cells within the CA1 subfield of the hippocampus, the number of TUNEL-positive neurons was counted in three adjacent ×400 microscopic images.
Statistical analysis
All data were represented as mean ± SEM. (standard error of mean) and processed by the software Graph Pad Prism® 5.0. One-way analysis of variance (ANOVA) followed by Turkey’s post hoc test were used for the behavioral test and molecular studies. Statistical significance is set at P < 0.05 (*P < 0.05, **P < 0.01, and ***P < 0.001).
Results
GA inhibits Hsp90 and upregulates Hsp70 in Aβ-injected rats
Hsps are upregulated following exposure to a variety of stresses and insults. Hsps, mainly Hsp70, have cytoprotective effects (Kelly and Yenari 2002; Yenari 2002). To determine whether GA affects the levels of Hsps, we measured the levels of Hsp90 and Hsp70 by Western blot. As illustrated in Fig. 1b, Aβ injection significantly increased Hsp90 level compared with control group (P < 0.05). However, GA pretreatment by dose of 1 μg/kg 24 h prior to Aβ injection caused a significant decline in Hsp90 level (P < 0.05) compared to the Aβ-injected group. Also, a higher GA dose (2 μg/kg) reduced Hsp90 level even further (P < 0.01). As it is evident in Fig. 1d, Hsp70 increased 1.28-fold in Aβ-injected rats compared to control rats, while GA (1 μg and 2 μg/kg) pretreatment resulted in a dose-dependent increase of Hsp70 level 1.21- and 1.35-fold, respectively, in relation to Aβ-injected only group.
Fig. 1.
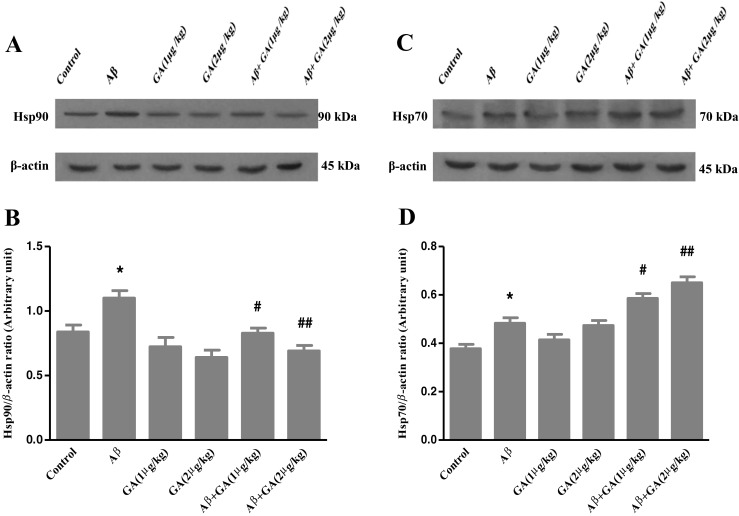
Western blot analyses to measure the effects of different concentrations of geldanamycin (GA), i.c.v. administration of 1 μg and 2 μg/kg, on Hsp90 and Hsp70 levels in the hippocampus of Aβ-injected rats. a, c Sixty micrograms of proteins were separated on SDS-PAGE, Western blotted, probed with specific primary antibodies and reprobed with anti β-actin antibody (Western blot was repeated two times for each animal, n = 3). b The densities of Hsp90 bands were measured and their ratios to β-actin bands were calculated. d The densities of Hsp70 bands were measured and their ratios to β-actin bands were calculated. Each point shows the mean ± SEM. *P < 0.05 versus control group. # P < 0.05; ## P < 0.01 versus the Aβ-injected group
Aβ-injected rats do not show deficit in learning of PA task
Animals in all experimental groups learned the passive avoidance task. As shown in Fig. 2, there was no significant difference in the number of trials to acquisition in rats received Aβ either alone or with administration of various doses of GA compared to the control group [F (5, 42) = 0.076; P = 0.995]. This finding suggests that learning was not impaired by the intra-CA1 injection of Aβ.
Fig. 2.

Effect of Aβ and geldanamycin (GA) on PA acquisition. There was no significant difference in number of trials to acquisition (learning trial) in rats received Aβ either alone or with administration of various doses of GA compared to the control group. Each point shows the mean ± SEM (n = 8)
GA improves Aβ-induced memory deficits
Figure 3 illustrates the results of the retention test performed 24 h after training. Statistical analysis revealed that there were significant differences in the STL [F (5, 42) = 18.61, P < 0.001] and TDC [F (5, 42) = 32.88, P < 0.001] between groups. Post-test analyses showed a significantly lower STL (P < 0.001) in Aβ-injected rats, while GA (1 μg and 2 μg/kg) pretreatment 24 h prior to Aβ injection caused a significant increase in STL (P < 0.001) compared to the Aβ administrated group (Fig. 3a). Also, TDC significantly increased in Aβ-injected rats (P < 0.001). However, GA pretreatment resulted in a marked decrease in TDC (P < 0.001) compared to the Aβ-injected group (Fig. 3b). Furthermore, there were no significant differences in PA retention between GA-only treatment and the control group. These findings indicate that GA suppresses memory deficits in Aβ-injected rats.
Fig. 3.

Effect of Aβ and geldanamycin (GA) on PA retention. Animals received intra-CA1 injection of Aβ (30 ng/3 μl PBS per side) or PBS (3 μl/side) as a vehicle, after pretreating by GA. Step-through latency (a) and time spent in dark compartment (b) during the retrieval test performed 24 h after PA acquisition. There was a significant decrease in STD and a marked increase in TDC in Aβ-injected animals compared to control group, whereas GA pretreatment (1 μg and 2 μg/kg) significantly prevented memory deficits in rats receiving Aβ. Each point shows the mean ± SEM (n = 8). ***P < 0.001 versus control group. ### P < 0.001 versus the Aβ-injected group
GA decreases the phosphorylation of p70S6K in Aβ-injected rats
We measured the level of phosphorylated form of p70S6K by Western blot. As shown in Fig. 4, phosphorylation of p70S6K decreased in the presence of Aβ (P < 0.05), while pretreatment with GA (1 μg/kg) caused a significant increase in phosphorylation of p70S6K compared to the Aβ-injected rats (P < 0.05). GA at 2 μg/kg dose further increased the phosphorylation of p70S6K (P < 0. 01).
Fig. 4.

Western blot analyses to measure the effects of different concentrations of geldanamycin (GA), i.c.v. administration of 1 μg and 2 μg/kg, on phospho-p70S6K level in the hippocampus of Aβ-injected rats. a Sixty micrograms of proteins were separated on SDS-PAGE, Western blotted, probed with anti p-p70S6K antibody and reprobed with anti p70S6K antibody (Western blot was repeated two times for each animal; n = 3). b The densities of p-p70S6K bands were measured and their ratios to p70S6K bands were calculated. Each point shows the mean ± SEM. *P < 0.05 versus control group. # P < 0.05; ## P < 0.01 versus the Aβ-injected group
GA restores GSH level in Aβ-injected rats
Aβ injection caused a significant decreased in GSH level (P < 0.01) compared with control group, while GA (1 μg/kg) pretreatment increased GSH level 1.34-fold compared with the Aβ group. By increasing the GA dose (2 μg/kg), GSH level further increased to 1.48-fold (Fig. 5).
Fig. 5.

The effect of geldanamycin (GA) at the dose of 1 μg and 2 μg/kg (i.c.v.) on changes in GSH level in the hippocampus of Aβ-injected rats. Each value represents the mean ± SEM (n = 3). **P < 0.01 significantly different from control group. # P < 0.05; ## P < 0.01 significantly different from Aβ-injected group
Aβ-mediated elevation of Bax/Bcl-2 ratio is modulated by GA
As shown in Fig. 6, Aβ injection significantly increased Bax level and caused a decrease in Bcl-2 level. The ratio of Bax/Bcl-2 increased by 1.95-fold in the Aβ-injected group compared to the control group. However, the Bax/Bcl-2 ratio in animals pretreated with 1 μg and 2 μg/kg of GA was reduced compared to the Aβ-injected rats to 1.33- and 1.65-fold, respectively.
Fig. 6.

Western blot analysis to measure the effects of different concentrations of geldanamycin (GA), i.c.v. administration of 1 μg and 2 μg/kg, on Bax and Bcl-2 levels in the hippocampus of Aβ-injected rats. a Sixty micrograms of proteins were separated on SDS-PAGE, Western blotted, probed with anti Bax and/or anti Bcl-2 antibodies and reprobed with anti β-actin antibody (Western blot was repeated two times for each animal; n = 3). b The densities of corresponding bands were measured and the ratio of Bax to Bcl-2 was evaluated. Each point shows the mean ± SEM. ***P < 0.001 different from the control group. # P < 0.05; ## P < 0.01 different from the Aβ-injected group
GA modulates caspase-3 and cleavage of PARP
Western blot showed that the cleavage of caspase-3 increased about 3.4-fold in Aβ-injected rats compared to control group, while pretreatment with GA at doses of 1 μg and 2 μg/kg caused a significant decrease of Aβ-induced caspase-3 cleavage by, respectively, 1.23- and 1.35-fold, compared with the Aβ-injected group (Fig. 7b). The cleavage of PARP significantly increased in Aβ-injected rats compared to the control group (P < 0.01). However, GA (1 μg and 2 μg/kg) treatment resulted in a marked dose-dependent decrease in cleaved PARP level compared with Aβ-injected rats (P < 0.05 and P < 0.01) (Fig. 7c).
Fig. 7.

Western blot analysis to measure the effects of different concentrations of geldanamycin (GA), i.c.v. administration of 1 μg and 2 μg/kg, on caspase-3 and PARP cleavage in the hippocampus of Aβ-injected rats. a Sixty micrograms of proteins were separated on SDS-PAGE, Western blotted, probed with specific primary antibodies and reprobed with anti β-actin antibody (Western blot was repeated two times for each animal; n = 3). b The densities of caspase-3 bands were measured and their ratios to β-actin bands were calculated. c The densities of PARP bands were measured and their ratios β-actin bands were calculated. Each point shows the mean ± SEM. **P < 0.01; ***P < 0.001 compared to control group. # P < 0.05; ## P < 0.01 compared to the Aβ-injected group
GA decreases TUNEL-positive cells induced by Aβ
As shown in Fig. 8, apoptotic cells identified by TUNEL staining displayed an increase in the hippocampus of Aβ-injected rats versus control groups (P < 0.001). In the GA pretreated groups, the number of TUNEL-positive cells significantly decreased compared to Aβ-injected group (P < 0.001), confirming the neuroprotective role of this compound against Aβ-induced apoptosis.
Fig. 8.

Hippocampal TUNEL-positive neurons, 7 days after stereotaxic injection of Aβ. a Sections indicate several TUNEL-positive neurons (arrows) were observed in Aβ-injected group, 1 week after Aβ injection, while in geldanamycin (GA) treated rats, the number of TUNEL-positive neurons has been reduced; light microscope (×400) was utilized for photography. b Represents the quantitative analysis of the data, n = 4. [**P < 0.01; ***P < 0.001 different from the vehicle group. ### P < 0.001 different from the Aβ-injected group]
Discussion
Although various genetic, biochemical, and pathological causes have been discussed, the most commonly believed cause and most frequently studied aspect of AD is the aggregation of Aβ (Karran et al. 2011). Our administration of intra-hippocampal Aβ and i.c.v. GA indicated GA-mediated improvement of memory in PA task, following Aβ injection. This was accompanied with upregulation of Hsp70 and recovery of p70S6K that consequently modulated apoptosis.
High levels of hippocampal Aβ have been associated with early memory decline and mild cognitive impairment (McLarnon et al. 2008). Since the first exogenous administration of Aβ to partially model pathology of AD, there has been a great improvement in methodology. Some of these improvements include shifting from i.c.v. to intraparenchymal Aβ administration and moving from short-term acting oligomers to long-term affecting fibril forms. As such, it has been observed that intracerebral Aβ administration exert short-term memory deficits and also has persistent/long-term synaptic effect on hippocampal network. Activation of oxidative and/or the inflammatory reactions, resembling the AD pathology, have been also shown in the mentioned models. However, the intracerebral Aβ approach has its own limitations, such as lack of generalized pattern and utilizing invasive procedure that have been partly resolved by introduction of transgenic models. It should be mentioned that genetically mutated models have shown a weak inflammatory state and the lack of overt neurodegeneration, which contradict human AD pathology (Stephan and Phillips 2005; Bales 2012). Our data indicated that 7 days following intra-hippocampal fibril Aβ injection, there was a significant memory decline that was assessed by PA task with no alteration in learning ability. PA predominantly evaluates long-term memory as a mainly hippocampal test (Akbari et al. 2008; Davoodi et al. 2011), so that a decrease in the STL indicates impairment of memory retrieval. GA is a small lipophilic molecule capable of penetrating the blood–brain barrier with a substantial toxicity that induces apoptosis in the high doses (Supko et al. 1995; Zagzag et al. 2003; Nimmanapalli et al. 2001). Therefore, we performed our experiments by relatively low doses which solely showed mild inductional effects. GA pretreatment, 24 h before Aβ injection, protected memory parameters up at control level. Consistent with our findings, Chen et al. (2014) have reported recently the modulation of Aβ-induced memory impairment by 17-allylamino-17-demethoxygeldanamycin (17-AAG), a GA derivative. Previously, GA and other Hsp90 inhibitors were shown to activate the proteasome pathway, which degrades intracellular misfolded Tau, but not Aβ, suggesting a means by which Hsp90 inhibition potentially relieves AD manifestations (Opattova et al. 2013).
Although the mechanism by which cognitive impairment develops is still elusive, a widely favored viewpoint implicates oxidative stress and its effects as the leading cause of Aβ neurotoxicity and memory decline (Cai et al. 2011). Evidence for activation of the oxidative stress pathway starts from CA3 sector in the hippocampus and leads to a progressive hippocampal apoptosis and atrophy (Cruz-Sánchez et al. 2010). The dual effect of Hsps in execution of Aβ-induced, oxidative stress-mediated apoptosis has been demonstrated (Abdul et al. 2006). Here, we observed that Aβ-depleted GSH, an indicator of oxidative stress, caused a mild increase in Hsp90 and Hsp70 levels. It is tempting to speculate that the mild increase of level of Hsps in Aβ-injected group may be due to previously described reactive oxygen species-induced dissociation and the activation of HSF-1 (Zhang et al. 2011). GA pretreatment increased Hsp70 more significantly along with modulation of Hsp90 hippocampal level. A well-known activity of GA is the inhibition of Hsp90 by binding to its N-terminal ATP-binding pocket, resulting in HSF-1 release and consequently induction of Hsps, such as Hsp70 (Zou et al. 1998; Lu et al. 2002). In vitro cell models have shown that Hsp70 overexpression attenuates neuronal apoptosis evoked by various stress stimuli such as oxidative stress or through inhibiting proapoptotic molecules like c-jun N-terminal kinase (Sabirzhanov et al. 2012; Meriin and Sherman 2005). This antiapoptotic impact, at least in part, has been attributed to antioxidative action of Hsp70, rather than its consensus protein quality control (Miyata et al. 2012). In support of this conclusion, it was reported that Hsp70 protects against oxidative-induced cellular injury through improving cell endogenous antioxidant stability (Afolayan et al. 2014). As another contributing factor, Solier et al. (2012) provided evidence for proapoptotic phosphorylation and activation of Hsp90 in an in vitro context. Moreover, altered kinase activity has been subjected to pathologic events in Aβ toxicity, which is tightly regulated by Hsp90 (Luo et al. 2008). Intriguingly, the protective role of Hsp90 against neuronal apoptosis also has been reported in a cellular model of Huntington disease (Lee et al. 2001). Our data suggest that Hsp70 upregulation following GA-mediated Hsp90 inhibition not only elevated intracellular antioxidant level, but also reduced significantly apoptotic markers. It is the hippocampal apoptosis that has been most often associated with progressive memory and cognitive decline.
P70S6K activity is known to correlate with enhanced protein synthesis, which has a close association with Hsp90. Zitzmann et al. (2013) showed that the lower concentration of Hsp90 inhibitors induced p70S6K signaling correlated with stimulation of compensatory pathways. Furthermore, p70S6K-deficient mice had distinct impacts on learning, memory, and hippocampal synaptic plasticity with a compromised hippocampal LTP (Antion et al. 2008). We found that hippocampal p70S6K level suppressed following Aβ injection that was prevented by GA pretreatment, concurrent with improvement in PA memory. In this manner, both in vitro and in vivo administration of Aβ by Lafay-Chebassier et al. (2005) resulted in significant decrease in p70S6K signaling. Although in another independent study of AD patients there was an aberrant activation of p70S6K, it was attributed to neurofibrillary pathology and tau phosphorylation, not Aβ (An et al. 2003). As a point of interest, a growing number of studies show that p70S6K survival signaling is a key antiapoptotic factor, by various pathways. From a molecular point of view, autophagy induction, decreased oxidative stress, and inactivation of proapoptotic molecules, such as Bcl-2-associated death promoter, are commonly described downstream of p70S6K signaling (Faghiri and Bazan 2010; Saiki et al. 2011; Harada et al. 2001).
Taken together, our study further demonstrates that GA administration preserves Aβ-mediated memory and cognitive impairment in rats through induction of Hsps and p70S6K, which subsequently results in oxidative stress reduction and restrained apoptosis (Fig. 9). 17-AAG, an analogue of GA, shows less hepatotoxicity in vivo, and also effectively activates HSF-1 by inhibiting Hsp90. Interestingly, i.p. administration of 17-AAG improved cognitive process and upregulated Hsp70 and Hsp27 expression. A possible therapeutic approach to the use of GA should lead to discovery and use of its less toxic derivatives, such as 17-AAG (Ortega et al. 2014). Although the mechanism through which Aβ toxicity generates progressive cognitive and memory decline is explored extensively, the precise molecular pathway to explain the whole Aβ toxicity manifestations remains an open question. GA, mainly its derivatives, provides a tool in which to identify the pathways as well as a potential direction of at least delaying the Aβ-induced pathology.
Fig. 9.

GA-inhibited Hsp90 protective pathways in Aβ-induced neuronal toxicity and long-term memory impairment. GA-induced Hsp90 inhibition leads to HSF-1 release that subsequently transactivates Hsps, such as Hsp70 and Hsp90. Upregulated Hsp70 have the potential to counter Aβ-induced oxidative stress by several antioxidative mechanisms that ultimately reduce Aβ-induce apoptosis. GA also activates p70s6k, a key factor in mTOR signaling pathway, which results in long-term memory improvement, disrupted by exogenous Aβ. P70s6k activation also reduces neuronal apoptosis by activation of autophagy, reduction of oxidative stress and the activation of antiapoptotic agents, such as Bcl-2
Acknowledgments
This work is part of the PhD student’s thesis of N. Zare at the Shahid Beheshti University of Medical Sciences.
Conflict of interest statement
The authors have no conflict of interest.
References
- Abdul HM, Calabrese V, Calvani M, Butterfield DA. Acetyl-L-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against amyloid-beta peptide 1–42-mediated oxidative stress and neurotoxicity: implications for Alzheimer's disease. J Neurosci Res. 2006;84:398–408. doi: 10.1002/jnr.20877. [DOI] [PubMed] [Google Scholar]
- Afolayan AJ, Teng RJ, Eis A, Rana U, Broniowska KA, Corbett JA, Pritchard K, Konduri GG. Inducible HSP70 regulates superoxide dismutase-2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. Am J Physiol Lung Cell Mol Physiol. 2014;306:L351–L360. doi: 10.1152/ajplung.00264.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbari E, Motamedi F, Naghdi N, Noorbakhshnia M. The effect of antagonization of orexin 1 receptors in CA1 and dentate gyrus regions on memory processing in passive avoidance task. Behav Brain Res. 2008;187:172–177. doi: 10.1016/j.bbr.2007.09.019. [DOI] [PubMed] [Google Scholar]
- An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Winblad B, Pei JJ. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol. 2003;163:591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antion MD, Merhav M, Hoeffer CA, Reis G, Kozma SC, Thomas G, Schuman EM, Rosenblum K, Klann E. Removal of S6K1 and S6K2 leads to divergent alterations in learning, memory, and synaptic plasticity. Learn Mem. 2008;15:29–38. doi: 10.1101/lm.661908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bales KR. The value and limitations of transgenic mouse models used in drug discovery for Alzheimer's disease: an update. Expert Opin Drug Discov. 2012;7:281–297. doi: 10.1517/17460441.2012.666234. [DOI] [PubMed] [Google Scholar]
- Cai Z, Zhao B, Ratka A. Oxidative stress and β-amyloid protein in Alzheimer’s disease. Neuromolecular Med. 2011;13:223–250. doi: 10.1007/s12017-011-8155-9. [DOI] [PubMed] [Google Scholar]
- Chen QS, Kagan BL, Hirakura Y. Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J Neurosci Res. 2000;60:65–72. doi: 10.1002/(SICI)1097-4547(20000401)60:1<65::AID-JNR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang B, Liu D, Li JJ, Xue Y, Sakata K, Liao FF. Hsp90 chaperone inhibitor 17-AAG attenuates Aβ-induced synaptic toxicity and memory impairment. J Neurosci. 2014;34:2464–2470. doi: 10.1523/JNEUROSCI.0151-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Sánchez FF, Girones X, Ortega A, Alameda F, Lafuente JV. Oxidative stress in Alzheimer's disease hippocampus: a topographical study. J Neurol Sci. 2010;299:163–167. doi: 10.1016/j.jns.2010.08.029. [DOI] [PubMed] [Google Scholar]
- Davoodi FG, Motamedi F, Akbari E, Ghanbarian E, Jila B. Effect of reversible inactivation of reuniens nucleus on memory processing in passive avoidance task. Behav Brain Res. 2011;221:1–6. doi: 10.1016/j.bbr.2011.02.020. [DOI] [PubMed] [Google Scholar]
- Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- Faghiri Z, Bazan NG. PI3K/Akt and mTOR/p70S6K pathways mediate neuroprotectin D1-induced retinal pigment epithelial cell survival during oxidative stress-induced apoptosis. Exp Eye Res. 2010;90:718–725. doi: 10.1016/j.exer.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo KA. Targeting HSP90 to halt neurodegeneration. Chem Biol. 2006;13:115–116. doi: 10.1016/j.chembiol.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci U S A. 2001;98:9666–9670. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- Kelly S, Yenari MA. Neuroprotection: heat shock proteins. Curr Med Res Opin. 2002;18:s55–s60. doi: 10.1185/030079902125000732. [DOI] [PubMed] [Google Scholar]
- Lafay-Chebassier C, Paccalin M, Page G, Barc-Pain S, Perault-Pochat MC, Gil R, Pradier L, Hugon J. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer's disease. J Neurochem. 2005;94:215–225. doi: 10.1111/j.1471-4159.2005.03187.x. [DOI] [PubMed] [Google Scholar]
- Lee MW, Park SC, Chae HS, Bach JH, Lee HJ, Lee SH, Kang YK, Kim KY, Lee WB, Kim SS. The protective role of HSP90 against 3-hydroxykynurenine-induced neuronal apoptosis. Biochem Biophys Res Commun. 2001;284:261–267. doi: 10.1006/bbrc.2001.4938. [DOI] [PubMed] [Google Scholar]
- Lu A, Ran R, Parmentier-Batteur S, Nee A, Sharp FR. Geldanamycin induces heat shock proteins in brain and protects against focal cerebral ischemia. J Neurochem. 2002;81:355–364. doi: 10.1046/j.1471-4159.2002.00835.x. [DOI] [PubMed] [Google Scholar]
- Luo W, Rodina A, Chiosis G. Heat shock protein 90: translation from cancer to Alzheimer's disease treatment? BMC Neuroscidoi. 2008;9(2):S7. doi: 10.1186/1471-2202-9-S2-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLarnon JG, Ryu JK. Relevance of abeta1-42 intrahippocampal injection as an animal model of inflamed Alzheimer's disease brain. Curr Alzheimer Res. 2008;5:475–480. doi: 10.2174/156720508785908874. [DOI] [PubMed] [Google Scholar]
- McLean PJ, Klucken J, Shin Y, Hyman BT. Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity in vitro. Biochem Biophys Res Commun. 2004;321:665–669. doi: 10.1016/j.bbrc.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Meriin AB, Sherman MY. Role of molecular chaperones in neurodegenerative disorders. Int J Hyperthermia. 2005;21:403–419. doi: 10.1080/02656730500041871. [DOI] [PubMed] [Google Scholar]
- Miyata Y, Rauch JN, Jinwal UK, Thompson AD, Srinivasan S, Dickey CA, Gestwicki JE. Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chem Bio. 2012;l19:1391–1399. doi: 10.1016/j.chembiol.2012.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, Koeppe RA, Mathis CA, Weiner MW, Jagust WJ. Episodic memory loss is related to hippocampal-mediated β-amyloid deposition in elderly subjects. Brain. 2009;132:1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmanapalli R, O’Bryan E, Bhalla K. Geldanamycin and its analogue 17-allylamino-17-demethoxygeldanamycin lowers Bcr-Abl levels and induces apoptosis and differentiation of Bcr-Abl-positive human leukemic blasts. Cancer Res. 2001;61:1799–1804. [PubMed] [Google Scholar]
- Ohji G, Hidayat S, Nakashima A, Tokunaga C, Oshiro N, Yoshino K, Yokono K, Kikkawa U, Yonezawa K. Suppression of the mTOR-raptor signaling pathway by the inhibitor of heat shock protein 90 geldanamycin. J Biochem. 2006;139:129–135. doi: 10.1093/jb/mvj008. [DOI] [PubMed] [Google Scholar]
- Opattova A, Filipcik P, Cente M, Novak M. Intracellular degradation of misfolded tau protein induced by geldanamycin is associated with activation of proteasome. J Alzheimers Dis. 2013;33:339–348. doi: 10.3233/JAD-2012-121072. [DOI] [PubMed] [Google Scholar]
- Ortega L, Calvillo M, Luna F, Pérez-Severiano F, Rubio-Osornio M, Guevara J, Limón ID. 17-AAG improves cognitive process and increases heat shock protein response in a model lesion with Aβ25-35. Neuropeptides. 2014;48:221–232. doi: 10.1016/j.npep.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson CR. The rat brain in stereotaxic coordinates. San Diego: Elsevier Academic Press; 2007. [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neuro. 2011;l7:137–152. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reix S, Mechawar N, Susin SA, Quirion R, Krantic S. Expression of cortical and hippocampal apoptosis-inducing factor (AIF) in aging and Alzheimer's disease. Neurobiol Aging. 2007;28:351–356. doi: 10.1016/j.neurobiolaging.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Sabirzhanov B, Stoica BA, Hanscom M, Piao CS, Faden AI. Over-expression of HSP70 attenuates caspase-dependent and caspase-independent pathways and inhibits neuronal apoptosis. J Neurochem. 2012;123:542–554. doi: 10.1111/j.1471-4159.2012.07927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki S, Sasazawa Y, Imamichi Y, Kawajiri S, Fujimaki T, Tanida I, Kobayashi H, Sato F, Sato S, Ishikawa K, Imoto M, Hattori N. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 2011;7:176–187. doi: 10.4161/auto.7.2.14074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Bio. 2004;l6:1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- Shen HY, He JC, Wang Y, Huang QY, Chen JF. Geldanamycin induces heat shock protein70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem. 2005;280:39962–39969. doi: 10.1074/jbc.M505524200. [DOI] [PubMed] [Google Scholar]
- Solier S, Kohn KW, Scroggins B, Xu W, Trepel J, Neckers L, Pommier Y. Heat shock protein 90α (HSP90α), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc Natl Acad Sci U S A. 2012;109:12866–12872. doi: 10.1073/pnas.1203617109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stéphan A, Phillips AG. A case for a non-transgenic animal model of Alzheimer's disease. Genes Brain Behav. 2005;4:157–172. doi: 10.1111/j.1601-183X.2004.00113.x. [DOI] [PubMed] [Google Scholar]
- Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmaco. 1995;l36:305–315. doi: 10.1007/BF00689048. [DOI] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: folding, refolding, and degrading proteins. Science. 1999;286:1888–1893. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- Yenari MA. Heat shock proteins and neuroprotection. Adv Exp Med Biol. 2002;513:281–299. doi: 10.1007/978-1-4615-0123-7_10. [DOI] [PubMed] [Google Scholar]
- Zagzag D, Nomura M, Friedlander DR, Blanco CY, Gagner JP, Nomura N, Newcomb EW. Geldanamycin inhibits migration of glioma cells in vitro: a potential role for hypoxia-inducible factor (HIF-1α) in glioma cell invasion. J Cell Physiol. 2003;196:394–402. doi: 10.1002/jcp.10306. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ahn YH, Benjamin IJ, Honda T, Hicks RJ, Calabrese V, Cole PA, Dinkova-Kostova AT. HSF1-dependent upregulation of Hsp70 by sulfhydryl-reactive inducers of the EAP1/NRF2/ARE pathway. Chem Biol. 2011;18:1355–1361. doi: 10.1016/j.chembiol.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitzmann K, Ailer G, Vlotides G, Spoettl G, Maurer J, Göke B, Beuschlein F, Auernhammer CJ. Potent antitumor activity of the novel HSP90 inhibitors AUY922 and HSP990 in neuroendocrine carcinoid cells. Int J Oncol. 2013;43:1824–1832. doi: 10.3892/ijo.2013.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/S0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]