

Abstract
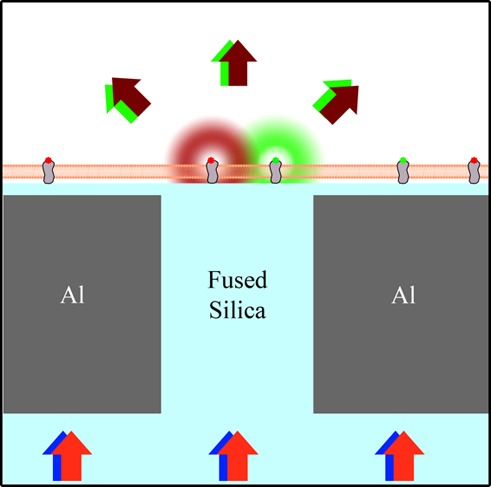
The organization and dynamics of plasma membrane components at the nanometer scale are essential for biological functions such as transmembrane signaling and endocytosis. Planarized nanoscale apertures in a metallic film are demonstrated as a means of confining the excitation light for multicolor fluorescence spectroscopy to a 55 ± 10 nm beam waist. This technique provides simultaneous two-color, subdiffraction-limited fluorescence correlation spectroscopy and fluorescence cross-correlation spectroscopy on planar membranes. The fabrication and implementation of this technique are demonstrated for both model membranes and live cells. Membrane-bound proteins were observed to cluster upon the addition of a multivalent cross-linker: On supported lipid bilayers, clusters of cholera toxin subunit B were formed upon cross-linking by an antibody specific for this protein; on living cells, immunoglobulin E bound to its receptor (FcεRI) on the plasma membranes of RBL mast cells was observed to form clusters upon exposure to a trivalent antigen. The formation of membrane clusters was quantified via fluorescence intensity vs time and changes in the temporal auto- and cross-correlations above a single nanoscale aperture. The illumination profile from a single aperture is analyzed experimentally and computationally with a rim-dominated illumination profile, yielding no change in the autocorrelation dwell time with changes in aperture diameter from 60 to 250 nm. This near-field fluorescence cross-correlation methodology provides access to nanoscale details of dynamic membrane interactions and motivates further development of near-field optical methods.
Keywords: near-field, membrane dynamics, fluorescence correlation spectroscopy (FCS), fluorescence cross-correlation spectroscopy (FCCS), nanofabrication, membrane cross-linking, subdiffraction-limited
Biological membranes are increasingly understood to be organized, dynamic, and functional at the nanometer scale. Complex interactions between membrane-bound proteins, carbohydrates, and lipids are of growing interest, as the mesoscale effects of their molecular-scale interactions have only recently become experimentally observable. In particular, the processes of antigen-induced receptor clustering and lipid-mediated transmembrane signaling are of fundamental immunological importance, but are dominated by interactions below the diffraction limit of light (∼200 nm). Thus, conventional optical attempts to observe membrane receptor clustering and lipid reorganization have been hampered by experimental resolution limits.
Two examples of dynamic, nanoscale membrane processes are the multivalent binding of cholera toxin subunit B (CTxB) to the ganglioside GM1 and the clustering of transmembrane immunoreceptors by multivalent antigens. Both of these phenomena involve ordered lipid domains composed of transient nanoscopic assemblies of proteins, glycolipids, and cholesterol.1 Further, CTxB and antigen binding have been shown to alter the membrane organization by inducing lipid phase separation in previously mixed model membrane systems2 and by affecting the local environment of key membrane proteins in living cells.3 CTxB is the membrane-binding component of cholera toxin and is commonly used as a marker for the ordered regions of the plasma membrane due to the order-preferring nature of GM1.2 On cells, the CTxB-GM1 complex undergoes rapid loss of mobility and internalization via mechanisms that are actively under investigation.4 CTxB is of key biological importance due to its frequent use as a marker for ordered lipid domains and its involvement in cholera infections. CTxB has previously been studied with subdiffraction-limited resolution on fixed and live cells.5 However, the transmembrane signaling caused by CTxB binding and the initiation of intracellular processes remains poorly defined, which reveals a critical lack of understanding of fundamental membrane biophysics.
Within the immune system, membrane receptor clustering by multivalent antigen binding is commonly the first stage in signal transduction and the consequent cellular response to allergen exposure. The signaling cascades induced by multivalent antigen binding to immunoglobulin E (IgE) associated with its immunoreceptor (FcεRI) cause granule exocytosis, which is primarily responsible for allergic responses mediated by mast cells and basophils that can lead to inflammation and anaphylaxis. Cross-linking of IgE-FcεRI causes its association with detergent-resistant, liquid-ordered membrane domains, and it is hypothesized to stabilize domains of order-preferring lipids that modulate the local concentration of membrane-bound kinases and phosphatases leading to signal propagation.6 The nanoscale organization and dynamics of IgE-FcεRI upon antigen binding have been studied with scanning electron microscopy (SEM),7−10 localization microscopy,10,11 and single-particle tracking (SPT).11−13 However, nanoscale heterogeneity in membrane domains in the vicinity of cross-linked receptors has yet to be observed directly due to experimental limitations.
The development of novel experimental techniques continues to be of value to furthering the understanding of these phenomena. In particular, subdiffraction-limited techniques to reveal temporal correlations of molecular trajectories over a fixed illumination spot provide key information on molecular dynamics and clustering. Reducing the size of the illumination spot enables a reduction of the number of molecules simultaneously observed and an increase in signal-to-noise for single-molecule measurements. Stimulated emission depletion (STED) microscopy utilizes laser-induced fluorescence depletion to collect fluorescence emission from only the center of a diffraction-limited fluorescence excitation spot of the sample with high-powered lasers.14 Near-field scanning optical microscopy (NSOM) utilizes metallic scanning probes that must be maintained in close proximity to an unperturbed membrane.15 Zero mode waveguides (ZMWs) require the detection area be within a hollow <100 nm diameter metallic aperture.16 STED, NSOM, and ZMWs have been used for fluorescence correlation spectroscopy (FCS) at length scales below the typical diffraction limit.17−21 However, the use of STED on biological samples has been limited by the need for intense irradiation of the sample (>150 mW/μm2), the use of scanning probes on soft samples poses challenges for maintaining the distance between probe and the sample, and the use of ZMWs requires the sample to penetrate into a narrow aperture.
Fluorescence cross-correlation spectroscopy (FCCS) provides information on the coordinated diffusion and cluster formation of fluorescently labeled molecules. Subdiffraction-limited illumination regions are especially important for the resolution of cross-correlations of weakly binding proteins of interest or in densely labeled samples due to the potentially large numbers of background molecules that could obscure the signal. Subdiffraction-limited FCCS has only previously been performed with ZMWs.22 However, our attempts to explore membrane domains with ZMWs have given inconsistent results presumably due to the inherent sample geometry on ZMWs. Membranes on ZMWs experience sharp curvature into the illumination volume within the hollow aperture,19,23 which may alter the diffusion of membrane domains with variable bending rigidity and curvature preference.24,25 The planarized aperture-based technique presented here overcomes these challenges to provide subdiffraction-limited spatial resolution and microsecond temporal resolution for simultaneous observation of chromatically distinct fluorophores on planar membranes.
Herein we present an experimental methodology for observing real-time, nanoscale membrane organization and dynamics in live cells and model membranes. FCS and two-color FCCS with a 55 nm illumination width are performed with planarized apertures within a two-objective microscope. Cross-correlations observed upon cross-linking membrane-bound proteins demonstrate the codiffusion and interactions at subdiffraction-limited length scales. The near-field excitation profile is analyzed both experimentally and computationally. The experimental system presented here provides significant advantages compared to other super-resolution techniques, including (1) the use of conventional fluorophores, (2) the lack of a scanning probe, (3) low laser powers, (4) no pulse timing or laser phase considerations, (5) minimal postacquisition data processing as compared to single-molecule localization methods, and (6) the capability, with future developments, to simultaneously observe from numerous apertures in a planarized array. The proof-of-principle results presented here demonstrate a 55 nm illumination width for FCCS on planar membranes for the detection of nanoscale molecular cross-linking and mobility.
Results
Experimental Setup
Our planarized apertures are 40–250 nm diameter glass pillars surrounded by a thin, opaque metal and coated by 10 nm of SiO2. Excitation light was transmitted through the aperture and confined to the subdiffraction-limited dimensions directly above the aperture. Only fluorophores directly above an aperture became excited and fluoresced (Figure 1), resulting in a subdiffraction-limited illumination profile, as previously reported.26 While this scheme cannot generate an image of the sample, it provides fast temporal resolution (limited by the APD to 108 Hz) and subdiffraction-limited illumination width (55 ± 10 nm) of isolated points on the membrane. A membrane sample was placed on the apertures. Adherent living cells were grown directly on the apertures for at least 4 h, or supported lipid bilayers were formed via vesicle fusion, as described in the Materials and Methods. Both supported lipid bilayers and adherent living cells are expected to conformally coat the aperture surface with minimal variations in time.27,28 This setup yielded minimal sample perturbations (i.e., membrane curvature, laser-induced heating, or probe pressure) and provided a conventional glass surface to support the membranes.
Figure 1.
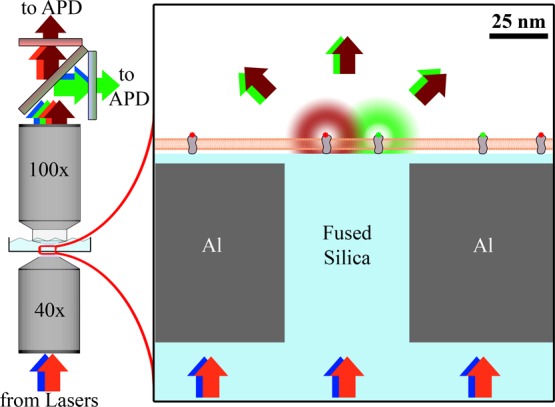
Planarized apertures restrict the excitation light to transmit through the aperture to excite only those fluorophores that are directly above the aperture. Multiple colors of excitation light can be simultaneously used to monitor chromatically distinct membrane-bound fluorophores. The emission was split by a dichroic mirror and chromatically filtered before being collected by an APD for each color channel.
The sample was then placed within the two-objective microscope with laser excitation focused on the underside of an aperture by a 40× objective below the substrate, and fluorescence emission was collected by a 100× dipping objective above the sample (Figure 1). The emission from fluorophores directly above a single aperture was measured by single-photon-counting avalanche photodiodes (APDs) to yield intensity vs time traces with 10 ns resolution.
Illumination Profile
To guide the fabrication of the planarized apertures, the illumination profile of the transmitted excitation light was measured computationally with finite element analysis (Figure 2, S1). Simulations were performed on apertures of 50 nm diameter (D50) and 100 nm diameter (D100) with 10 nm of SiO2 coating the Al film. The opaque Al film restricted the light to be primarily within and above the aperture, causing the transmitted illumination profile to be determined by the aperture properties rather than by the diffraction limit. Simulations revealed that when linear polarized light was incident upon the underside of the apertures, there was a strong enhancement of the electric fields at the aperture rim perpendicular to the polarization (Figure 2A, E) and no field enhancement at the corners parallel to the polarization (Figure 2B, F) (i.e., at different locations around the top rim of the glass-filled metal aperture). Averaging of these results yielded the expected results for unpolarized or time-averaged circularly polarized illumination (Figure 2C, D, G, H), as was used in the fluorescence experiments.
Figure 2.
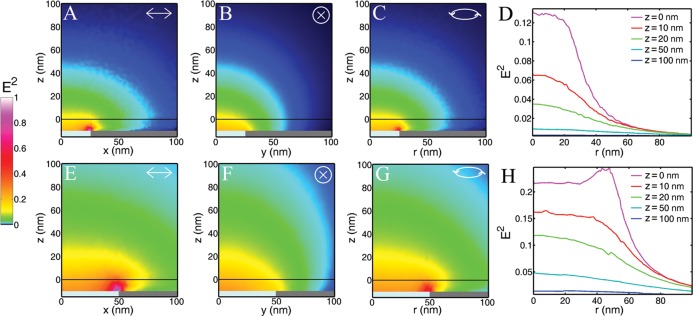
Finite element analysis was performed to computationally predict the illumination profile above (A–D) 50 nm and (E–F) 100 nm diameter apertures. Normalizations were performed on each aperture diameter independently so that the brightest spots in A–D and E–H were separately set to unity, with 54× greater maximum intensity for the 100 nm vs 50 nm apertures. Results for (A, B, E, F) linear and (C, D, G, H) circularly polarized incident light are shown. The vertical scale (z) was set equal to zero at the top of the SiO2 coating of the apertures; the Al film and the SiO2 pillar exist at z < −10 nm.
At the top of the SiO2 coating (z = 0), the maximum light intensity was found above the center for D50. However, for D100, the edge enhancement causes the greatest light intensity above the aperture edge at z = 0. For z ≥ 10 nm, the strongest light intensity was found above the center of the aperture for both D50 and D100 (Figure 2D, H).
The width of the illumination increased with increasing z (Figure 2D, H). The narrowest illumination was closest to the apertures, with a full width at half-maximum (fwhm) at z = 0 of 64 and 121 nm for D50 and D100, respectively. At z = 20, the fwhm increased to 80 and 124 nm for D50 and D100, respectively; at z = 50 nm, the fwhm increased to 130 and 135 nm for D50 and D100, respectively. These numbers illustrate how the illumination profiles spread more quickly after being confined to smaller apertures. For example, as z increased from 0 to 50 nm, the fwhm increased by 103% for D50 and 12% for D100. Similarly, the maximum intensity of the illumination at different z planes decreased quicker for smaller diameter apertures (Figure 2D, H). The peak intensity at z = 0 was double that found at z = 11 nm and z = 20 nm for D50 and D100, respectively. These numbers display the very strong z dependence on the illumination intensity, as can be seen by comparing the y-intercepts in Figure 2D, H. The z confinement of the illumination after passing through these nanoscale apertures was 10–20 times smaller than that for total internal reflection fluorescence (TIRF) microscopy.
For further evaluation, finite element analysis was used to compute the effects of varying the aperture properties. The aperture diameter, the index of refraction of the material within the aperture, the index of refraction of the material covering the aperture, and the type of metal surrounding the aperture were tested, and the resulting illumination profiles are shown in the Supporting Information (Figure S1).
Aperture Fabrication and Characterization
On a 500 μm thick fused silica substrate, 40–250 nm diameter fused silica pillars were defined by variable exposure duration of electron beam lithography and CF4 reactive ion etching (Figures S2, S3). Pillars were surrounded by 100 nm thick, sputtered Al alloy film. The sample was planarized with chemical mechanical polishing (Figures S2,S5), and 10 nm of SiO2 was deposited on the planarized substrate via atomic layer deposition (ALD) to ensure biological compatibility.
Demonstration of the variation in aperture diameters, the control over the aperture spacing, and the minimal number of potentially disruptive pinholes through the metal could be quickly assessed through color images of transmitted light (Figure 3). Typically there were >1000 fabricated apertures per pinhole, and the apertures were easily identified via their consistent brightness and fabrication on a square lattice. Light of longer wavelengths experiences greater attenuation through smaller diameter apertures, in agreement with Bethe–Bouwkamp theory.29,30 Larger apertures transmit a greater intensity of light, and the light they do transmit contains longer wavelengths compared to the light transmitted through smaller apertures. Apertures were fabricated on a square lattice with variable spacing: 1 μm spacing enabled easier observation of multiple apertures with atomic force microscopy (AFM) and SEM, and 10 μm spacing ensured there would be no cross-talk between the apertures in the resulting transmitted illumination profiles for fluorescence measurements.31
Figure 3.
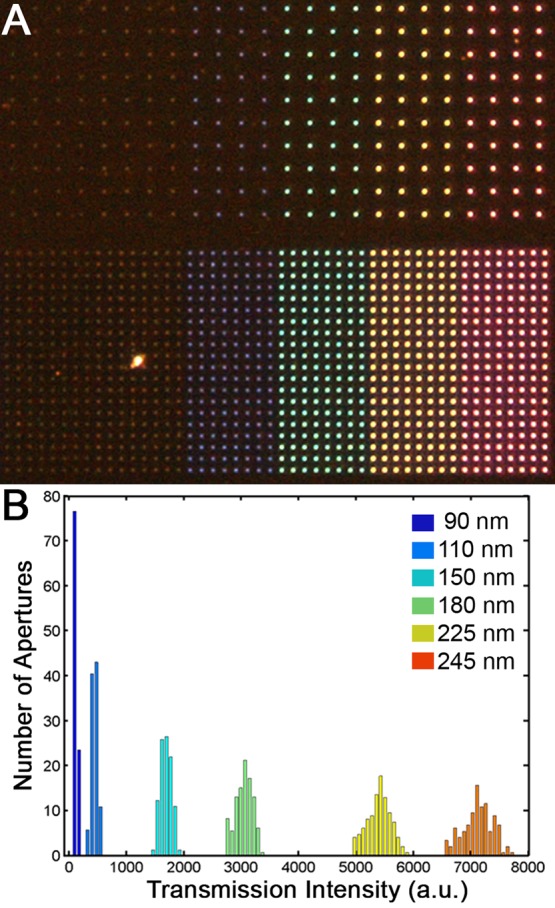
Far-field images of the transmission of white light through apertures reveal the variation in transmission intensity as dependent on aperture diameter and light wavelength. (A) Color micrographs reveal brighter and redder transmission through largest apertures (right, 245 nm diameter) vs smallest apertures (left, 90 nm diameter). Apertures are spaced 10 μm apart (top) or 5 μm apart (bottom) in this field of view. An image such as this enables the detection of pinholes. (B) The quantification of total transmission intensity from each aperture reveals a narrow distributions for each size aperture, with standard deviations 3% of the average intensity.
The height of the apertures relative to the surrounding metal film was measured with AFM (Figure 4). The final surface of the apertures had an RMS roughness of 1.2 nm with an aperture protrusion of less than 3 nm. The height and size of the aperture were comparable to the grains of Al alloy and ALD-deposited SiO2 surrounding the apertures.
Figure 4.
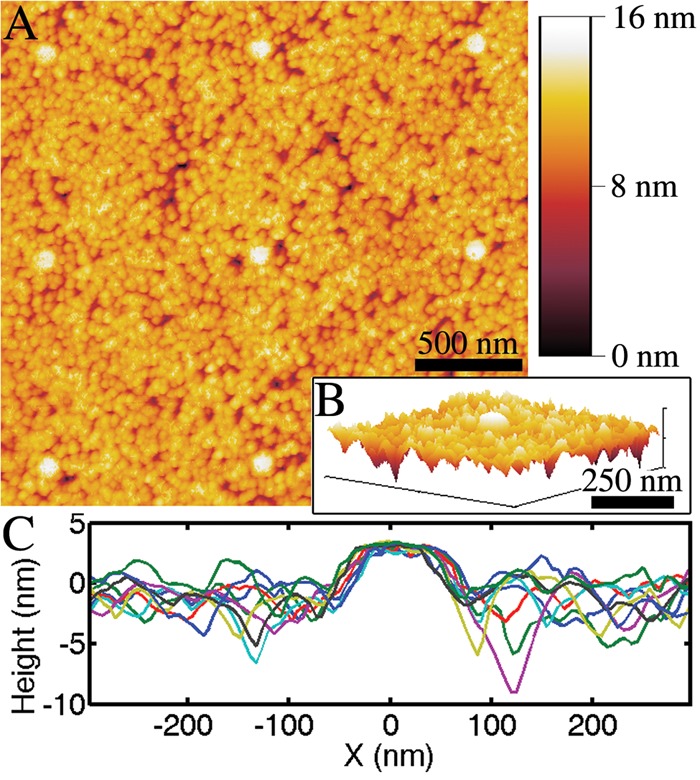
Atomic force microscopy was used to measure the flatness of the planarized apertures coated with 10 nm of SiO2 following chemical mechanical polishing and atomic layer deposition. (A) Eight apertures are shown, each with a height <3 nm above the surrounding film. (B) A 3D rendering of a single aperture and (C) line scans over nine different apertures show the film roughness as compared to the protrusions of the SiO2-filled apertures.
Near-Field FCCS on Model Membranes
The diffusion of CTxB labeled with either Alexa Fluor 488 (AF488) or Alexa Fluor 647 (AF647) was measured on a supported lipid bilayer of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) with 0.1% GM1 over the planarized apertures (Figure 5). After formation of the supported lipid bilayer and incubation with CTxB-AF488 and CTxB-AF647, near-field FCS and FCCS were performed to examine the lateral diffusion of labeled CTxB above 50 nm diameter planarized apertures. The density of independent diffusers was assessed by examining the correlations at short lag times (G0,eq 3) prior to the addition of a cross-linking antibody. Upon addition of a bivalent, polyclonal antibody specific for CTxB (anti-CTxB), G0,AF488, G0,AF647, and G0,Cross increased by factors of 1.8, 2.8, and 8.3, respectively (Figure 5E–G). The mean dwell time (τD, eq 7) of CTxB in autocorrelations prior to cross-linking was 6 ± 1 ms. After cross-linking, the average τD of the autocorrelations increased to 11 ± 2 ms, while τD from the cross-correlation was 40 ± 4 ms. The observed degree of clustering of the separately labeled populations of CTxB (FC, eq 8) increased from 0.03 to 0.12 upon cross-linking by anti-CTxB.
Figure 5.
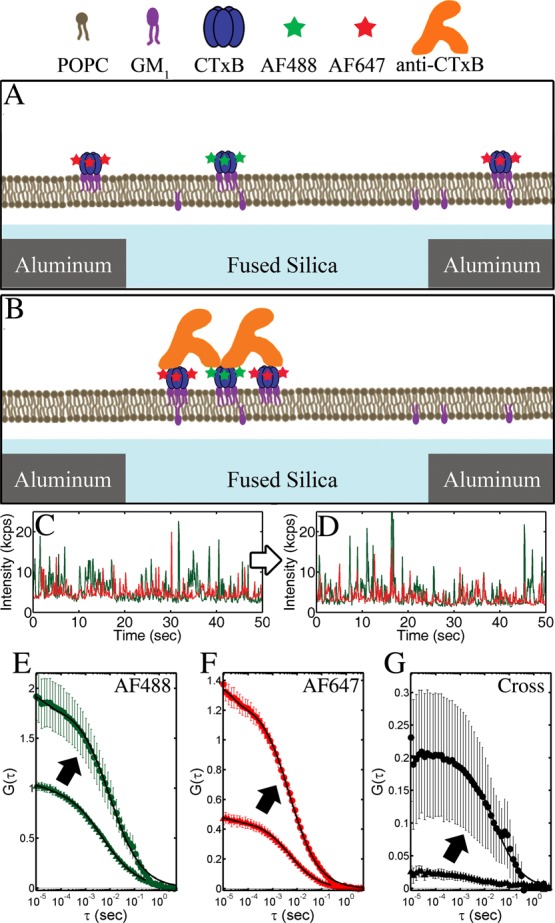
Cross-linking of CTxB-AF488 (A) and CTxB-AF647 (B) by a CTxB-specific antibody on a model membrane represented schematically and observed through changes to the (C, D) I vs t, (E, F) autocorrelations, and (G) cross-correlations. (C–F) Data from AF488 and AF647 are depicted as green and red, respectively. (A, C) CTxB initially diffused independently, but (B, D) the addition of anti-CTxB results in the formation of larger, slower, multicolored clusters of CTxB. Cross-linking resulted in an increase in G0 and τD of the (E, F) autocorrelations and (G) cross-correlation. Correlations before cross-linking are indicated with a triangle (▲) and after cross-linking are indicated with a circle (●), as shown by arrows. Fits of eq 7 to the correlations are shown in solid black lines. Error bars are the standard error of the mean of 10 sequential 30 s FCCS measurements.
Fitting these results to eq 7 quantifies the diffusion anomaly and the fluorophore blinking. However, these parameters were not significantly affected by the cross-linking antibody. For example, the autocorrelations for both color channels and for both before and after cross-linking yielded α = 0.68 ± 0.06, τT = 0.2 ± 0.01 ms, and FT = 0.07 ± 0.03. These supported lipid bilayer results were repeated on five different days with different apertures with quantifiably reproducible results (±15%).
Near-Field FCCS on Live Cells
FcεRI in the plasma membrane of mast cells were bound to fluorescently labeled IgEs, which were bivalent for 2,4-dinitrophenyl (DNP). The IgEs were covalently labeled with either AF488 or AF647, and their diffusion while bound to FcεRI was measured by FCS and FCCS above 50 nm diameter planarized apertures. The lateral diffusion of FcεRI-IgE-AF488 and FcεRI-IgE-AF647 in the plasma membrane was observed to be independent in the absence of a cross-linker, with very weak cross-correlation; FC = 0.03 (Figure 6). Cross-linking of FcεRI-IgE was performed with 4.2 nM of a trivalent, Y-shaped DNA complex (Y16-DNP3). This DNA complex is formed by three complementary single strands (16 bases each) that are each conjugated with a DNP to yield 5.2 ± 0.9 nm between each pair of DNPs. It stimulates a robust signaling response in IgE-sensitized RBL mast cells.32 Upon cross-linking of the FcεRI-IgE with Y16-DNP3, the autocorrelations became slower and a cross-correlation became prominent (Figure 6C, D). Upon cross-linking, the average autocorrelation τD increased from 11 ± 2 ms to 62 ± 10 ms, while the cross-correlation τD after cross-linking was 114 ± 20 ms; the diffusion of cross-linked receptors was 10 times slower than receptors prior to cross-linking. Cross-linking also resulted in an increase of G0,AF488, G0,AF647, and G0,Cross, with FC increasing to 0.33. These results were repeated on four different days with different individual apertures and quantitatively similar results (±20%).
Figure 6.
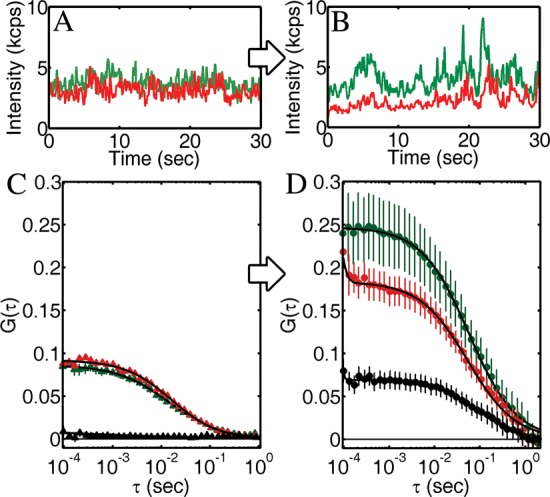
Cross-linking of IgE-FcεRI by a trivalent antigen on the plasma membrane of living mast cells was observed through changes in the auto- and cross-correlations. (A) I vs t before cross-linking shows many small, uncorrelated peaks. (B) After cross-linking, I vs t for the two color channels shows correlated peaks. These results were quantified in the auto- and cross-correlations (C) before and (D) after cross-linking. Before cross-linking, autocorrelations for IgE-A488 and IgE-A647 were clear, while the cross-correlation was not. After cross-linking for 10 min with 4.2 nM Y16-DNP3, G0 and τD for both the auto- and cross-correlations increased. All correlations were fit to eq 7, and error bars are the standard error of the mean of four sequential measurements.
Varying concentrations of Y16-DNP3 and the highly multivalent DNP-bovine serum albumin (BSA) with 10–20 DNP groups per BSA were tested. Both higher concentrations of Y16-DNP3 and a range of concentrations of DNP-BSA appropriate for stimulation yielded correlations that were very noisy, and the extraction of fit parameters was unreliable. For lower concentrations of Y16-DNP3 (<3 nM), autocorrelations continued to be clear for both color channels and FC did not increase consistently, suggesting that this low cross-linker concentration was insufficient to cluster FcεRI-IgE.
Discussion
FCS and FCCS data are notoriously difficult to interpret due to similar autocorrelation shapes for various diffusion processes, the inherent signal averaging in correlation functions, and the requirement of large numbers of single-particle trajectories within a single measurement as to achieve reproducible results. Fitting parameters as described in eq 7 provide a basis for data interpretation. Traditionally, G0 quantifies the density of diffusers and τD quantifies the characteristic duration over the illumination area. As described below, we have utilized these fit parameters to reveal the distribution of cluster sizes. The values of τT and FT are a measure of the particular fluorophore properties (e.g., blinking rate) as affected by the total illumination intensities and buffer conditions and are not expected to reveal insights into the molecular mobility or clustering. Future studies will assess the cause of the apparent anomalous diffusion of CTxB on GM1 in these supported POPC bilayers (i.e., α ≠ 1), which could be the result of substrate–membrane interactions, transient associations between molecules within the membrane, or a non-Gaussian illumination profile, as also discussed below.
Sub-Diffraction-Limited Illumination Profile
By measuring the dwell time of a diffuser with a known diffusion coefficient (D), we estimate the characteristic size of the illumination profile. The diffusion coefficient (D) of CTxB in a POPC-supported lipid bilayer has been measured by both SPT via localization microscopy and fluorescence recovery after photobleaching (FRAP) to be 0.12 ± 0.03 μm2/s (Figure S6). The mean dwell time (τD, eq 7) of CTxB in autocorrelations prior to cross-linking above planarized apertures was 6 ± 1 ms. 2D Brownian diffusers are expected to diffuse a mean squared distance, ⟨r2⟩, in the time Δt according to ⟨r2⟩ = 4DΔt. Applying the measured value of D for CTxB and the measured dwell time with planarized apertures, this yields a characteristic illumination of 55 ± 10 nm. This is approximately one-quarter that expected for a diffraction-limited system and consistent with computational simulations for 50 nm diameter apertures.
Similar analysis to estimate the size of the illumination profile was performed for planarized aperture illumination of live cell membranes. D of FcεRI-IgE in the plasma membrane has been previously measured to be 0.10 ± 0.05 μm2/s in the absence of cross-linking,11 and we observe a dwell time of 11 ± 2 ms for FcεRI-IgE before cross-linking. Therefore, the characteristic illumination width from a planarized aperture on live cells was 66 ± 15 nm. This observed size of the illumination profile on live cell membranes was marginally larger than that observed for supported lipid bilayers and may be attributed to the different separation distances from the substrates to the membranes; however this difference in apparent illumination profile was not statistically significant. Further, τD slowed by 10 times upon cross-linking FcεRI-IgE with Y16-DNP3 to 114 ± 20 ms with the same illumination profile, indicating a 10 times decrease in D of FcεRI-IgE after cross-linking to 0.010 ± 0.005 μm2/s.
Detecting Membrane Cross-Linking
Larger G0 values indicate that an increased fraction of the intensity signal comes from each independent diffuser. Thus, a larger G0 value indicates that there are fewer independent diffusers simultaneously present in the observation region on average, and this is commonly interpreted as a decrease in the density of diffusers. Upon cross-linking, previously independent diffusers are combined into fewer, larger clusters, and this alone reduces the density of independent diffusers without necessarily reducing the number of fluorophores or proteins on the membrane.
In the presence of stochastic cross-linking without fixed stoichiometries, it is feasible to simultaneously have monomeric, single-colored diffusers and multimeric, multicolored diffusers on the membrane. These two species can be distinguished through comparison of the autocorrelations with the cross-correlations. Both the single-colored and multicolored diffusers will contribute to each autocorrelation, while only the multicolored diffusers will contribute to the cross-correlations.
On both model membranes and live cells, clear autocorrelations were observed in both color channels, while minimal cross-correlation was observed before the addition of cross-linkers (Figures 5E–G and 6C). The presence of a nonzero G0,Cross before cross-linking may have been due to chromatic bleed-through between the two color channels or due to a small amount of spontaneous aggregation of the proteins on the membrane in the absence of cross-linker. Identical values of FC on both live cells and model membranes before cross-linking support chromatic bleed-through as the explanation. When CTxB on model membranes was cross-linked with anti-CTxB, clusters of multiple CTxB formed on the membrane, including both CTxB-AF488 and CTxB-AF647 within the same clusters. This was measured by the increase in FC from 0.03 to 0.12, the increase in G0,488 and G0,647 by an average of 2.1 times, and analysis of the correlation dwell times as discussed below. Similarly, when FcεRI-IgE on live cells were cross-linked with Y16-DNP3, clusters containing both FcεRI-IgE-AF488 and FcεRI-IgE-AF647 were formed. These clusters were identified by the large changes in the cross-correlations observed upon cross-linking (Figures 5G and 6C, D). On live cells, FC increased from 0.03 to 0.33 and G0,488 and G0,647 increased by an average of 2.4 times upon cross-linking with Y16-DNP3.
At the cross-linker concentrations used, wide-field images of both the model and live cell membranes appeared to have a uniform fluorescence in both the AF488 and AF647 color channels before and after cross-linking. This indicated that the fluorophore density was roughly consistent on all diffraction-limited length scales (i.e., >200 nm) and that clustering caused no large CTxB or FcεRI-IgE density differences within the time scale of these measurements. At the concentration used in our experiments (4.2 nM), Y16-DNP3 was shown to stimulate 40% of the maximal degranulation response at 42 nM Y16-DNP3 in RBL mast cells with minimal receptor internalization despite having only 10% of the cross-linker concentration at an optimal dose.32 These data contribute to the body of data that suggests clusters of membrane receptors need not be massively cross-linked or immobilized to initiate downstream signaling.11 Further analysis of the relative potency of signaling clusters with varying size and mobility will be a focus of future studies.
Varying the cross-linker concentration or valency altered the observed clustering. Concentrations of anti-CTxB and Y16-DNP3 that were 3 times lower or 3 times higher than reported here yielded cross-linking that is difficult to resolve. Similarly, increasing the valency of the IgE cross-linker via the use of DNP-BSA resulted in a greater extent of cross-linking of the FcεRI-IgE, receptor immobilization, and internalization. Both immobilization and internalization would contribute to a greater noise within the FCS and FCCS data as fewer diffusers would be sampled during a single 30 s observation.
Dwell Time Changes Reveal Fractional Clustering
As individual membrane-bound proteins are cross-linked into larger clusters of multiple proteins diffusing together, their diffusion is slowed. Previous studies have suggested diffusion on membranes should slow proportionally to the log of the inverse of the protein radius,33,34 while others find slowing proportional to the inverse of the protein radius.35 Challenges measuring the membrane viscosity, adhesion to the substrate, and interactions with the fluid surrounding the membrane make direct determination of the clustered fractions from the changes in diffusion rates difficult. However, comparison of the cross-correlation vs autocorrelation dwell times upon cross-linking provides a means of assessing the relative density of monomeric vs clustered diffusers.
Upon cross-linking model membrane-bound CTxB with anti-CTxB, τD from the autocorrelations increased from 6 ms to 11 ms (Figure 5E, F). τD for the cross-correlation after cross-linking was 40 ms (Figure 5G), 4 times longer than τD for the post-cross-linking autocorrelations. τD for the cross-correlation before cross-linking could not be fit with certainty. The difference between the auto- and cross-correlations after cross-linking is that autocorrelations measure both multicolored and single-colored diffusers, whereas the cross-correlations measure only the multicolored diffusers. The difference in diffusion rates between single CTxB and clusters of CTxB was not sufficient with respect to the noise level of the autocorrelations to warrant fitting the autocorrelations to a functional form for two or more unknown diffusion rates. However, an approximate ratio of un-cross-linked CTxB (as determined by pre-cross-linked autocorrelation) vs cross-linked clusters of CTxB (as determined by the post-cross-linked cross-correlation) was determined by fitting the post-cross-linking autocorrelation to a linear combination of the pre-cross-linking autocorrelation τD and the post-cross-linking cross-correlation τD. Through fitting a linear combination of these fits to the post-cross-linking autocorrelation, it was determined that 67% of the post-cross-linking autocorrelation was from monomeric CTxB and 33% was from clustered diffusers.
A Poisson distribution of cluster sizes is expected if individual receptors and clusters of receptors both diffuse over the membrane and cross-link to any other receptors they contact. The fraction of clusters with n proteins is given by a diffuser size distribution (DSD) according to
| 1 |
where a is the mean cluster size. The contribution to the autocorrelation functions from each cluster is proportional to the number of proteins (and thus the number of fluorophores) in the cluster. Binomial statistics indicate that for clusters with two CTxB, for example, 25% would have two CTxB-AF488, 25% would have two CTxB-AF647, and 50% would have one CTxB-AF488 and one CTxB-AF647. Thus, the average brightness in one channel for a cluster of two CTxB would be 1.33 times the brightness of a single CTxB since the cluster entirely composed by CTxB of the other color would not contribute to the signal. In a single-color channel, a cluster of n proteins would have a brightness B(n)-fold greater than a monomeric CTxB in that color channel, where
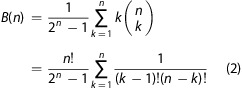 |
2 |
As expected, B(n) quickly approaches n/2, as a large cluster will be on averaged equal numbers of each color of CTxB. Thus, the predicted fraction of the post-cross-linking autocorrelation that comes from independently diffusing CTxB as a function of the average diffuser size would be
 |
3 |
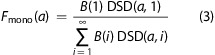
Numerically solving Fmono for the observed 67% yields a mean of 1.3 CTxB per diffuser after cross-linking, which translates to 74% of the CTxB were diffusing independently, 22% were diffusing bound in pairs, 3.4% were diffusing in clusters of three CTxB, etc., from eq 1.
We can apply this same analysis to the correlations from IgE-FcεRI on living cells. The live cell autocorrelations after cross-linking had a faster dwell time than the cross-correlation as result of autocorrelations having measured both monomeric receptors and clusters of receptors, whereas cross-correlations measured only clusters of receptors. Fitting the post-cross-linking autocorrelation data to a linear combination of the dwell times from the pre-cross-linking autocorrelation (to represent the monomer fraction) and the post-cross-linking cross-correlation (to represent the clustered fraction) shows that 28% of the post-cross-linking autocorrelation came from monomeric FcεRI-IgE diffusers and 72% from receptors in larger clusters. Solving eq 3 for Fmono(a) = 28% yields an average of 2.0 IgE-FcεRI per cluster, which translates to 38% of diffusers were monomeric IgE-FcεRI, 37% of diffusers were composed of two IgE-FcεRI, 18% of diffusers included three IgE-FcεRI, etc., from eq 1.
Dwell Time vs Aperture Diameter
Planarized apertures could provide a means of controlling the sub-diffraction-limited illumination through control of the aperture diameter. As light was limited to transmission through the apertures, those with smaller diameters could yield a smaller illumination area at the top of the aperture, as predicted with computational simulations (Figures 2 and S1). This could provide a means for examining nanoscale non-Brownian diffusion through analysis of τDvs illumination area.36,37 However, we have not observed variation in τD with aperture diameter for planar membranes above current apertures varying from 60 to 250 nm diameter (Figure 7). The dwell time for GM1-Bodipy in a POPC bilayer was 0.40 ± 0.1 ms regardless of aperture diameter. D for GM1-Bodipy in a POPC bilayer has been previously measured to be 2.0 ± 0.2 μm2/s.26 The characteristic illumination width was calculated to be 56 ± 12 nm without an observable dependence on aperture diameter.
Figure 7.
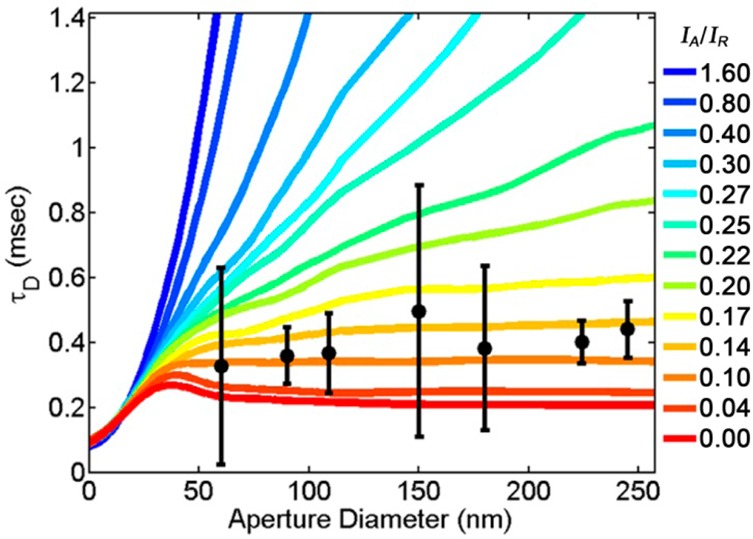
Dwell times for GM1-Bodipy in a POPC-supported lipid bilayer on apertures are consistent across apertures of diameter varying from 60 to 250 nm (black circles). Simulations of a Brownian diffuser over a rim-affected illumination profile (eq 4) display differing dwell times vs aperture diameters for varying ratios of IA/IR (colored lines). Error bars represent the standard deviation of sequential measurements distributed between numerous apertures. Simulations were matched to experimental data by setting D = 2 μm/s and wR = 17 nm.
We hypothesize that the illumination profile on the membrane was affected by the plasmonic effects at the top rim of the aperture. This near-field rim effect could reduce the dependence of the illumination size on the diameter of the aperture. The corners of metal that define the top rim of the aperture are a source of significant enhancement of the excitation light, as seen within finite element analysis (FEA) simulations (Figure 2). The rim effect could be greater than observed in FEA simulations due to nanoscale roughness in the fabricated apertures that could magnify the field enhancement. Previous studies have demonstrated the presence of a rim effect in single-fluorophore emission brightness,38 polarization dependence,39 lifetime,40 angular emission,41 and computational analysis.21,42 Our attempts to measure the illumination profile from the planarized aperture directly with near-field scanning optical microscopy proved to be uninformative (data not shown). Presumably the convolution of the nontrivial NSOM point spread function with that from the planarized apertures resulted in a highly convoluted NSOM image. Previous uses of ZMWs with membranes have demonstrated a variation in dwell time with aperture diameter,21,37 although these studies incorporated neither a solid filling of their aperture, leaving uncertainty as to the penetration of the membrane into the aperture, nor planarization, which may sharpen the aperture rim and enhance the edge effect. Further, Leutengger etal. demonstrated a suppressed increase in dwell time with increasing aperture diameter, which they justified as “surface effects” of the fluorophores in solution.21 The traditional interpretation of cross-correlations and clustering continues to apply to a rim-affected illumination profile; if different fluorophores are clustered so that they regularly enter and leave the illumination profile together, then a cross-correlation will be evident regardless of the shape of the illumination profile. Many prior studies with fluorescence from apertures suggest a strong influence of aperture edges on fluorescence excitation,38−42 and we have performed simulations to estimate the effects of a rim-dominated illumination profile.
To model how a rim-affected illumination profile would influence the apparent dwell time vs varying aperture radius, we simulated a 2D Brownian diffuser within an illumination profile defined as
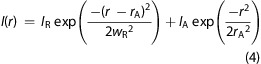 |
4 |
Here, r is the distance between the diffuser and the center of the aperture, rA is the aperture radius, IR is the intensity of illumination enhanced by the aperture rim (of width wR), and IA is the intensity of the illumination that spans the width of the aperture. To compare the simulation of a Brownian diffuser with the observed data of GM1-Bodipy in a POPC-supported lipid bilayer, we set D = 2 μm2/s and wR = 17 nm while simulating varying values for rA and the ratio of IA/IR (Figure 7). Individual plots of the dwell time vs aperture size for different IA/IR ratios could fit the experimental data set better with different values of wR; however, setting wR = 17 nm provided a suitable compromise for the different IA/IR ratios that demonstrated an appropriately low increase in dwell time vs aperture size.
The ratio of IA/IR determines the contribution of each component to the net illumination profile. When IA/IR = 0, the illumination profile is solely determined by the aperture rim and the dwell time stops increasing with aperture radius for rA > 1.2 wR. When IA/IR > 1, the illumination profile contains negligible contribution from the aperture rim and the dwell time increases proportionally to the square of the aperture radius, as expected. When IA/IR = 1, wR = 17 nm, and rA = 50 nm, the power of illumination from the aperture rim accounts for 43% of the net illumination power.
Comparison of the experimentally observed changes in dwell time vs aperture size to the Brownian diffusion simulations reveals the observed contribution of the rim to the net illumination profile (Figure 7). Despite the large uncertainty in the experimental data, agreement between the experimental and simulated data requires IA/IR < 0.2, indicating that at least 81% of the net illumination power is coming from the field enhancement at the aperture rim when wR = 17 nm and rA = 50 nm. The FEA simulations presented earlier suggest that 100 nm diameter apertures (rA = 50 nm) have a IA/IR = 2 at z = 0 (Figure 2H). However, nanoscale roughness in the fabricated apertures may greatly enhance the field enhancement at the aperture rim, which were not incorporated into the FEA simulations of perfectly flat and round apertures. Further, it is likely that the ratio of IA/IR could vary with aperture radius, as the larger diameter apertures would be less attenuating of the incident fluorescence excitation. These Brownian diffusion simulations demonstrate the consistency between a rim-affected illumination profile and the observed variation in dwell time with aperture diameter, but additional experiments would be necessary to elucidate the details.
Conclusions
Planarized nanoscale apertures have been be used to create simultaneous sub-diffraction-limited illumination with two excitation wavelengths for FCS and FCCS on membranes. The illuminated region of the sample is determined by the aperture rather than by the diffraction limit. With two microscope objectives, the incident excitation light passes through the aperture to excite only fluorophores in close proximity to the aperture, and the fluorophore emission was collected from above. These planarized apertures are advantageous because they do not induce membrane curvature and maintain close proximity to the membrane without a scanning probe. The functionality of this method has been demonstrated on both model membranes and plasma membranes of living cells. In both cases, two chromatically distinct and independently diffusing populations of membrane-bound proteins were cross-linked, and their cross-correlation became prominent. Through analysis of the change in the correlations with cross-linking, we have quantified the change in mobility and the cluster sizes of membrane-bound proteins. Analysis of the change in diffusion dwell time with aperture diameter has suggested the presence of a rim-affected illumination profile where the electric field enhancement at the aperture rim dominates the fluorophore excitation regardless of aperture diameter.
This technique holds promise for the nanoscale study of signaling cascades and protein recruitment to nanoscale signaling complexes. It is particularly well suited for the study of protein–protein and protein–lipid associations that may be dependent on sub-micrometer-scale membrane domains. For this application, the capacity to simultaneously monitor multiple near-field apertures would offer the potential to map membrane heterogeneities at physiologically relevant length scales. Future measurements of the correlated diffusion of interacting membrane-bound signal-transmitting lipids and proteins with sub-diffraction-limited resolution are expected to provide novel insights into the fundamental biophysical processes of nanoscale membrane phenomena.
Materials and Methods
FCS and FCCS Analysis
FCS and FCCS are powerful techniques for extracting mobility and binding information on mobile fluorescent objectives (“diffusers”) traversing through a detection region. FCS is an analysis of the temporal autocorrelation of intensity (I) vs time (t) in a single-color channel according to
| 5 |
where ⟨⟩ indicates the time average, τ is the lag time, and δI = I – ⟨I⟩. FCCS is the examination of the temporal cross-correlation of two different intensity signals (I1 and I2), reflecting the correlated diffusion of two species that are labeled with two chromatically separate fluorophores, according to
| 6 |
Both autocorrelations and cross-correlations on membranes can be fit to the functional form expected for two-dimensional anomalous diffusion in the presence of fluorophore blinking, according to
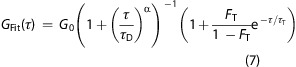 |
7 |
where τD is the characteristic dwell time of the diffuser, α quantifies the degree of diffusion anomaly, FT is the fraction of diffusers within the dark triplet state, and τT is the triplet blinking correlation time.17,26
G0 reflects the ratio in intensity of individual intensity fluctuations to the total detected intensity and is often interpreted as the inverse of the number of independent diffusers simultaneously within the detection region (N). However, G0 equals 1/N only in the absence of background or chromatic bleed-through within the fluorescence signal. More accurately, the cross-linked fraction of diffusers (FC) is quantified as the ratio of the G0 values for the cross-correlation vs the average of the autocorrelations, according to
| 8 |
where G0,1, G0,2, and G0,C represent the values of G0 fit from eq 7 to the autocorrelation of the first and second color channels and the cross-correlation between color channels, respectively. Upon cross-linking, the total number of fluorescent molecules on the membrane may not have changed, while G0 values increase since the fluorescent molecules became clustered and stopped diffusing independently. This results in fewer, brighter independent diffusers, each with multiple fluorescent molecules.
Fabrication
Pillars of negative resist (Sumitomo Chemical Co., Ltd., Sumiresist NEB-31A2) were patterned by electron beam lithography (JEOL USA Inc., JBX-9300FS) on a 100 mm diameter, 500 μm thick fused silica wafer to be 150 nm tall with diameters between 80 and 500 nm. The resist pillars masked the fused silica during a CF4 reactive ion etch (Oxford Instruments plc, PlasmaLab 80+) for the creation of pillars of fused silica. The resist was removed with O2 reactive ion etch (Oxford Instruments plc, PlasmaLab 80+), and the pillars of fused silica were further etched with buffered oxide etch (6:1 volume ratio of 40% NH4F in water to 49% HF in water) to yield fused silica pillars as small as 40 nm diameter. WARNING: HF is very dangerous and precautions must be taken. The wafer was then uniformly coated with 200 nm of sputtered Al alloy, Al95%Cu4%Si1% (Cascade Vacuum Consulting, 601), and planarized with chemical mechanical polishing (Strasbaugh, 6EC) to remove all but 100 ± 20 nm of the Al alloy and expose the tops of the fused silica pillars. The shape of the illumination profile is not expected to vary with the thickness of the metal films, but the intensity of the transmitted light did vary with film thickness. Metal films thinner than 80 nm provided insufficient blocking of light around the aperture, while metal films thicker than 120 nm resulted in too little illumination transmitted through the aperture. Metal deposition via sputtering films yielded a more conformal coating of the fused silica pillars and fewer pinholes than evaporated Al films. The polished apertures were coated with 10 nm of SiO2 through atomic layer deposition (Oxford Instruments plc, FlexAL) to ensure biological compatibility, consistency with other experiments performed on glass coverslips, and greater consistency in supported lipid bilayer formation. Thirty-two independent 14 mm × 14 mm dies, each with nine different sizes of apertures, were created from each 100 mm diameter wafer. Each 14 mm × 14 mm die was glued over a 10 mm diameter hole on the underside of a 35 mm diameter plastic Petri dish for containment of the buffer or biological media. The devices were examined optically (Olympus Corp., IX71, BX51), with SEM (Carl Zeiss Microscopy LLC, Supra 55), and with AFM (Veeco Instruments Inc., Dimension Icon).
Finite Element Analysis
Three-dimensional finite element analysis simulations were performed in the electromagnetic module of COMSOL Multiphysics (COMSOL Inc.) with a perfectly matched boundary layer condition.43,44 Simulations included transmission of linearly polarized light with a free-space wavelength of 488 nm through a nanoaperture in a 100 nm thick metal film that was supported by, filled with, and coated with SiO2. Above the apertures, water was assumed to be covering the substrate, as would be the case in biological experiments. Simulation results for unpolarized or the time averaged circularly polarized incident light were calculated as an average over the azimuthal angle, yielding cylindrical symmetry. Complex indices of refraction used to simulate SiO2, Al, and H2O were 1.46, 0.73–5.9i, and 1.34, respectively. In the Supporting Information, simulations of other aperture designs are described. Details of the simulated material properties are shown in Table S1 and Figure S1.
Simulated Diffusion
A Brownian diffusing particle was simulated over the illumination profile given in eq 4. The intensity vs time was calculated, and a temporal autocorrelation was performed. Size and length scale of the simulation was determined through comparison of the simulation results to the experimental data. The particle was observed over 12.5 s with time steps of 0.19 μs and D = 2 μm2/s with periodic boundary conditions within a square box of >4 μm width and repeated 30 times for r0 varying from 0 to 2.5 μm and w0 = 17 nm. The mean dwell times vs r0 are plotted in Figure 7D, and standard error of the mean of repeated simulations was approximately the width of the dashed line. Since an analytically predicted functional form of the autocorrelation results of these simulations was not available, the lag time at which the autocorrelation decayed to half of its maximum value was used as an estimate of the dwell time.
Optical Setup
A two-objective, transillumination microscope was developed for near-field FCS and FCCS with planarized apertures that incorporated both an inverted microscope (Olympus Corp., IX71) and an upright microscope (Olympus Corp., BX51). A 40× objective (NA = 0.65) for focusing the excitation light on the underside of a single aperture and a 100× dipping objective (NA = 1.0) above the sample for collecting the fluorophore emission were used (Figure 1). The top objective was necessary to collect fluorophore emission; requiring emission to transmit through the aperture for collection by the bottom objective would have resulted in the reduction of signal due to the restricted transmission through the aperture. Only fluorophores that were in close proximity to the aperture absorbed the excitation light and emitted at longer wavelengths, λex and λem, respectively. AF488 (λex = 488 nm, λem = 505–540 nm) and AF647 (λex = 647 nm, λem = 650–690 nm) were excited with 1 mW of circularly polarized excitation light on the underside of the aperture. Approximately 1% of the incident excitation light was transmitted through the aperture, with variations depending on aperture diameter and λex, yielding a peak excitation intensity of approximately 2 kW/cm2 on the membrane and 104 counts/s/fluorophore of collected emission. Excitation intensities were varied to ensure that there was not significant photobleaching or photodamage to the sample at this illumination intensity. The results presented here are quantitatively similar to illumination intensities as low as 0.2 kW/cm2, albeit with greater signal-to-noise at 2 kW/cm2. The emission was divided into two color channels by a dichroic mirror (Semrock Inc., FF562-Di02) and individually chromatically filtered (Semrock Inc., FF01-512/25 or Chroma Technology Corp., ET655lp). Each color channel was directed to an avalanche photodiode (PerkinElmer, SPCM-AQRH) via a 50 μm core diameter single-mode optical fiber and correlated by a high-resolution multiple-tau correlator (Correlator.com Flex02-12D). Details of the procedure used to achieve alignment of the optical system are provided in the Supporting Information.
FCS Analysis
Sequential I vs t traces, each lasting 30 s, were acquired and correlated with eqs 1 and 2. Resulting correlations were averaged and fit to eq 7 (Figures 5E–G and 6C, D); the error bars represent the standard error of the mean of sequential measurements. The I vs t plots were evaluated to ensure there was no significant decrease in I at large t, which could imply photobleaching or alignment drift. Often individual peaks in I vs t could be identified for analysis (Figures 5C, D and 6A, B).
Supported Lipid Bilayers
Supported lipid bilayers were prepared according to standard techniques on recently plasma cleaned, SiO2-coated planarized apertures by vesicle fusion. For experiments with CTxB, small unilaminar vesicles (SUVs) were created by first mixing 1 mg of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids Inc.) and 1 μg of GM1 (Avanti Polar Lipids Inc.), each initially in chloroform, in a glass vial. For experiments with GM1-Bodipy, 1 mg of POPC and 1 μg of GM1-Bodipy (Life Technologies Inc.) were added to the glass vial. The mixtures were dried of all chloroform for >20 min under an N2 stream and >10 min under vacuum. The resulting film was rehydrated by the addition of 1 mL of phosphate-buffered saline solution (PBS) and 20 min sonication with a probe sonicator (Thermo Fisher Scientific Inc., model 50 Sonic Dismembrator). The resulting SUV solutions were then spun in a microcentrifuge at 16000g to pellet the metallic contaminates from the sonicator. The resulting supernatant was stored for up to a week at 4 °C as a 1 mg/mL mixture of SUVs.
Within 10 min prior to the supported lipid bilayer formation, the substrate was exposed to 20 s of air plasma (Harrick Plasma PCD-001) to make the glass surface hydrophilic. Each supported lipid bilayer was created by the incubation of 100 μL of the 1 mg/mL SUV solution on a 100 mm2 hydrophilic glass surface. The exposure of the SUVs to the hydrophilic surface resulted in vesicle fusion and the formation of a continuous planar bilayer. Excessive SUVs were aggressively rinsed away with 10 mL of PBS. When applicable, the resulting membrane was incubated in PBS with 0.1 μg/mL each of CTxB-AF488 and CTxB-AF647 for 20 min, and unbound CTxB was rinsed away with excess PBS. The lateral diffusion of CTxB bound to the membrane or GM1-Bodipy within the membrane was measured at room temperature (21 °C). For the cross-linking of the CTxB, 1 μL of the anti-CTxB (Calbiochem, 227040 goat pAb) was diluted into 2 mL of PBS, and cross-correlations were apparent within 5 min at room temperature and consistent for over 30 min. Determination of the diffusion coefficient of CTxB in a POPC-supported lipid bilayer was performed with FRAP45 and SPT via STORM ((direct) stochastic optical reconstruction microscopy),11 as described in detail in the Supporting Information, making use of PALM3D46 (fluorescence photoactivated localization microscopy) and Track.pro for MATLAB.47
Live Cells
Rat basophilic leukemia (RBL-2H3) mast cells48 were cultured and harvested as described previously.49 For near-field spectroscopy, the cells were incubated for at least 4 h and generally overnight on the glass-coated planarized apertures in a cell growth medium (minimal essential medium with 20% fetal bovine serum and 10 μg/mL gentamicin sulfate) at 37 °C and 5% CO2. These adherent cells were sensitized with 0.5 μg/mL IgE-AF488 and 0.5 μg/mL IgE-AF647 in HEPES-buffered MEM for 20 min at room temperature on a shaker. Excess IgE was removed through rinsing, and cells were imaged at room temperature within buffered saline solution (BSS, 135 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose, 20 mM HEPES, 1 mg/mL BSA, pH 7.4). Cells were stimulated with the addition of 1 mL of BSS premixed with DNA- or BSA-based multivalent antigens: Y16-DNP3 at final concentrations of 0.42, 4.2, and 42 nM and DNP-BSA at final concentrations of 0.1 and 1 μg/mL were tested. Y16-DNP332 and DNP-BSA50 were prepared as described previously.
Acknowledgments
C.V.K. was supported by an NIH Kirschstein National Research Service Award postdoctoral fellowship (F32-GM092106) and generous start-up funds from Wayne State University. Additional research support came from NIAID at the National Institutes of Health (R01-AI018306) and the Nanobiotechnology Center at Cornell University (NSF ECS-9876771). Device fabrication was performed in the Cornell Nanoscale Science and Technology Facility (NSF ECS-9731293). The authors thank Pangshn Zhu and Huizhong Xu for useful discussions concerning the computational model and Sarah A. Shelby for discussions concerning single-particle tracking and STORM.
Supporting Information Available
Finite element results with varying aperture properties (Figure S1); scanning electron microscopy images of fused silica pillars (Figures S2, S3), planarized apertures in Al (Figures S2, S4) and Al95%Si4%Cu1% (Figures S2, S5); fluorescence correlation after photobleaching and single-particle tracking results for CTxB diffusion (Figure S6); and a table of parameters used in finite element simulations (Table S1). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Simons K.; Gerl M. J. Revitalizing Membrane Rafts: New Tools and Insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 688–699. [DOI] [PubMed] [Google Scholar]
- Hammond A. T.; Heberle F. A.; Baumgart T.; Holowka D.; Baird B.; Feigenson G. W. Crosslinking a Lipid Raft Component Triggers Liquid Ordered-Liquid Disordered Phase Separation in Model Plasma Membranes. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 6320–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holowka D.; Gosse J. A.; Hammond A. T.; Han X.; Sengupta P.; Smith N. L.; Wagenknecht-Wiesner A.; Wu M.; Young R. M.; Baird B. Lipid Segregation and IgE Receptor Signaling: A Decade of Progress. Biochim. Biophys. Acta 2005, 1746, 252–259. [DOI] [PubMed] [Google Scholar]
- Day C. A.; Kenworthy A. K. Mechanisms Underlying the Confined Diffusion of Cholera Toxin B-Subunit in Intact Cell Membranes. PLoS One 2012, 7, e34923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zanten T. S.; Gomez J.; Manzo C.; Cambi A.; Buceta J.; Reigada R.; Garcia-Parajo M. F. Direct Mapping of Nanoscale Compositional Connectivity on Intact Cell Membranes. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 15437–15442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machta B. B.; Veatch S. L.; Sethna J. P. Critical Casimir Forces in Cellular Membranes. Phys. Rev. Lett. 2012, 109, 138101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump R. F.; Pfeiffer J. R.; Seagrave J.; Oliver J. M. Mapping Gold-Labeled IgE Receptors on Mast Cells by Scanning Electron Microscopy: Receptor Distributions Revealed by Silver Enhancement, Backscattered Electron Imaging, and Digital Image Analysis. J. Histochem. Cytochem. 1988, 36, 493–502. [DOI] [PubMed] [Google Scholar]
- Veatch S. L.; Chiang E. N.; Sengupta P.; Holowka D. A.; Baird B. A. Quantitative Nanoscale Analysis of IgE-FcεRI Clustering and Coupling to Early Signaling Proteins. J. Phys. Chem. B 2012, 116, 6923–6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B. S.; Pfeiffer J. R.; Surviladze Z.; Gaudet E. A.; Oliver J. M. High Resolution Mapping of Mast Cell Membranes Reveals Primary and Secondary Domains of Fc Epsilon RI and LAT. J. Cell Biol. 2001, 154, 645–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S. L.; Machta B. B.; Shelby S. A.; Chiang E. N.; Holowka D. A.; Baird B. A. Correlation Functions Quantify Super-Resolution Images and Estimate Apparent Clustering Due to over-Counting. PLoS One 2012, 7, e31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelby S. A.; Holowka D.; Baird B.; Veatch S. L. Distinct Stages of Stimulated FcεRI Receptor Clustering and Immobilization Are Identified through Superresolution Imaging. Biophys. J. 2013, 105, 2343–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spendier K.; Lidke K. A.; Lidke D. S.; Thomas J. L. Single-Particle Tracking of Immunoglobulin E Receptors (FcεRI) in Micron-Sized Clusters and Receptor Patches. FEBS Lett. 2012, 586, 416–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews N. L.; Lidke K. A.; Pfeiffer J. R.; Burns A. R.; Wilson B. S.; Oliver J. M.; Lidke D. S. Actin Restricts FcepsilonRI Diffusion and Facilitates Antigen-Induced Receptor Immobilization. Nat. Cell Biol. 2008, 10, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell S. W. Far-Field Optical Nanoscopy. Science 2007, 316, 1153–1158. [DOI] [PubMed] [Google Scholar]
- Hinterdorfer P.; Garcia-Parajo M. F.; Dufrene Y. F. Single-Molecule Imaging of Cell Surfaces Using Near-Field Nanoscopy. Acc. Chem. Res. 2012, 45, 327–336. [DOI] [PubMed] [Google Scholar]
- Levene M. J.; Korlach J.; Turner S. W.; Foquet M.; Craighead H. G.; Webb W. W. Zero-Mode Waveguides for Single-Molecule Analysis at High Concentrations. Science 2003, 299, 682–686. [DOI] [PubMed] [Google Scholar]
- Eggeling C.; Ringemann C.; Medda R.; Schwarzmann G.; Sandhoff K.; Polyakova S.; Belov V. N.; Hein B.; von Middendorff C.; Schonle A.; etal. Direct Observation of the Nanoscale Dynamics of Membrane Lipids in a Living Cell. Nature 2009, 457, 1159–1121. [DOI] [PubMed] [Google Scholar]
- Manzo C.; van Zanten T. S.; Garcia-Parajo M. F. Nanoscale Fluorescence Correlation Spectroscopy on Intact Living Cell Membranes with NSOM Probes. Biophys. J. 2011, 100, L8–L10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samiee K. T.; Moran-Mirabal J. M.; Cheung Y. K.; Craighead H. G. Zero Mode Waveguides for Single-Molecule Spectroscopy on Lipid Membranes. Biophys. J. 2006, 90, 3288–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutenegger M.; Ringemann C.; Lasser T.; Hell S. W.; Eggeling C. Fluorescence Correlation Spectroscopy with a Total Internal Reflection Fluorescence STED Microscope (TIRF-STED-FCS). Opt. Express 2012, 20, 5243–5263. [DOI] [PubMed] [Google Scholar]
- Leutenegger M.; Gosch M.; Perentes A.; Hoffmann P.; Martin O. J. F.; Lasser T. Confining the Sampling Volume for Fluorescence Correlation Spectroscopy Using a Sub-Wavelength Sized Aperture. Opt. Express 2006, 14, 956–969. [DOI] [PubMed] [Google Scholar]
- Wenger J.; Gerard D.; Lenne P.; Rigneault H.; Dintinger J.; Ebbesen T.; Boned A.; Conchonaud F.; Marguet D. Dual-Color Fluorescence Cross-Correlation Spectroscopy in a Single Nanoaperture: Towards Rapid Multicomponent Screening at High Concentrations. Opt. Express 2006, 14, 12206–12216. [DOI] [PubMed] [Google Scholar]
- Moran-Mirabal J. M.; Torres A. J.; Samiee K. T.; Baird B. A.; Craighead H. G. Cell Investigation of Nanostructures: Zero-Mode Waveguides for Plasma Membrane Studies with Single Molecule Resolution. Nanotechnology 2007, 18, 195101. [Google Scholar]
- Parthasarathy R.; Yu C.; Groves J. T. Curvature-Modulated Phase Separation in Lipid Bilayer Membranes. Langmuir 2006, 22, 5095–5099. [DOI] [PubMed] [Google Scholar]
- Tian A.; Baumgart T. Sorting of Lipids and Proteins in Membrane Curvature Gradients. Biophys. J. 2009, 96, 2676–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly C. V.; Baird B. A.; Craighead H. G. An Array of Planar Apertures for Near-Field Fluorescence Correlation Spectroscopy. Biophys. J. 2011, 100, L34–L36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun D.; Fromherz P. Fluorescence Interferometry of Neuronal Cell Adhesion on Microstructured Silicon. Phys. Rev. Lett. 1998, 81, 5241–5244. [Google Scholar]
- Suzuki K.; Masuhara H. Groove-Spanning Behavior of Lipid Membranes on Microfabricated Silicon Substrates. Langmuir 2005, 21, 6487–6494. [DOI] [PubMed] [Google Scholar]
- Bethe H. A. Theory of Diffraction by Small Holes. Phys. Rev. 1944, 66, 163–182. [Google Scholar]
- Novotny L.; Hecht B.. Principles of Nano-Optics, 2nd ed.; Cambridge University Press: Cambridge, UK, 2012. [Google Scholar]
- Ebbesen T. W.; Lezec H. J.; Ghaemi H. F.; Thio T.; Wolff P. A. Extraordinary Optical Transmission through Sub-Wavelength Hole Arrays. Nature 1998, 391, 667–669. [Google Scholar]
- Sil D.; Lee J. B.; Luo D.; Holowka D.; Baird B. Trivalent Ligands with Rigid DNA Spacers Reveal Structural Requirements for IgE Receptor Signaling in RBL Mast Cells. ACS Chem. Biol. 2007, 2, 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffman P. G.; Delbrück M. Brownian Motion in Biological Membranes. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 3111–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadurai S.; Holt A.; Krasnikov V.; van den Bogaart G.; Killian J. A.; Poolman B. Lateral Diffusion of Membrane Proteins. J. Am. Chem. Soc. 2009, 131, 12650–12656. [DOI] [PubMed] [Google Scholar]
- Gambin Y.; Lopez-Esparza R.; Reffay M.; Sierecki E.; Gov N. S.; Genest M.; Hodges R. S.; Urbach W. Lateral Mobility of Proteins in Liquid Membranes Revisited. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 2098–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenne P. F.; Wawrezinieck L.; Conchonaud F.; Wurtz O.; Boned A.; Guo X. J.; Rigneault H.; He H. T.; Marguet D. Dynamic Molecular Confinement in the Plasma Membrane by Microdomains and the Cytoskeleton Meshwork. EMBO J. 2006, 25, 3245–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger J.; Conchonaud F.; Dintinger J.; Wawrezinieck L.; Ebbesen T. W.; Rigneault H.; Marguet D.; Lenne P. F. Diffusion Analysis within Single Nanometric Apertures Reveals the Ultrafine Cell Membrane Organization. Biophys. J. 2007, 92, 913–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E.; Chichester R. J. Single Molecules Observed by Near-Field Scanning Optical Microscopy. Science 1993, 262, 1422–1425. [DOI] [PubMed] [Google Scholar]
- Veerman J. A.; Otter A. M.; Kuipers L.; van Hulst N. F. High Definition Aperture Probes for near-Field Optical Microscopy Fabricated by Focused Ion Beam Milling. Appl. Phys. Lett. 1998, 72, 3115–3117. [Google Scholar]
- Bian R. X.; Dunn R. C.; Xie X. S.; Leung P. T. Single Molecule Emission Characteristics in near-Field Microscopy. Phys. Rev. Lett. 1995, 75, 4772–4775. [DOI] [PubMed] [Google Scholar]
- Gersen H.; García-Parajó M. F.; Novotny L.; Veerman J. A.; Kuipers L.; van Hulst N. F. Influencing the Angular Emission of a Single Molecule. Phys. Rev. Lett. 2000, 85, 5312–5315. [DOI] [PubMed] [Google Scholar]
- Wu J. H. Modeling of near-Field Optical Diffraction from a Subwavelength Aperture in a Thin Conducting Film. Opt. Lett. 2011, 36, 3440–3442. [DOI] [PubMed] [Google Scholar]
- Xu H. Z.; Zhu P. S.; Craighead H. G.; Webb W. W. Resonantly Enhanced Transmission of Light through Subwavelength Apertures with Dielectric Filling. Opt. Commun. 2009, 282, 1467–1471. [Google Scholar]
- Jin J.-M.The Finite Element Method in Electromagnetics, 2nd ed.; Wiley: New York, 2002. [Google Scholar]
- Axelrod D.; Koppel D. E.; Schlessinger J.; Elson E.; Webb W. W. Mobility Measurement by Analysis of Fluorescence Photobleaching Recovery Kinetics. Biophys. J. 1976, 16, 1055–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York A. G.; Ghitani A.; Vaziri A.; Davidson M. W.; Shroff H. Confined Activation and Subdiffractive Localization Enables Whole-Cell PALM with Genetically Expressed Probes. Nat. Methods 2011, 8, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Track.m, 1999; The Matlab Particle Tracking Code Repository: http://physics.georgetown.edu/matlab/code.html (accessed Jul 7, 2014).
- Barsumian E. L.; Isersky C.; Petrino M. G.; Siraganian R. P. IgE-Induced Histamine Release from Rat Basophilic Leukemia Cell Lines: Isolation of Releasing and Nonreleasing Clones. Eur. J. Immunol. 1981, 11, 317–323. [DOI] [PubMed] [Google Scholar]
- Gosse J. A.; Wagenknecht-Wiesner A.; Holowka D.; Baird B. Transmembrane Sequences Are Determinants of Immunoreceptor Signaling. J. Immunol. 2005, 175, 2123–2131. [DOI] [PubMed] [Google Scholar]
- Larson D. R.; Gosse J. A.; Holowka D. A.; Baird B. A.; Webb W. W. Temporally Resolved Interactions between Antigen-Stimulated IgE Receptors and Lyn Kinase on Living Cells. J. Cell Biol. 2005, 171, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.