

Abstract
Growth hormone (GH) and insulin-like growth factor (IGF) signaling regulates lifespan in mice. The modulating effects of genetic background gained much attention because it was shown that life-prolonging effects in Snell dwarf and GH receptor knockout vary between mouse strains. We previously reported that heterozygous IGF-1R inactivation (IGF-1R+/−) extends lifespan in female mice on 129/SvPas background, but it remained unclear whether this mutation produces a similar effect in other genetic backgrounds and which molecules possibly modify this effect. Here, we measured the life-prolonging effect of IGF-1R+/− mutation in C57BL/6J background and investigated the role of insulin/IGF signaling molecules in strain-dependent differences. We found significant lifespan extension in female IGF-1R+/− mutants on C57BL/6J background, but the effect was smaller than in 129/SvPas, suggesting strain-specific penetrance of longevity phenotypes. Comparing GH/IGF pathways between wild-type 129/SvPas and C57BL/6J mice, we found that circulating IGF-I and activation of IGF-1R, IRS-1, and IRS-2 were markedly elevated in 129/SvPas, while activation of IGF pathways was constitutively low in spontaneously long-lived C57BL/6J mice. Importantly, we demonstrated that loss of one IGF-1R allele diminished the level of activated IGF-1R and IRS more profoundly and triggered stronger endocrine feedback in 129/SvPas background than in C57BL/6J. We also revealed that acute oxidative stress entails robust IGF-1R pathway activation, which could account for the fact that IGF-1R+/− stress resistance phenotypes are fully penetrant in both backgrounds. Together, these results provide a possible explanation why IGF-1R+/− was less efficient in extending lifespan in C57BL/6J compared with 129/SvPas.
Keywords: Genetic background, gene knockout, IGF-I, IRS, lifespan, stress resistance
Introduction
Somatotropic hormones and their downstream signaling components regulate lifespan in animals (reviewed in Kenyon, 2005, 2010; Berryman et al., 2008). Mice with mutations in genes encoding key proteins of somatotropic and metabolic endocrine pathways show extended lifespan. This is the case for Ames dwarfs, Snell dwarfs, and Little mice (Flurkey et al., 2001, 2002), GH receptor null mutants (GHRKO, Coschigano et al., 2000), heterozygous IGF-1R knockout (IGF-1R+/−; Holzenberger et al., 2003), several insulin receptor and IRS knockout mice (Blüher et al., 2003; Taguchi et al., 2007; Selman et al., 2008, 2011), and mutants with inactivation of downstream molecules like S6K1 (Selman et al., 2009). Low circulating levels of IGF-I obtained by liver-specific IGF-I inactivation (LI-IGF−/−; Svensson et al., 2011) or brain-specific IGF-1R knockout (Kappeler et al., 2008) also extend lifespan in mice. We demonstrated that the longevity effect of IGF-1R+/− mutation on 129/SvPas genetic background is sexually dimorphic, because lifespan was extended by 33% in females, while the effect was not significant in males (Holzenberger et al., 2003). Similarly, the longevity effect of low IGF-I in LI-IGF−/− mice was seen in females, but not in males (Svensson et al., 2011).
Recently, Yuan et al. (2009) showed that longevity of mouse strains is inversely correlated with circulating levels of IGF-I. Moreover, this group identified several quantitative trait loci (QTL) that are linked to circulating IGF-I level, one of which co-localizes with the igf1 gene (Leduc et al., 2010), underlining the role of genetic background in longevity determination. Meanwhile, it has been demonstrated that the effect of Pitdw/dw (Snell dwarf mutation) and GHR knockout on lifespan varies conspicuously with genetic background (Coschigano et al., 2000, 2003; Flurkey et al., 2001, 2002). In addition, the robust effect of dietary restriction, which is linked to low circulating levels of IGF-I, also depends on the mouse strain (Liao et al., 2010). This raises the question whether and how longevity effects of IGF-1R loss-of-function mutation depend on genetic background. This debate was fueled when Bokov et al. (2011) reported only 5% extension of lifespan in female IGF-1R+/− mice on C57BL/6J genetic background and unchanged lifespan in mutant males. While their results suggest that sexual dimorphism of IGF-1R lifespan regulation is conserved between 129/SvPas and C57BL/6J genetic background, they also point to possible genetic modifiers that reduce lifespan-extending effects of IGF-1R inactivation in C57BL/6J mice. We speculated that background-specific differences in IGF pathway signaling could underlie the variable penetrance of IGF-1R+/− longevity phenotypes. We hypothesized that the consequences of a signal transduction bottleneck created by heterozygous IGF-1R knockout depends on the strain-specific activity routed through this pathway. Intriguingly, impaired growth and enhanced resistance to oxidative stress, which are also characteristics for IGF-1R+/− mutants, do not vary with genetic background (Holzenberger et al., 2003; Bokov et al., 2011). To find an explanation for this dissociation of phenotypes, we investigated activation of IGF signaling under acute oxidative stress.
Results
Extended lifespan by IGF-1R+/− mutation in C57BL/6J females
We used mice with heterozygous IGF-1R knockout (IGF-1R+/−) backcrossed to C57BL/6J (B6) genetic background. Mutant males and females showed a moderate growth deficit that started at puberty (Fig. 1A), very similar to the growth trajectories previously observed in 129/SvPas (129S2) background (Holzenberger et al., 2003). At 12 weeks of age, male and female IGF-1R+/− mice were 9% lighter than their respective wild-type (WT) controls (P < 0.001, Student’s t-test). At 1 year of age, body weight was 8% lower in IGF-1R+/− females (IGF-1R+/−: 22.8 ± 0.4 vs. controls: 24.6 ± 1.2 g), and 9% lower in IGF-1R+/− males (IGF-1R+/−: 28.2 ± 0.5 vs. control: 30.9 ± 0.4 g) (both P < 0.001, Student’s t-test). Growth and final body size depend on pubertal peaks of endocrine IGF-I, when plasma IGF-I is two to three times higher than either immediately after birth or later during adulthood (Kappeler et al., 2008). Accordingly, growth trajectories of IGF-1R+/− and IGF-1R+/+ groups start to diverge at the age of pubertal growth spurt, not before (Fig. 1A–C; Holzenberger et al., 2005).
Figure 1.
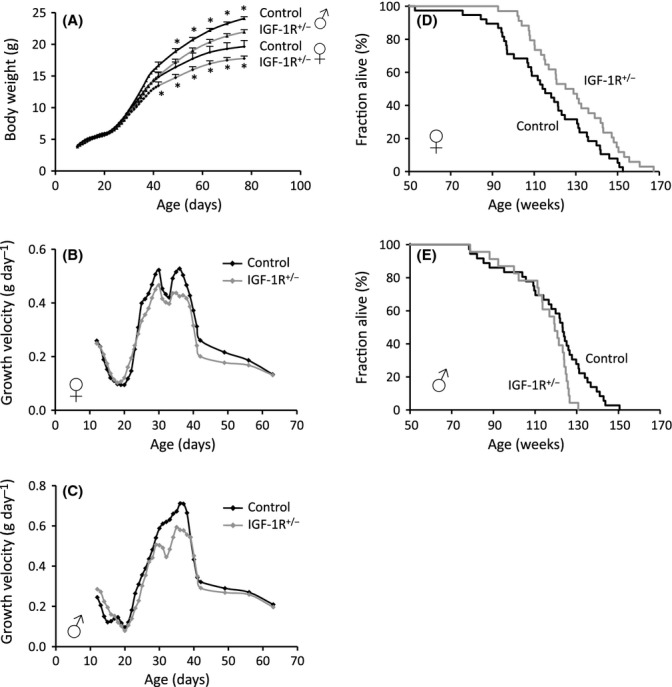
Heterozygous knockout of IGF-1R in C57BL/6J genetic background inhibits growth in both sexes and extends lifespan in females. (A) Postnatal growth in male and female IGF-1R+/− mice. Significant differences existed from 6 weeks of age onwards in females (P < 0.05) and from 7 weeks onwards in males (P < 0.01; Student’s t-test, N = 18–24, error bars represent SEM). A star indicates difference between genotypes within the same sex (P values between 0.036 and < 0.001). (B and C) Male and female growth velocity was calculated as weight gain per day using data from (A). Peaks of growth velocity at 4 weeks of age are blunted in IGF-1R+/− mutants. (D) Heterozygous knockout of IGF-1R extended lifespan in female mice. IGF-1R+/− females (gray line) lived 11% longer than controls (black) (896 ± 23 vs. 805 ± 26 day; P = 0.023, Cox regression test; P = 0.021, log-rank test). (E) IGF-1R+/− males (gray line) showed reduced maximum lifespan (803 ± 20 vs. 831 ± 22 day; P = 0.027, Cox regression test; P = 0.025, log-rank test). Detailed lifespan data and descriptive statistics are provided in Tables S1 and S2. For comparison, mean lifespan in B6 control groups from the recent literature is on average 811 days in females and 822 days in males (Table S3).
IGF-1R+/− females on B6 genetic background showed a significant 11% extension of lifespan (IGF-1R+/−: 896 ± 23 day vs. controls: 805 ± 26 day, P = 0.023; Fig. 1D). Male IGF-1R+/−, in contrast, showed no extension and in fact had shorter maximum lifespan (Fig. 1E). This demonstrated that a sexual dimorphism of IGF-1R+/− longevity effect existed also in B6 background. However, the 11% increase in lifespan observed here in B6 IGF-1R+/− females was markedly smaller than the 33% increase previously found in 129S2 females (Holzenberger et al., 2003).
Differential IGF pathway activation between strains
To investigate genetic background-specific differences in IGF signaling pathway, we first compared IGF-1R signaling between adult (3-month-old) WT mice of 129S2 and B6 genetic background. We found that circulating levels of IGF-I were significantly higher in WT 129S2 mice than in B6 mice (Fig. 2A). This difference was present in both males and females. Body length showed concordant differences between 129S2 and B6 mice (Fig. 2B), with B6 being significantly shorter. Adult females also showed consistent differences in body weight (Fig. 2C). Moreover, we detected elevated plasma levels of acid-labile subunit (ALS) in 129S2 mice (Fig. 2D), consistent with their high IGF-I. Plasma GH in contrast was lower in 129S2 mice compared with B6 (Fig. 2E), and we concluded that low endocrine IGF-I levels in B6 mice were due to constitutive differences in IGF-I expression.
Figure 2.
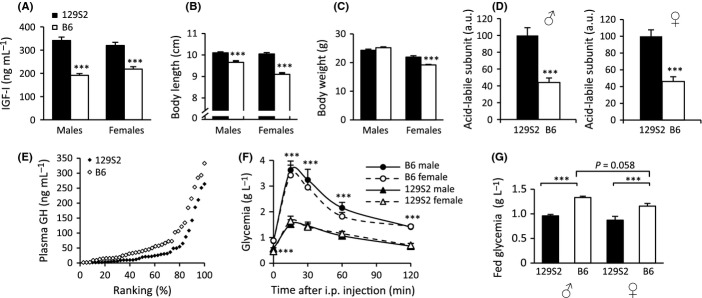
Comparing somatotropic hormones, adult body size and glucose homeostasis between wild-type (WT) B6 and 129S2 mice. (A) Plasma IGF-I concentration. ***P < 0.001, Student’s t-test; N = 18–19 per group. (B) Body length (naso-anal distance). (C) Body weight. N = 11–13 per group. (D) Plasma acid-labile subunit (ALS). ***P < 0.001, Student’s t-test; N = 9 per group. (E) Comparing plasma GH concentration by rank plot analysis (Xu et al., 2011) between WT females from B6 (open marks) and 129S2 (black marks) background (P = 0.012, log-rank test). (F) Glucose tolerance test (GTT) in male and female mice. Glucose (2 g kg−1 body weight) was intraperitoneally (i.p.) injected at T0 and glycemia measured at T0, 15, 30, 60, and 120 min. Note that all groups rapidly regained control over glycemia. ***P < 0.002 (N = 7–8 per group) comparing B6 with 129S2. Individual area under curve (AUC) between both strains is significantly different (P < 0.001, one-way ANOVA with Bonferroni’s post hoc test). (G) Glycemia in the fed state. ***P < 0.001 (N = 6–10 per group) (one-way ANOVA with Bonferroni’s post hoc test). A-E and G were performed in 11- to 13-week-old mice. Tests in panel F were performed in 2-month-old mice. Error bars indicate SEM.
IGF-I participates in metabolic regulation, namely through crosstalk with insulin signaling pathways. These pathways show strain-specific differences in glucose homeostasis marked by poor glucose tolerance in males (Kulkarni et al., 2003; Goren et al., 2004). Here, we showed strong differences in glucose homeostasis between B6 and 129S2 mice. Glucose tolerance test (GTT) revealed elevated glycemia profiles in B6 mice compared with 129S2 (Fig. 2F). Similarly, glucose levels in the fed state were significantly higher in B6 mice, whereas the highest levels occurred in B6 males (Fig. 2G). Because IGF-I is able to reduce glycemia, it is conceivable that low IGF-I in B6 contributes to the elevated blood glucose profiles.
To better understand how the observed strain-related differences in endocrine IGF-I translate into signaling activation of IGF-1R, we analyzed receptor abundance and activation in tissues of ad libitum-fed mice from WT B6 and 129S2 backgrounds using Western blot (WB). In muscle, the amount of IGF-1R protein was very similar in both strains (Fig. 3A), and this was also true for IRS-1 and IRS-2 (data not shown). However, the degree of tyrosine phosphorylation of IGF-1R, IRS-1, and IRS-2 was distinctly lower in B6 mice (Fig. 3B–D). This was the case for females (B6 to 129S2–ratio of 35, 53, and 38% for P-IGF-1R, P-IRS-1, and P-IRS-2, respectively) as well as for males (B6 to 129S2–ratio of 62, 40, and 44% for P-IGF-1R, P-IRS-1, and P-IRS-2, respectively) (Fig. 3B–D), all consistent with the low levels of IGF-I in B6 mice. We confirmed these findings in brain and lung tissues (B6 to 129S2–ratio for P-IGF-1R in the brain: females 61%, males 44%; in lung tissue from females: 43%; all results P < 0.01; N = 6 per group). Brain and lung are also major targets for IGF signaling and are implicated in somatotropic development, stress response, and survival (Ahamed et al., 2005; Kappeler et al., 2008). Taken together, we found similar IGF-1R and IRS protein abundance but significant differences in IGF-I/IGF-1R pathway activation between strains. This suggested that high circulating levels of IGF-I resulted in significantly high IGF ligand–receptor interaction and downstream signaling activity in 129S2 mice.
Figure 3.
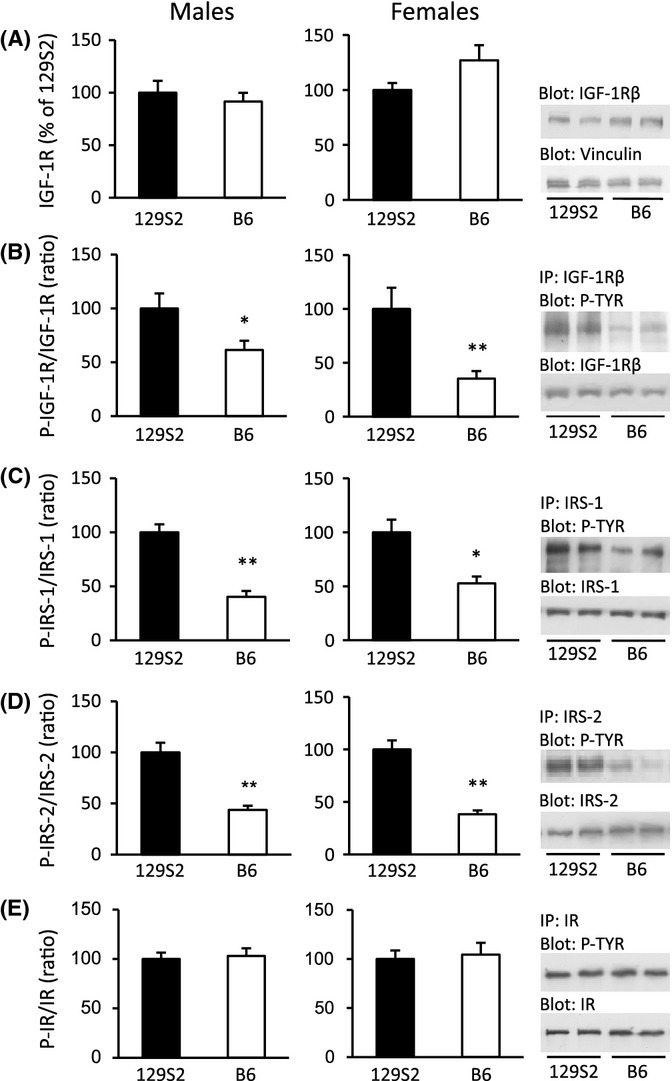
Activation of IGF-1R and substrates IRS-1 and IRS-2 is higher in WT mice of 129S2 than in WT mice of B6 genetic background, in males and females. Ad libitum-fed mice (11–13 week old) were used to recapitulate pathway activation under physiological conditions. Results shown are from skeletal muscle. Representative Western blots (WB) are displayed on the right. (A) Total IGF-1R; vinculin was used as loading control. (B) Phospho-IGF-1R (P-IGF-1R), (C) P-IRS-1, (D) P-IRS-2, and (E) P-IR were detected by immunoprecipitation (IP) using specific antibodies, followed by WB using an anti-phosphotyrosine (P-TYR) antibody. Signals of activated protein were expressed relative to total protein. N = 6 per group, in males and females; *P < 0.05, **P < 0.01, Student’s t-test; error bars represent SEM.
IGF-1R is structurally and functionally related to insulin receptor (IR) and both share substrates (LeRoith & Yakar, 2007). To check whether insulin receptors also show differential activation, we measured IR abundance and prevalence of P-IR, but found both to be unchanged in WT B6 and 129S2 mice (Fig. 3E). Moreover, plasma insulin in the fed state was similar in females of both strains (129S2: 0.85 ± 0.19 vs. B6: 0.98 ± 0.17 ng mL−1, NS; N = 16–18 per group) and was elevated in B6 males (129S2: 0.65 ± 0.11 vs. B6: 1.43 ± 0.18 ng mL−1, P < 0.001; N = 15–16 per group). Thus, insulin receptors and insulin were unlikely to contribute significantly to the observed strain-specific activation of IGF-1R and downstream pathways.
Differential effect of IGF-1R+/− mutation on IGF signaling in B6 and 129S2 background
We then asked how experimentally induced decrease in IGF-1R abundance interacts with the strain-specific molecular settings of the IGF axis. For this, we used the heterozygous IGF-1R+/− mutants maintained on B6 and 129S2 genetic background and compared them with their respective IGF-1R+/+ littermate controls. IGF-1R signaling was analyzed using immunoprecipitation (IP) of the receptor followed by WB, in 11- to 13-week-old males and females separately (Fig. 4A). As expected, IGF-1R abundance was identical in both backgrounds, and heterozygous knockout of IGF-1R diminished the receptor abundance to half of the wild-type levels (Fig. 4B). No difference between genetic backgrounds existed in this regard. We then measured the phosphotyrosine activation of IGF-1R in these mice (Fig. 4C). Activation of IGF-1R in WT mice of 129S2 background was stronger compared with B6, confirming the findings in Fig. 3B. Importantly, inactivation of one IGF-1R allele entailed a two to three times stronger reduction in phospho-IGF-1R levels in 129S2 background compared with the corresponding reduction in B6 background (Fig. 4C). This effect occurred very similarly in males and in females. We then calculated the degree of phosphorylation of IGF-1R (Fig. 4D). This showed that mutant and control groups in 129S2 genetic background had a significantly higher P-IGF-1R/IGF-1R ratio than mutant and control groups in B6 genetic background. However, no significant differences existed between mutant and control groups within the same background.
Figure 4.
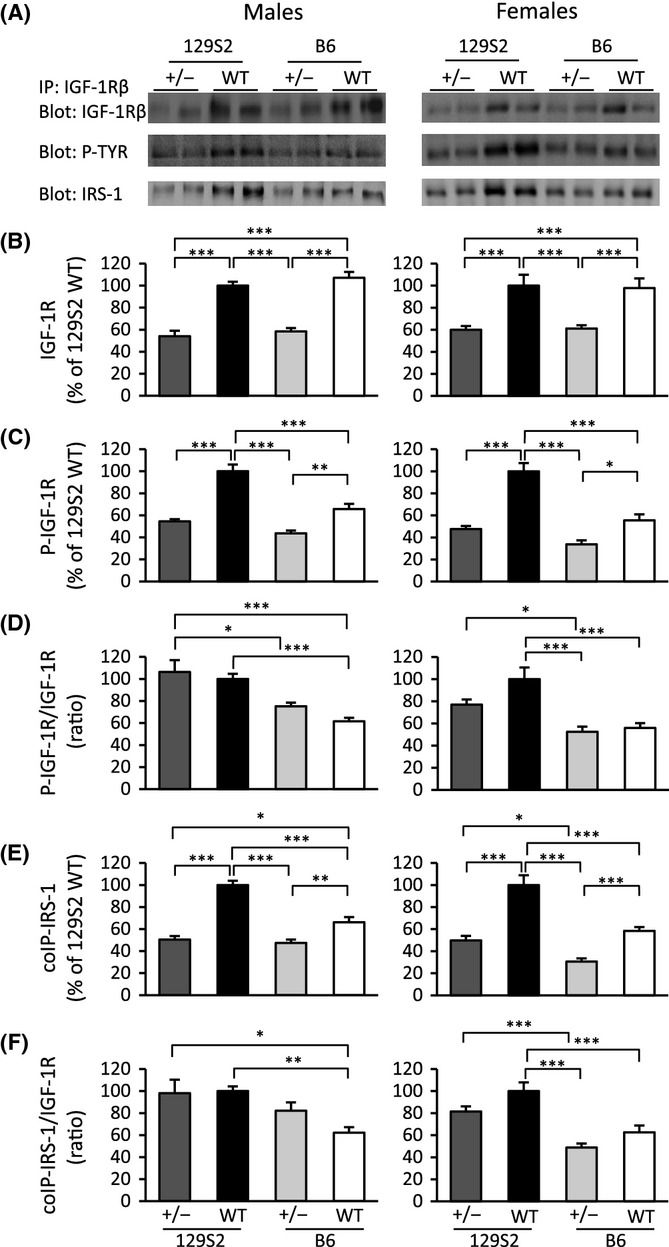
IGF-1R phosphotyrosine activation and IRS-1 recruitment differ significantly between 129S2 and B6 mice of IGF-1R+/− (+/−) and WT genotype. Ad libitum-fed 11- to 13-week-old mice were used. (A) Representative WB results. (B) Prevalence of IGF-1R revealed by IP and subsequent WB. (C) Phospho-IGF-1R (P-IGF-1R) detected from total immunoprecipitated IGF-1R using phosphotyrosine-specific antibody (P-TYR). (D) P-IGF-1R relative to total immunoprecipitated IGF-1R. (E) IRS-1 co-immunoprecipitating with IGF-1R shows a profile similar to (C). (F) Expressing co-immunoprecipitated IRS-1 relative to IGF-1R shows a profile similar to (D). In each graph, the mean of WT 129S2 mice was arbitrarily set to 100, and results from other groups expressed relative to that. N = 7–12 per group; *P < 0.05, **P < 0.01, ***P < 0.001, in one-way ANOVA with Bonferroni’s post hoc test; error bars represent SEM.
Activated IGF-1R recruits IRS to trigger downstream signal transduction cascades. This interaction can be revealed by measuring the amount of IRS-1 co-immunoprecipitating (coIP) with IGF-1R. The IRS-1 coIP pattern among heterozygous and WT mice (Fig. 4E,F) plainly confirmed the phospho-IGF-1R pattern (Fig. 4C,D) in both sexes. Collectively, these results showed that quantitative aspects of IGF-1R pathway activation depend on genetic background. Data also suggested that interaction between IGF-1R abundance and activation of signaling differed slightly between males and females, although these sex-specific differences did not reach levels of significance.
Data in Figs 3 and 4 showed that IGF-1R pathway activation changed with endocrine IGF-I levels, which were high in 129S2 and low in B6 mice (Fig. 2A). We concluded that under high IGF-1R stimulation, heterozygous receptor knockout strongly decreases phosphotyrosine-activated IGF-1R. In contrast, when IGF-1R stimulation is low, heterozygous knockout engenders much smaller, although still significant decreases in activated IGF-1R. Similar to our previous findings in 129S2 background, 4-month-old IGF-1R+/− mutants on B6 background showed upregulated circulating IGF-I (+9%, P = 0.032; Student’s t-test; N = 15 for each group), yet the effect was smaller than previously reported in 129S2, suggesting reduced endocrine feedback in IGF-1R+/− mutants on B6 background. All together, these findings suggest that activation of IGF signaling is less affected in B6 IGF-1R+/− mice than in 129S2 IGF-1R+/− mice, which can explain the observed differences in lifespan extension between backgrounds.
Response to acute oxidative stress involves peak levels of IGF pathway activation
While longevity effects vary, somatic growth and stress resistance phenotypes of IGF-1R+/− are fully penetrant in both backgrounds: measured 72 h after paraquat injection, survival to paraquat increased from 52% in WT to 75% in IGF-1R+/− on 129S2 background (P < 0.05; Holzenberger et al., 2003) and from 50 to 74% on B6 background (P < 0.001; Bokov et al., 2011). As shown in Fig. 4, reduction in receptor levels in IGF-1R+/− mutants has limited inhibitory effect on downstream signaling in B6 mice under basal conditions, but we speculated that peak concentrations in IGF-I hormone are able to reveal differences in capacity of pathway activation between IGF-1R+/− and WT mice. Postnatal growth spurt in juvenile mice is induced by maximal IGF-1R pathway activation, but it is not known whether this is also the case for the response to oxidative stress. To test this, we challenged 3-month-old wild-type mice with paraquat and analyzed plasma IGF-I and downstream signaling (Fig. 5A). Within 48 h under acute oxidative stress, endocrine IGF-I tripled, total IGF-1R abundance increased significantly in lung tissue (IP: +48%, WB: +54%; P < 0.05), and levels of phosphotyrosine-activated IGF-1R increased as well (+155%) (P < 0.05; Fig. 5B). Downstream of IGF-1R, both Akt and Erk levels increased significantly, while IRS-1 showed moderate depletion. We detected several-fold increases in phospho-activation of IRS-1 (+108%), Akt (+152%), and Erk (+370%) after 48 h of stress, proof that strong IGF-1R pathway activation is a hallmark of response to oxidative stress. In addition, low activation of p38 facilitates IGF-1R signaling (Dávila & Torres-Aleman, 2008), and we showed here that levels of activated P-p38 diminished under paraquat (Fig. 5). We obtained similar results in muscle (not shown). Collectively, we demonstrated that IGF-1R signaling was strongly activated under oxidative stress, a situation similar to IGF pathway activation during juvenile growth spurt.
Figure 5.
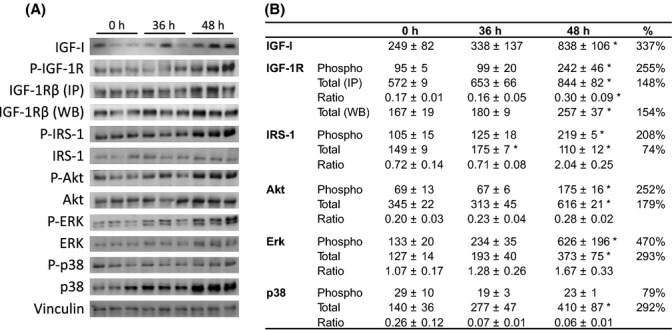
Activation of IGF signaling pathways 36 and 48 h after i.p. injection of paraquat. (A) WB detection of IGF-I from blood and of key signal transduction proteins in IGF pathways (P-tyrosine-activated forms and total protein) from lung tissue. Gel loading was controlled by vinculin. (B) Quantification of WB by chemiluminescence. Phosphotyrosine-IGF-1R (P-IGF-1R) and P-IRS-1 were detected using a phosphotyrosine-specific antibody after IP. Note that increase in IGF-1R abundance over time was very similar whether detected from IP samples or by direct WB. Tests were performed in 11- to 13-week-old mice. N = 3 per group; mean ± SEM, expressed in arbitrary units. *P < 0.05, Mann–Whitney U-test.
Finally, with respect to metabolic phenotype, we confirmed that GTT and ITT profiles are normal in adult (4- to 6-month-old) IGF-1R+/− males and females on B6 background (Supplementary Fig. S1), aspects that have been thoroughly investigated by Bokov et al. (2011) and Garg et al. (2011). Fed glycemia and insulin levels in control males were high compared with females, indicating insulin resistance in males on B6 background.
Discussion
We showed here that heterozygous knockout of IGF-1R on B6 background extended female lifespan significantly, albeit to a lesser degree than on 129S2 background (Holzenberger et al., 2003). This finding is in line with previous reports, showing that diminished endocrine IGF-I favors longer lifespan (Kappeler et al., 2008; Svensson et al., 2011; Yuan et al., 2012). We also showed that partial IGF-1R inactivation is less efficient in inhibiting IGF-1R signaling in B6 than in 129S2 background. Moreover, comparing the mean lifespans of mice of B6 and 129S2 backgrounds suggested that IGF-1R activation level is inversely related to strain-specific longevity. This result is compatible with the work by Yuan et al. (2009) revealing an inverse correlation between adult (6-month-old) endocrine IGF-I levels and mean lifespan in a panel of mouse strains. Data from Yuan et al. also confirm sexual dimorphism, as the correlation between IGF-I and lifespan is significant for females, but not for males. These authors also studied B6 mice that carried an IGF-I allele backcrossed from C3H genetic background (Yuan et al., 2012). High IGF-I levels from this allele decreased lifespan in B6 females, but not in males. Flurkey et al. (2001, 2002) and Coschigano et al. (2000, 2003) had shown that genetic background modulates the life-prolonging effect of mutations in somatotropic genes upstream of IGF-I. Notably, the increase in lifespan in GHRKO mice relative to the control group was roughly twice as strong on mixed 129/Ola-Balb/cJ background than on B6 (Coschigano et al., 2000, 2003). At the same time, authors found 60% higher plasma IGF-I in control groups of 129/Ola-Balb/cJ background compared with B6. Here, we demonstrated that the 129S2 strain has high circulating IGF-I levels. Conversely, comparing 129S2 longevity (Holzenberger et al., 2003) with other strains, Yuan et al. (2009) showed that 129S2 longevity is at the lower quantile. Thus, it appears that a short-lived strain with high endocrine IGF-I and elevated IGF-1R activation has a higher potential for lifespan extension than a spontaneously long-lived strain with low IGF-I. Yuan et al. (2012) showed that B6 is one of, if not, the most long-lived inbred strains, enhancing the import of the observed 11% increase in B6 lifespan by IGF-1R+/− mutation.
While present results show that partial IGF-1R inactivation extends lifespan in females on a long-lived genetic background, they also allow us to draw conclusions with respect to endocrine mechanisms. Genes in the GH/IGF axis act in networks, such that longevity effects of mutations inhibiting this pathway likely depend on strain-specific molecular context. Our finding that diminishing IGF-1R levels had stronger effects on IGF signaling in 129S2 than in B6 background suggested that signaling activity in this pathway is primarily determined by the prevalence of activated forms of receptor and substrates, but not by their total abundance. Our finding that IGF-1R+/− mutation was less efficient in extending longevity in a low IGF-I background is in accord with the fact that both IGF-I and its receptor regulate lifespan. Indeed, the mechanisms of genetic epistasis predict that for genes acting in the same pathway, the combined effects of inactivating mutations are smaller than the sum of effects when they are measured for each component separately (reviewed in Cordell, 2002). Consequently, in a strain with low IGF-I, the loss of one IGF-1R allele does not represent the same bottleneck effect as in a mouse strain with high circulating IGF-I. This seems to also apply to decreases in IGF-I provoked by dietary restriction. Combined effects of dietary restriction and GHRKO on lifespan are much smaller than the sum of their separate effects (Bonkowski et al., 2006). Finally, while it seems natural that in IGF-1R+/− mice, IGF-I is involved in differential activation of IGF-1R, other effectors such as protein tyrosine phosphatases (PTPs) and IGF binding proteins (IGFBP-1 to IGFBP-6) could also play a role. However, it should be noted that none of these possibilities seems as powerful as the regulation by the cognate ligand IGF-I itself.
Using an equivalent IGF-1R+/− knockout mouse (Liu et al., 1993) on B6 genetic background, Bokov et al. (2011) reported that female mutants lived longer than controls (+5%, P = 0.02 by log-rank test). However, the increase in lifespan was noticeably smaller than our original observation on 129S2 background. Bokov et al. mistakenly compared our 129S2 longevity data with a different strain, 129/SvImJ (129S1), and speculated that our original survival experiments were carried out under stressful conditions, presumably favoring premature death. This explanation is unproven and is unlikely. Our 129S2 and B6 experiments were performed under similar conditions to ensure comparability, and lifespan of our B6 control population is similar to data from other laboratories (see Table S3 and methods for details). Admittedly, a problem arises from the fact that no reference data are available for 129S2 and that many genetically divergent substrains were developed from the original 129 mouse strain through extensive use, notably for ES-cell derivation and KO mouse production. These strains currently present with different lifespans (Table S4) (Simpson et al., 1997; Threadgill et al., 1997; Petkov et al., 2004; Yuan et al., 2009, 2012). Yuan et al. (2009) showed that circulating IGF-I varies up to 3-fold among mouse strains and that this is correlated with strain-specific longevity, which varies from < 300 days to more than 900 days. Median lifespan, even in closely related strains, can differ by more than a 100 days.
Besides genetic background, differences in longevity among laboratories can originate from environmental factors, for example breading protocols, diet, or litter size (Taguchi & White, 2008; Kappeler et al., 2009). We showed that it is possible to modify the development and adult function of somatotropic hormone axis through early life nutrition (Kappeler et al., 2009). Interestingly, the control females from the Bokov study lived on average 923 days (males: 983 days), which is considerably longer than B6 lifespan reported by most laboratories (Table S3). One may ask whether controls in the Bokov study exhibit a particularly low somatotropic tone. That would explain both their outstanding longevity and also the limited beneficial effect of IGF-1R+/− mutation. A direct comparison of IGF-1R activation from their controls could clarify this issue. A useful clue may also be the age at weaning, which is 21 days in the Bokov study, while we keep litters with mothers until day 30, as recommended for offspring with growth deficiency. Early weaning can impair learning of eating behavior and slow down somatic development, especially during periods of maximum growth in juvenile animals. Diet composition is another important factor, and note should be taken that chow we provided contains higher proportion of protein (22%) than NIH-31 (18%), used by Bokov et al. (2011). Further inspection reveals that Bokov’s and our findings do fit into a bigger picture (Fig. 6), regardless of animal husbandry or health status, as Richardson and colleagues speculated (Liang et al., 2003; Ladiges et al., 2009; Bokov et al., 2011). In supplementary results, Bokov et al. present additional survival data from females on a hybrid B6/129 background, where median lifespan was 1009 days for controls, and no effect of IGF-1R+/− mutation was observed. In Fig. 6, this result marks the point where heterozygous inactivation of IGF-1R would no longer produce any life-prolonging effect in females. Bokov et al. predicted a ‘stressor’ that would explain the comparatively low mean lifespan of our 129S2 controls. Evidence from present work suggests that this ‘stressor’ is in fact endocrine IGF-I and IGF-1R activation.
Figure 6.
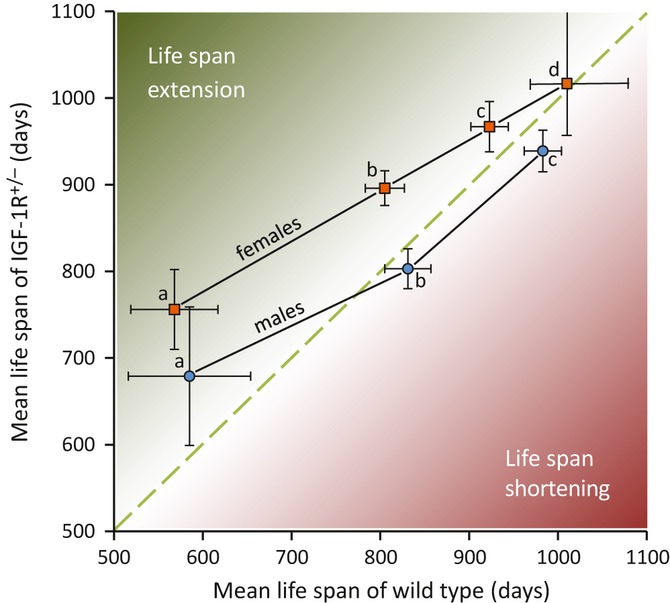
Schematic illustration suggesting that heterozygous IGF-1R knockout extends lifespan depending on longevity of genetic background and on sex. Data are from Holzenberger et al., 2003 (a, 129S2), Xu et al. (b, B6; this paper), and Bokov et al. (2011) (c, B6; d, hybrid B6/129). Data are used to compare mean lifespan of IGF-1R+/− mutant populations (y-axis) with mean lifespan of corresponding WT control populations (x-axis). In d, median lifespan was used. Error bars represent SEM in a, b, and c, and 95% confidence interval in d. In females, lifespan-extending effect of heterozygous IGF-1R knockout tends to diminish with increasing mean lifespan of WT control. Male populations show a similar trend, but seem to be exposed to adverse, life-shortening effects of IGF receptor inactivation as mean lifespan increases. This schematic representation raises the question whether stronger IGF-1R gene inactivation further extends longevity, in spontaneously long-lived strains or under environmental conditions that already favor long lifespan. It also suggests that DR may extend lifespan more efficiently in 129S2 than in B6 (see also Swindell, 2012).
Another pertinent question is how differences in glucose metabolism between B6 and 129S2 strains can interfere with longevity effects. Intriguingly, we found that maximum lifespan was reduced in IGF-1R+/− males with B6 background. We showed previously that heterozygous IGF-1R knockout decreases glucose tolerance in males, while it ameliorates glucoses tolerance in females (Holzenberger et al., 2003). Here, B6 males showed high glycemia and high insulin levels indicating insulin resistance, and it seems possible that male IGF-1R+/− mutants are more susceptible to diabetic dysregulation, especially as they grow older, which may account for their longevity phenotype (Holzenberger et al., 2003; Garg et al., 2011; and this manuscript). Insulin receptor (IR) activation on the other hand was visibly unaffected (Fig. 3E). Withers et al. (1999) showed by intercrossing mice heterozygous for null alleles of Igf1r and Irs-2 that Igf1-Irs2 signaling is a key in the development and maintenance of pancreatic β cell mass and endocrine function. Consequently, a downregulation of IRS-2 signaling pathway or a reduced expression of Igf1r can explain peripheral insulin resistance. Kido et al. (2002) showed that IGF-1R+/− did not alter glucose homeostasis as long as insulin receptor signaling remained intact. Together, this may explain why deleterious effects of IGF-1R inactivation develop progressively in aged animals. It might be interesting to perform caloric restriction in IGF-1R mutants and investigate IGF-I signaling during refeeding, even though short-term fasting and refeeding does not normally affect IGF-I levels (Lewitt et al., 2001). In mice with muscle-specific GHR knockout, GH signaling plays a role in muscle lipid oxidation in basal conditions, but not after fasting–refeeding (Vijayakumar et al., 2012). One may ask whether a similar mechanism is at work in IGF-1R inactivation. High GH levels in contrast may interfere with the metabolic and longevity phenotype of IGF-1R+/− mice. Both IGF-1R+/− and liver-specific IGF−/− mice (Sjögren et al., 1999; Yakar et al., 1999) show increased circulating GH levels and at the same time develop a sexual dimorphism in terms of longevity, where males benefit less or not at all from gene mutation. Yakar et al. (2004) reported that inhibition of GH action improves insulin sensitivity in liver-specific IGF-I-deficient mice, and it is therefore possible that metabolic and longevity phenotypes are connected via enhanced GH action on glucose homeostasis. Finally, male and female secretory patterns of GH determine sex-dependent gene expression in the rodent liver that may explain adverse effects of IGF-1R+/− mutation on male survival at advanced ages (Ahluwalia et al., 2004; Waxman & Holloway, 2009).
We found it intriguing that IGF-1R+/− mutation induces stress resistance and inhibits somatic growth to a similar extent in both B6 and 129S2 genetic backgrounds, while the longevity phenotypes were quite different. Increased stress resistance is a common feature of mutants with attenuated IGF signaling, from worms to mice (Berryman et al., 2008), but the mechanism is still not clear. We showed that IGF signaling is hyperactive under oxidative stress, and thus, it seems that IGF-1R+/− mutation limits damage and ameliorates survival by blocking acute overreaction to IGF-I. Similarly, growth spurts are blunted in IGF-1R+/− mutants because the pubertal peak of IGF-I is no longer able to stimulate maximal growth, thereby forcing individuals to leave their growth trajectory (Holzenberger et al., 2005). Thus, stress response and somatic growth are both determined by markedly high IGF-I levels. In contrast to promote organismal longevity, it seems pivotal to reduce metabolic activity and to keep protein turn over and cell renewal low. Hence, a perfect IGF-I longevity allele would be a weak one that ensures low basal hormone levels and limited inducibility, probably at the expense of maximum somatic growth potential. We showed here that IGF-1R+/− mutation inhibits signaling less efficiently under low-level IGF stimulation than during high IGF-I, which would explain the dissociation of the mutant phenotype between genetic backgrounds. Together, our results suggest that stress resistance may be uncoupled from longevity and that it may be the low basal levels of IGF-1R activation that determine longevity, yet this awaits experimental confirmation.
In conclusion, several independent experiments showed that IGF-I and IGF-1R regulate lifespan in mammals. In particular, the dose-dependent effects illustrated in Fig. 6 confirm that the IGF-1R+/− mouse is a model of increased longevity as predicted by mutations of IGF-1R homologues in invertebrates. Here, we point out the modulating effect of genetic background on IGF-1R signaling in gene knockout experiments. In view of these results, we suggest a more systematic look into IGF-1R activation, as a complement to measuring endocrine IGF-I levels. Investigating the role of the mammalian IGF system in longevity is of prime importance because heterozygous mutations have been identified in human centenarians (Suh et al., 2008), and several studies found association between exceptionally long human lifespan and gene variants of AKT1, FOXO1A, and FOXO3A (Willcox et al., 2008; Flachsbart et al., 2009; Li et al., 2009; Pawlikowska et al., 2009; Soerensen et al., 2010), together suggesting that these pathways also control human aging.
Experimental procedures
Mouse genetics and husbandry
IGF-1R+/− mutant mice were produced and maintained on 129/SvPas genetic background (Holzenberger et al., 2000, 2003) and backcrossed to wild-type (WT) C57BL/6J mice for > 15 generations. WT mice of pure C57BL/6J (B6) and 129/SvPas (129S2) genetic background were from Charles River Laboratories (L’Arbresle, France). For lifespan studies and analysis of growth, IGF-1R+/− mice and their littermate controls were produced by mating B6 IGF-1R+/− males to WT B6 females. For signaling studies and biochemistry, IGF-1R+/− mice and littermate controls were produced by mating B6 IGF-1R+/− males to WT B6 females and by mating 129S2 IGF-1R+/− males to WT 129S2 females. Litter size was trimmed to 5–8 pups (mean 6.6 ± 1.5 SD). We separated mice from mothers on day 30 and housed six males or six females per cage, each containing control and mutant animals. Mice lived under SPF conditions in a barrier facility in individually ventilated cages (Tecniplast, Milan, Italy). Sentinel mice (8–12 per room and year) were tested at Harlan Laboratories following FELASA recommendations. No infections or pathogens were detected. Mice were kept at 23°C, with a 14/10-h light/dark cycle, and had free access to water and rodent chow, containing 22% proteins and 4% lipids (Special Diet Services, Essex, UK). Cages were equipped with mouse houses (Tecniplast) to prevent male aggressiveness. All animal experiments were conducted according to the EC Council Directive (86/609/EEC).
Lifespan studies
Mice in the lifespan cohort were checked daily, but otherwise left undisturbed until they died from natural causes. Single surviving females were housed with neighbors. Importantly, WT littermates served as age- and sex-matched internal controls. Three individuals (2.3%) died at young age: one WT male at 21 days and two WT females at 80 and 119 days, respectively. This early mortality was supposedly caused by developmental defects or accident, and not included in lifespan analyses. Lifespan studies in 129S2 (Holzenberger et al., 2003) and B6 mice (this report) were performed in the same facility under identical conditions with respect to air conditioning, temperature, luminosity, light–dark cycle, cage size and occupation, ad libitum access to food and water, age of weaning, and animal care. Differences between studies were use of conventional cages (129S2) vs. IVC (B6), 24% (129S2) vs. 22% (B6) protein in diet, and more uniform litter size in B6. In 129S2 mice, lifespan was measured under clean conventional conditions (Holzenberger et al., 2003), while B6 lifespan was measured under SPF barrier. With respect to reproducibility of lifespan measurements in the facility, mean lifespan in control groups of long-lived strains (B6 and B6/129S2 F1 hybrids) has been stable over the last 10 years in male (831 ± 22 day, 853 ± 43 day, 851 ± 27 day) and female populations (805 ± 26 day, 821 ± 36 day, 789 ± 27 day) (see also Supporting Information).
Postnatal growth
To measure growth, mice were weighed daily until postnatal day 42 and sliding means from current weight and weight on preceding and subsequent day computed. Mice were anesthetized by 40 mg/kg pentobarbital i.p., to measure nose-rump length at 11–13 weeks.
Blood sampling and hormone determinations
Blood was collected from ocular sinus in conscious 11- to 13-week-old mice using topical anesthetic and EDTA, cooled on ice, centrifuged within 15 min, and plasma was frozen. For GH detection, we used the protocol of Xu et al. (2011). Samples were drawn immediately after moving the cage (within 20 sec) to minimize the effect of stress. Plasma GH was measured using ELISA (DSL-10-72100, Beckman Coulter, Brea CA, USA). Plasma IGF-I was measured by a single-plex immunoassay (RMIGF187K, Millipore, Billerica MA, USA) and mouse IGF-I Quantikine ELISA (MG100; R&D Systems, Minneapolis MN, USA). Plasma insulin was measured by ELISA (EZRMI-13K, Millipore).
Glucose metabolism
Glycemia was measured in tail blood using OneTouch Ultra (Lifescan, Issy-les-Moulineaux, France). For glucose tolerance test (GTT), animals were 14 h fasted and i.p. injected with 20% D-glucose (2 g kg−1 body weight). For insulin tolerance test (ITT), fed animals were i.p. injected with 1 U insulin (I9278, Sigma) per kg body weight.
Tissue sampling
IGF-1R pathway activation was analyzed under physiological conditions in ad libitum-fed, 11- to 13-week-old mice, when IGF-I levels are stable (Kappeler et al., 2008). For protein extraction, tissue samples from quadriceps muscle, cerebral cortex and lung from IGF-1R+/− mice and WT littermate controls, on B6 and 129S2 genetic background, respectively, were quickly dissected and immediately snap-frozen over liquid nitrogen and stored at −80°C. To study IGF-1R pathway activation under oxidative stress, 11- to 13-week-old mice received i.p. injection of 60 mg paraquat (Sigma) per kg body mass. Mice were sacrificed 36 and 48 h after injection and compared with noninjected controls. Tissues were sampled as described.
Immunoblotting
Western blot (WB) and immunoprecipitation (IP) from tissue were performed as described in Dupont et al. (2000). Activated forms of IGF-1R, IR, IRS-1, and IRS-2 were detected after IP using anti-IGF-1R β-subunit (C-20, Santa Cruz Biotechnology, Santa Cruz CA, USA), anti-IR (Transduction Laboratories, BD Biosciences, Franklin Lakes NJ, USA), anti-IRS-1, and anti-IRS-2 (Upstate Biotechnology Inc, Lake Placid NY, USA) antibodies, followed by WB with anti-phosphotyrosine antibodies (PY20, Transduction Laboratories). P-Akt, Akt, and P-Erk (Cell Signaling Technology, Danvers MA, USA), Erk and p38 (Santa Cruz Biotechnology), and P-p38 (Cell Signaling Technology) were determined by WB. Equal loading was controlled by determining vinculin using antivinculin antibody (Sigma). For all WB, bound antibodies were revealed using peroxidase-conjugated secondary antibodies and ECL (Amersham Pharmacia Biotech, Orsay, France). Signals were quantified using MacBas 2.5 (Fujifilm, Bois d’Arcy, France).
For WB of IGF-I (Fig. 5), 1.0 μL plasma samples diluted in reducing Laemmli buffer was resolved by PAGE (4–20% Criterion TGX; Bio-Rad) and transferred to PVDF membranes. Blocked membranes were incubated with anti-IGF-I primary antibody (AF791, 1:1000; R&D systems) for 3 h at room temperature (RT), washed in TBS, and incubated for 60 min at RT with horseradish peroxidase-labeled secondary rabbit anti-goat antibody (A5420, 1:5000; Sigma). Blots were analyzed using ECL (RPN2232, GE Healthcare), ChemiDoc and Quantity One 4.2.1 (Bio-Rad, Hercules CA, USA). Plasma ALS was assayed using WB and anti-ALS antibody (AF1436, R&D Systems) as described by Kappeler et al. (2008). Signals were revealed with ECL (Novex, Invitrogen, Cergy Pontoise, France).
Statistical analysis
Two-tailed Student’s t-test, Mann–Whitney U-test, and one-way ANOVA with Bonferroni’s post hoc test were performed to determine statistical significance of differences between groups. GH pattern was analyzed by log-rank test. Survival curves were evaluated by Cox’s regression and log-rank test. Tests were performed using SPSS (SPSS Institute Inc., Chicago, USA). Results are presented as means ± SEM.
Acknowledgments and funding
We thank Julianne Rieders for language revision. ANR (grant NT05-3 42491) and EU NoE (036894) sponsored this study with grants to M.H. FRM and SFEDP supported J.X. MENRT supported G.G. AXA Research Fund supported Z.C.
Author contributions
J.X., J.D., and M.H. designed experiments. J.X., J.D., G.G., Z.C., P.L., and M.H. performed experiments and analyzed data. J.X. and M.H. wrote the manuscript. All authors discussed results and commented on the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
Fig. S1 Glucose homeostasis in 4- to 6-month-old B6 IGF-1R+/−mice.
Table S1 Data of Figure 1D and E (lifetime survival).
Table S2 Descriptive statistics of lifespan and survival data from male and female IGF-1R+/− and control mice in B6 genetic background (Data correspond to Figure 1, Panel D and E).
Table S3 Mean lifespan of B6 control mice in recent studies of mouse longevity genes. Highest and lowest means are in bold.
Table S4 Longevity of male and female mice from strains derived from original 129 mice.
References
- Ahamed K, Epaud R, Holzenberger M, Bonora M, Flejou JF, Puard J, Clement A, Henrion-Caude A. Deficiency in type 1 insulin-like growth factor receptor in mice protects against oxygen-induced lung injury. Respir. Res. 2005;6:31. doi: 10.1186/1465-9921-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahluwalia A, Clodfelter KH, Waxman DJ. Sexual dimorphism of rat liver gene expression: regulatory role of growth hormone revealed by deoxyribonucleic acid microarray analysis. Mol. Endocrinol. 2004;18:747–760. doi: 10.1210/me.2003-0138. [DOI] [PubMed] [Google Scholar]
- Berryman DE, Christiansen JS, Johannsson G, Thorner MO, Kopchick JJ. Role of the GH/IGF-1 axis in lifespan and healthspan: lessons from animal models. Growth Horm. IGF Res. 2008;18:455–471. doi: 10.1016/j.ghir.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blüher M, Kahn B, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- Bokov AF, Garg N, Ikeno Y, Thakur S, Musi N, DeFronzo RA, Zhang N, Erickson RC, Gelfond J, Hubbard GB, Adamo ML, Richardson A. Does reduced IGF-1R signaling in Igf1r+/− mice alter aging? PLoS ONE. 2011;6:e26891. doi: 10.1371/journal.pone.0026891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkowski MS, Rocha JS, Masternak MM, Al Regaiey KA, Bartke A. Targeted disruption of growth hormone receptor interferes with the beneficial actions of calorie restriction. Proc. Natl Acad. Sci. USA. 2006;103:7901–7905. doi: 10.1073/pnas.0600161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell HJ. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum. Mol. Genet. 2002;11:2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- Coschigano KT, Clemmons D, Bellush LL, Kopchick JJ. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology. 2000;141:2608–2613. doi: 10.1210/endo.141.7.7586. [DOI] [PubMed] [Google Scholar]
- Coschigano KT, Holland AN, Riders ME, List EO, Flyvbjerg A, Kopchick JJ. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased lifespan. Endocrinology. 2003;144:3799–3810. doi: 10.1210/en.2003-0374. [DOI] [PubMed] [Google Scholar]
- Dávila D, Torres-Aleman I. Neuronal death by oxidative stress involves activation of FOXO3 through a two-arm pathway that activates stress kinases and attenuates insulin-like growth factor I signaling. Mol. Biol. Cell. 2008;19:2014–2025. doi: 10.1091/mbc.E07-08-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont J, Karas M, LeRoith D. The potentiation of estrogen on insulin-like growth factor I action in MCF-7 human breast cancer cells includes cell cycle components. J. Biol. Chem. 2000;275:35893–35901. doi: 10.1074/jbc.M006741200. [DOI] [PubMed] [Google Scholar]
- Flachsbart F, Caliebe A, Kleindorp R, Blanché H, von Eller-Eberstein H, Nikolaus S, Schreiber S, Nebel A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl Acad. Sci. USA. 2009;106:2700–2705. doi: 10.1073/pnas.0809594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Life span extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc. Natl Acad. Sci. USA. 2001;98:6736–6741. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Harrison DE. The Snell dwarf mutation Pit1(dw) can increase life span in mice. Mech. Ageing Dev. 2002;123:121–130. doi: 10.1016/s0047-6374(01)00339-6. [DOI] [PubMed] [Google Scholar]
- Garg N, Thakur S, McMahan CA, Adamo ML. High fat diet induced insulin resistance and glucose intolerance are gender-specific in IGF-1R heterozygous mice. Biochem. Biophys. Res. Commun. 2011;413:476–480. doi: 10.1016/j.bbrc.2011.08.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goren HJ, Kulkarni RN, Kahn CR. Glucose homeostasis and tissue transcript content of insulin signaling intermediates in four inbred strains of mice: C57BL/6, C57BLKS/6, DBA/2, and 129X1. Endocrinology. 2004;145:3307–3323. doi: 10.1210/en.2003-1400. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Leneuve P, Hamard G, Ducos B, Perin L, Binoux M, Le Bouc Y. A targeted partial invalidation of the IGF-I receptor gene in mice causes a postnatal growth deficit. Endocrinology. 2000;141:2557–2566. doi: 10.1210/endo.141.7.7550. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates life span and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Kappeler L, De Magalhaes Filho C, Le Bouc Y. The GH/IGF axis: Insights from animal models. In: Carel J-C, Kelly PA, Christen Y, editors. Deciphering Growth. Berlin: Springer; 2005. pp. 41–51. [Google Scholar]
- Kappeler L, De Magalhaes Filho C, Dupont J, Leneuve P, Cervera P, Périn L, Loudes C, Blaise A, Klein R, Epelbaum J, Le Bouc Y, Holzenberger M. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008;6:2144–2153. doi: 10.1371/journal.pbio.0060254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappeler L, De Magalhaes Filho C, Leneuve P, Xu J, Brunel N, Chatziantoniou C, Le Bouc Y, Holzenberger M. Early postnatal nutrition determines somatotropic function in mice. Endocrinology. 2009;150:314–323. doi: 10.1210/en.2008-0981. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Kido Y, Nakae J, Hribal ML, Xuan S, Efstratiadis A, Accili D. Effects of mutations in the insulin-like growth factor signaling system on embryonic pancreas development and beta-cell compensation to insulin resistance. J. Biol. Chem. 2002;277:36740–36747. doi: 10.1074/jbc.M206314200. [DOI] [PubMed] [Google Scholar]
- Kulkarni RN, Almind K, Goren HJ, Winnay JN, Ueki K, Okada T, Kahn CR. Impact of genetic background on development of hyperinsulinemia and diabetes in insulin receptor/insulin receptor substrate-1 double heterozygous mice. Diabetes. 2003;52:1528–1534. doi: 10.2337/diabetes.52.6.1528. [DOI] [PubMed] [Google Scholar]
- Ladiges W, Van Remmen H, Strong R, Ikeno Y, Treuting P, Rabinovitch P, Richardson A. Lifespan extension in genetically modified mice. Aging Cell. 2009;8:346–352. doi: 10.1111/j.1474-9726.2009.00491.x. [DOI] [PubMed] [Google Scholar]
- Leduc MS, Hageman RS, Meng Q, Verdugo RA, Tsaih SW, Churchill GA, Paigen B, Yuan R. Identification of genetic determinants of IGF-1 levels and longevity among mouse inbred strains. Aging Cell. 2010;9:823–836. doi: 10.1111/j.1474-9726.2010.00612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoith D, Yakar S. Mechanisms of disease: metabolic effects of growth hormone and insulin-like growth factor 1. Nat. Clin. Pract. Endocrinol. Metab. 2007;3:302–310. doi: 10.1038/ncpendmet0427. [DOI] [PubMed] [Google Scholar]
- Lewitt MS, Brismar K, Wang J, Wivall-Helleryd IL, Sindelar P, Gonzalez FJ, Bergman T, Bobek GA. Responses of insulin-like growth factor (IGF)-I and IGF-binding proteins to nutritional status in peroxisome proliferator-activated receptor-alpha knockout mice. Growth Horm. IGF Res. 2001;11:303–313. doi: 10.1054/ghir.2001.0247. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang WJ, Cao H, Lu J, Wu C, Hu FY, Guo J, Zhao L, Yang F, Zhang YX, Li W, Zheng GY, Cui H, Chen X, Zhu Z, He H, Dong B, Mo X, Zeng Y, Tian XL. Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum. Mol. Genet. 2009;18:4897–4904. doi: 10.1093/hmg/ddp459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Masoro EJ, Nelson JF, Strong R, McMahan CA, Richardson A. Genetic mouse models of extended lifespan. Exp. Gerontol. 2003;38:1353–1364. doi: 10.1016/j.exger.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Liao CY, Rikke BA, Johnson TE, Diaz V, Nelson JF. Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell. 2010;9:92–95. doi: 10.1111/j.1474-9726.2009.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- Pawlikowska L, Hu D, Huntsman S, Sung A, Chu C, Chen J, Joyner AH, Schork NJ, Hsueh WC, Reiner AP, Psaty BM, Atzmon G, Barzilai N, Cummings SR, Browner WS, Kwok PY, Ziv E Study of Osteoporotic Fractures. Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging Cell. 2009;8:460–472. doi: 10.1111/j.1474-9726.2009.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkov PM, Ding Y, Cassell MA, Zhang W, Wagner G, Sargent EE, Asquith S, Crew V, Johnson KA, Robinson P, Scott VE, Wiles MV. An efficient SNP system for mouse genome scanning and elucidating strain relationships. Genome Res. 2004;14:1806–1811. doi: 10.1101/gr.2825804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C, Lingard S, Choudhury AI, Batterham RL, Claret M, Clements M, Ramadani F, Okkenhaug K, Schuster E, Blanc E, Piper MD, Al-Qassab H, Speakman JR, Carmignac D, Robinson IC, Thornton JM, Gems D, Partridge L, Withers DJ. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2008;22:807–818. doi: 10.1096/fj.07-9261com. [DOI] [PubMed] [Google Scholar]
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman C, Partridge L, Withers DJ. Replication of extended lifespan phenotype in mice with deletion of insulin receptor substrate 1. PLoS ONE. 2011;6:e16144. doi: 10.1371/journal.pone.0016144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat. Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- Sjögren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Törnell J, Isaksson OG, Jansson JO, Ohlsson C. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc. Natl Acad. Sci. USA. 1999;96:7088–7092. doi: 10.1073/pnas.96.12.7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soerensen M, Dato S, Christensen K, McGue M, Stevnsner T, Bohr VA, Christiansen L. Replication of an association of variation in the FOXO3A gene with human longevity using both case-control and longitudinal data. Aging Cell. 2010;9:1010–1017. doi: 10.1111/j.1474-9726.2010.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh Y, Atzmon G, Cho MO, Hwang D, Liu B, Leahy DJ, Barzilai N, Cohen P. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl Acad. Sci. USA. 2008;105:3438–3442. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson J, Sjögren K, Fäldt J, Andersson N, Isaksson O, Jansson J-O, Ohlsson C. Liver-derived IGF-I regulates mean life span in mice. PLoS ONE. 2011;6:e22640. doi: 10.1371/journal.pone.0022640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swindell WR. Dietary restriction in rats and mice: a meta-analysis and review of the evidence for genotype-dependent effects on lifespan. Ageing Res. Rev. 2012;11:254–270. doi: 10.1016/j.arr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi A, White MF. Response to Comment on brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2008;320:1012. doi: 10.1126/science.1152366. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317:369–372. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Yee D, Matin A, Nadeau JH, Magnuson T. Genealogy of the 129 inbred strains: 129/SvJ is a contaminated inbred strain. Mamm. Genome. 1997;8:390–393. doi: 10.1007/s003359900453. [DOI] [PubMed] [Google Scholar]
- Vijayakumar A, Wu Y, Buffin NJ, Li X, Sun H, Gordon RE, Yakar S, LeRoith D. Skeletal muscle growth hormone receptor signaling regulates basal, but not fasting-induced, lipid oxidation. PLoS ONE. 2012;7:e44777. doi: 10.1371/journal.pone.0044777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman DJ, Holloway MG. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol. Pharmacol. 2009;76:215–228. doi: 10.1124/mol.109.056705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, Masaki KH, Willcox DC, Rodriguez B, Curb JD. FOXO3A genotype is strongly associated with human longevity. Proc. Natl Acad. Sci. USA. 2008;105:13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat. Genet. 1999;23:32–40. doi: 10.1038/12631. [DOI] [PubMed] [Google Scholar]
- Xu J, Bekaert AJM, Dupont J, Rouve S, Annesi-Maesano I, De Magalhaes Filho C, Kappeler L, Holzenberger M. Exploring endocrine growth hormone regulation in mice using rank plot analysis and random blood samples. J. Endocrinol. 2011;208:119–129. doi: 10.1677/JOE-10-0317. [DOI] [PubMed] [Google Scholar]
- Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl Acad. Sci. USA. 1999;96:7324–7329. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakar S, Setser J, Zhao H, Stannard B, Haluzik M, Glatt V, Bouxsein ML, Kopchick JJ, LeRoith D. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J. Clin. Invest. 2004;113:96–105. doi: 10.1172/JCI200417763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan R, Tsaih S-W, Petkova SB, Evsikoca CMD, Xing S, Marion MA, Bogue MAB, Mills KD, Peters LL, Bult CJ, Rosen CJ, Sundberg JP, Harrison DE, Churchill GA, Paigen B. Aging in inbred strains of mice: study design and interim report on median lifespans and circulating IGF1 levels. Aging Cell. 2009;8:277–287. doi: 10.1111/j.1474-9726.2009.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan R, Meng Q, Nautiyal J, Flurkey K, Tsaih SW, Krier R, Parker MG, Harrison DE, Paigen B. Genetic coregulation of age of female sexual maturation and lifespan through circulating IGF1 among inbred mouse strains. Proc. Natl Acad. Sci. USA. 2012;109:8224–8229. doi: 10.1073/pnas.1121113109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Glucose homeostasis in 4- to 6-month-old B6 IGF-1R+/−mice.
Table S1 Data of Figure 1D and E (lifetime survival).
Table S2 Descriptive statistics of lifespan and survival data from male and female IGF-1R+/− and control mice in B6 genetic background (Data correspond to Figure 1, Panel D and E).
Table S3 Mean lifespan of B6 control mice in recent studies of mouse longevity genes. Highest and lowest means are in bold.
Table S4 Longevity of male and female mice from strains derived from original 129 mice.