

ABSTRACT
Cancer cells exhibit modifications in nuclear architecture and transcriptional control. Tumor growth and metastasis are supported by RUNX family transcriptional scaffolding proteins, which mediate the assembly of nuclear-matrix-associated gene-regulatory hubs. We used proteomic analysis to identify RUNX2-dependent protein–protein interactions associated with the nuclear matrix in bone, breast and prostate tumor cell types and found that RUNX2 interacts with three distinct proteins that respond to DNA damage – RUVBL2, INTS3 and BAZ1B. Subnuclear foci containing these proteins change in intensity or number following UV irradiation. Furthermore, RUNX2, INTS3 and BAZ1B form UV-responsive complexes with the serine-139-phosphorylated isoform of H2AX (γH2AX). UV irradiation increases the interaction of BAZ1B with γH2AX and decreases histone H3 lysine 9 acetylation levels, which mark accessible chromatin. RUNX2 depletion prevents the BAZ1B–γH2AX interaction and attenuates loss of H3K9 and H3K56 acetylation. Our data are consistent with a model in which RUNX2 forms functional complexes with BAZ1B, RUVBL2 and INTS3 to mount an integrated response to DNA damage. This proposed cytoprotective function for RUNX2 in cancer cells might clarify its expression in chemotherapy-resistant and/or metastatic tumors.
KEY WORDS: RUNX2, DNA damage response, Nuclear matrix, Proteomics, Cancer, Breast, Prostate, Osteosarcoma, INTS3, BAZ1B
INTRODUCTION
Nuclei are highly structured, and the spatial organization that supports nuclear metabolism is increasingly well characterized. This nuclear architecture encompasses two interconnected structures – chromatin and a nuclear matrix (Berezney et al., 1995; van Driel et al., 1995; Stenoien et al., 1998; Zink et al., 2004; Kubben et al., 2010; Markaki et al., 2010; Meldi and Brickner, 2011; Simon and Wilson, 2011). Molecular complexes that perform and regulate transcription, RNA processing, DNA replication, DNA repair and apoptosis, and that organize chromatin structure are localized within distinct nuclear domains. The ultrastructure of the nuclear matrix is well characterized and, in addition to protein and DNA, requires RNA to maintain its integrity (Nickerson et al., 1997). The protein composition of the isolated nuclear matrix was initially analyzed using two-dimensional gel electrophoresis (Fey and Penman, 1988; Dworetzky et al., 1990; Getzenberg et al., 1991). This approach indicated that the nuclear matrix is comprised of ≥200 proteins and that its composition is partially cell-type specific. Several dozen nuclear matrix proteins have been identified, including Numa, matrin, HnRNPs, NMP1 (also known as YY1) and NMP2 (also known as RUNX2) (Fey and Penman, 1988; Bidwell et al., 2001; Merriman et al., 1995; Javed et al., 2005), revealing that the nuclear matrix contains both structural and regulatory components.
Molecular characterization of the nuclear matrix protein NMP2 (Bidwell et al., 1993; Merriman et al., 1995) established that this protein is a member of the RUNX (AML/PEBP2alpha/CBFA) family of lineage-specific transcription factors, which control tissue development and have pathological roles in cancer (Zaidi et al., 2007a). RUNX proteins are localized at specific subnuclear domains through peptide-targeting sequences (Zeng et al., 1997; Zeng et al., 1998). They participate in the scaffolding of macromolecular protein–protein complexes that control gene transcription by supporting chromatin-related epigenetic mechanisms (Zaidi et al., 2001). These mechanisms involve formation of multiple complexes with proteins that mediate histone modifications or chromatin remodeling (Delcuve et al., 2009), including protein histone acetyl transferases (HATs, such as p300), histone deacetylases (e.g. HDACs), co-regulators (e.g. TLE-1, a homolog of groucho), YAP and SMADs (Zaidi et al., 2002; Zaidi et al., 2004) Disruption of RUNX protein subnuclear targeting alters transcriptional programs and compromises cell growth and differentiation (Zaidi et al., 2006), reflecting its pathological linkage to acute myelogenous leukemia and breast and prostate cancers (Zaidi et al., 2007b).
RUNX2 (NMP2) is a master regulator of skeletal development (Lian et al., 2004; Lian et al., 2006; Kuo et al., 2009), and has been linked to bone cancer and metastases from other cancers (Pratap et al., 2011). Because RUNX2 is a rate-limiting scaffolding protein that is crucial for the molecular organization of both transcriptional and epigenetic complexes at multiple target genes within matrix-associated subnuclear domains, establishing which nuclear matrix proteins are linked to the diverse biological functions of RUNX2 is a key objective. In this study, we used advanced protein mass spectrometry and proteomic analysis to identify proteins that are recruited to the nuclear matrix in a RUNX2-dependent manner and that associate with RUNX2 in bone, breast and prostate cancer cells. Strikingly, we found that three proteins with previously reported separate and distinct functions might associate with RUNX2 to form a novel, multifunctional integrated protein complex involved in the UV-induced DNA damage response. Our data indicate that RUNX2 supports the scaffolding of these proteins into an integrated complex that is coupled to the DNA damage response. This finding suggests that RUNX2 is a direct participant and regulator of the DNA damage response, thus providing a new molecular dimension in our understanding of DNA repair and its dysregulation in cancer.
RESULTS
Definition of a RUNX2-dependent proteome associated with the nuclear matrix
RUNX2 localizes in subnuclear domains (Fig. 1A) that are associated with the nuclear matrix (Zaidi et al., 2001). To investigate RUNX2-related protein complexes in this nuclease-resistant chromatin compartment by proteomic analysis and analyze the extent to which the protein profile of this compartment is RUNX2 dependent, we used siRNA-mediated knockdown of RUNX2 in Saos2 osteosarcoma cells. We transiently transfected cells with siRNA targeting RUNX2 and examined the subcellular localization of RUNX protein by confocal immunofluorescence microscopy. The number of RUNX2-containing nuclear foci was diminished in cells transfected with RUNX2-targeting siRNA (siRUNX2), but gross nuclear morphology was maintained (Fig. 1A). Cells were then fractionated and proteins from different subcellular compartments were analyzed by SDS-PAGE and western blotting, using markers to validate the fractionation of cytoplasmic, chromatin and nuclear matrix compartments (e.g. GAPDH was used as a cytoplasmic marker, acetylated histone H3 as a chromatin marker and lamin B as a nuclear matrix marker) (Fig. 1B). siRNA-mediated depletion of RUNX2 greatly reduced RUNX2 protein levels in whole-cell lysates and chromatin fractions, and rendered it undetectable in the nuclear matrix compartment. RUNX2 knockdown did not, however, alter the overall composition of the most abundant nuclear proteins residing in the nuclear matrix fraction, as visualized by staining the gel with Coomassie Blue (data not shown).
Fig. 1.

Proteomic analysis of RUNX2-related nuclear matrix proteins in RUNX2-knockdown cells. (A) Immunofluorescence staining of Saos2 cells transfected with either nontargeting siRNA (siNS) or RUNX2-targeting siRNA (siRUNX2). Insets show differential interference contrast (DIC, upper right) and DAPI images (lower right). (B) Proteins in whole-cell lysates, cytoplasmic extracts, DNase I/salt extracts and nuclear matrix fractions, were resolved by 15% SDS-PAGE and analyzed by western blotting using primary antibodies specific for the indicated proteins. GAPDH, histone H3 and lamin B were used as markers for cytoplasmic extracts, DNase I/salt extracts and nuclear matrix fractions, respectively. FBR (fibrillarin) and B23 (nucleophosmin) are nuclear matrix components that are expected to be recovered in both DNase I/salt extracts and the nuclear matrix fraction. (C) Workflow for the proteomic screening of RUNX2-dependent nuclear matrix proteins. A total of 1093 proteins were identified by mass-spectrometry-assisted fingerprinting of the nuclear matrix fraction prepared from Saos2 cells transfected with nontargeting siRNA or RUNX2-targeting siRNA. Of these, 721 were identified as nuclear proteins by Gene Ontology (GO) analysis. Spectral counting obtained from mass spectrometry was used to compare the relative fold-change of protein levels; 136 proteins out of 207 RUNX2-dependent nuclear proteins were identified as nuclear proteins that were downregulated by RUNX2 knockdown (see supplementary material Table S1). A functional subset of proteins, including chromatin remodelers, epigenetic regulators, transcriptional controllers and most RUNX2-dependent nuclear proteins, was selected by further screening for proteins identified through ≥5 peptides (supplementary material Table S2). (D) The graph shows the log (base 2) fold-change in protein levels between nuclear matrix fractions of cells transfected with RUNX2-targeting siRNA or nontargeting siRNA determined by spectral counting obtained from mass spectrometry analysis. Results for a functional subset of proteins, including transcription regulators, chromatin remodelers and histone modifiers is shown. The number of unique peptides identified by mass spectrometry is indicated, as are the biological functions of proteins – T, transcription regulator; C, chromatin remodeler; H, histone modifier. (E) Functions and fold decreases due to RUNX2 knockdown of representative proteins from the mass spectrometry analysis of nuclear matrix proteins from siRNA-transfected Saos2 cells. The fold decrease in protein levels was calculated by dividing the spectral counts for an identified protein by the sum of the spectral counts per sample.
Mass spectrometry analysis of the nuclear matrix proteome in bone cancer cells
Nanoliquid chromatography tandem mass spectrometry (nanoLC-MS/MS) was used to examine the molecular consequences of RUNX2 depletion on the proteomic profile of nuclear matrix proteins. Proteins in the nuclear matrix fraction from control and siRNA-transfected cells were separated by SDS-PAGE and processed for nanoLC-MS/MS (Dzieciatkowska et al., 2014). In total, >1000 proteins were identified from the product ion spectra using the Swissprot database (supplementary material Table S1), and >700 nuclear proteins were selected based on Gene Ontology (GO) analysis (Fig. 1C). To assess differences in the relative protein abundance in the nuclear matrix fraction between cells transfected with nontargeting control or RUNX2-targeting siRNA, we used semi-quantitative spectral counting (Zybailov et al., 2005; Lundgren et al., 2010). With this approach, we found that >100 proteins in the nuclear matrix fraction are downregulated >2-fold in cells transfected with siRNA targeting RUNX2 (Fig. 1C). Of this subset, we selected proteins that exhibited the greatest relative change when RUNX2 was depleted by siRNA-mediated knockdown or that are known to have a role in transcriptional regulation, chromatin remodeling and/or histone modification (Fig. 1D,E; supplementary material Table S2). For example, in cells with a 40-fold reduction in RUNX2 levels, INTS3 levels exhibit a >80-fold decrease.
Because the transcription factor RUNX2 might modulate the levels of distinct proteins in the nuclear matrix by regulating transcription or protein–protein interactions, we examined the mRNA expression of selected proteins by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis. As expected, RUNX2 mRNA levels in cells transfected with RUNX2 siRNA were decreased 3–4-fold compared to levels in cells transfected with nontargeting siRNA (Fig. 2A). However, mRNA expression levels corresponding to the proteins identified in our proteomic screen did not display significant changes due to transfection with siRNA targeting RUNX2 (Fig. 2A). This suggests that the differences in protein abundance observed by proteomic analysis (see Fig. 1D) are not due to RUNX2-dependent transcriptional regulation. RUNX2-dependent proteins that were detected by at least five unique peptides in nanoLC-MS/MS-based protein profiling and showed promise as regulatory contributors by preliminary bioinformatic analysis were confirmed to be localized in the nuclear matrix compartment by western blot analysis (Fig. 2B). Proteins of interest, PRMT1 and CTCF, were also included in the analysis. Collectively, these data indicate that changes in the nuclear matrix association of proteins upon RUNX2 depletion are mediated by post-transcriptional mechanisms (e.g. protein–protein interactions, subcellular compartmentalization).
Fig. 2.

Quantitative analysis of RUNX2-dependent proteins. (A) The levels of mRNA for the indicated genes from Saos2 cells treated with nontargeting or RUNX2-targeting siRNA (siRUNX2) were analyzed by quantitative real time polymerase chain reaction (qRT-PCR). Two independent biological duplicates were performed and the data show the mean±s.d. (B) Western blot analysis of the subcellular localization of proteins identified by mass spectrometry confirms that all are present in the nuclear matrix fraction (‘N’) of Saos2 cells. (C) Immunofluorescent staining of RUVBL2, INTS3 and BAZ1B proteins in Saos2 cells. Insets show differential interference contrast (DIC, upper right) and DAPI images (lower right).
We selected three RUNX2-interacting proteins, RUVBL2, INTS3 and BAZ1B, for further studies, because their functions are broadly related to chromatin organization, DNA repair and/or formation of protein–protein complexes that might be relevant to the molecular pathology of cancer cells. To support our proteomic identification, we examined the endogenous cellular localization of these proteins in Saos2 cells using immunofluorescence confocal microscopy (Fig. 2C). Nuclear foci were seen using primary antibodies that target RUVBL2, INTS3 and BAZ1B (Fig. 2C), consistent with the possibility that these proteins localize in the nuclear matrix in association with RUNX2.
Selective interactions of RUNX2 with RUVBL2, INTS3 and BAZ1B
We examined whether RUNX2 changes the nuclear matrix association of RUVBL2, INTS3 and BAZ1B proteins by their recruitment through protein–protein interactions (Fig. 3A). Co-immunoprecipitation experiments revealed that endogenous RUVBL2 and BAZ1B each exhibit interactions with RUNX2 in osteosarcoma cell lines, Saos2 and U2OS (Fig. 3B). Endogenous RUVBL2 protein present in lysates of Saos2 and U2OS cells was effectively co-immunoprecipitated with M70 antibody, which recognizes a C-terminal domain in RUNX2. BAZ1B was also co-immunoprecipitated using S19 antibody, which interacts with the N-terminal region of RUNX2 (Fig. 3B). We did not detect a clear interaction between endogenous RUNX2 and INTS3 using either the C- or N-terminally directed RUNX2 antibodies, but were able to readily detect RUNX2–INTS3-complexed proteins in RUNX2 and RUVBL2 co-expression analysis (see below).
Fig. 3.

Interaction of RUNX2 with RUVBL2, INTS3 and BAZ1B. (A) The functional domains of RUNX2, RUVBL2, INTS3 and BAZ1B, and the location of peptide sequences identified by mass spectrometry. The peptide fragment identified by mass spectrometry is indicated as a closed bar. Functional domains in each peptide are indicated. RHD, Runt homolog domain; NMTS, nuclear matrix targeting sequences; AAA, ATPase associated with a variety of cellular activities; KD, kinase domain; DDT, DNA binding homeobox and different transcription factors; PHD, plant homeodomain; BRD, bromodomain. (B) Co-immunoprecipitation of RUNX2 with interacting proteins was analyzed by western blotting. To detect RUNX2–RUVBL2, RUNX2–INTS3 or RUNX2–BAZ1B endogenous interactions, 5 mg of whole-cell lysates from Saos2 or U2OS cells were immunoprecipitated (IP) with 5 µg of anti-RUNX2 antibodies or 5 µg of normal rabbit IgG as a negative control. Immunoprecipitation products were then analyzed by western blotting, using anti-RUVBL2, anti-INTS3 or anti-BAZ1B antibodies. Note that no clear immunoprecipitation products were seen using anti-INTS3 antibodies and the results are not shown. (C) Co-immunoprecipitation of FLAG–RUVBL2 protein with full-length RUNX2 [wildtype (WT), amino acids 1–528] or C-terminally deleted mutant (ΔC, amino acids 1–376). U2OS cells were transiently co-transfected with a FLAG–RUVBL2 expression construct and either full-length or C-terminally deleted RUNX2 construct. Whole-cell lysates were incubated with anti-FLAG M2 agarose beads (Sigma). Washed beads were subjected to SDS-PAGE and analyzed by western blotting (WB) using specific antibodies against the indicated proteins. Asterisks (*) mark bands caused by nonspecific interactions. (D) Bacterially expressed GST (‘G’), GST fused to the Runt homolog domain of RUNX2 (amino acids 107–241; GST-R) or GST fused to the C-terminus of RUNX2 (amino acids 240–528; GST-C) proteins were immobilized on glutathione beads and incubated with whole-cell lysates from Saos2 cells. After extensive washing, proteins bound to the beads were eluted in protein sample buffer and analyzed by western blotting with antibodies against the indicated proteins.
Because RUVBL2 is a molecular chaperone (Izumi et al., 2010), we tested whether RUVBL2 can recruit RUNX2 and the other two identified proteins, INTS3 and BAZ1B in U2OS cells. We transiently co-transfected cells with plasmids expressing FLAG–RUVBL2 and either full-length (amino acids 1–528) or C-terminal-deleted (amino acids 1–376) RUNX2 protein. The exogenously expressed FLAG–RUVBL2 co-immunoprecipitated protein from both RUNX2 constructs, as well as endogenous INTS3 and BAZ1B (Fig. 3C). This result suggests that RUVBL2 can form a complex in vivo, and that the C-terminal region of RUNX2 might not be necessary for the interaction of RUNX2 with RUVBL2, INTS3 or BAZ1B. To further support the finding that the N-terminal region of RUNX2, containing the Runt homolog domain (RHD), is responsible for these interactions, we examined protein–protein interactions using glutathione-S-transferase (GST) pull-down assays. GST, GST plus the RUNX2 RHD or GST plus the RUNX2 C-terminal domain downstream of the RHD were immobilized on beads and incubated with Saos2 cell lysate. After washing, samples were recovered and separated using PAGE, then visualized by western blot analysis. The results indicate that RUVBL2, INTS3 and BAZ1B proteins each interact with the DNA-binding RHD (amino acids 107–241) of RUNX2, whereas no interaction was seen in assays that did not include this N-terminal RUNX2 domain (Fig. 3D). The signal from BAZ1B was stronger than those from INTS3 and RUVBL2; this could be due to technical variation in antibody interactions or biological differences in the ‘strength’ of protein interactions. These results indicate that the conserved DNA-binding domain, RHD, of RUNX2 is important for interactions with RUVBL2, INTS3 and BAZ1B.
The identification of RUNX2 complexes containing RUVBL2, INTS3 and BAZ1B in osteosarcoma cells, which express RUNX2 endogenously, raises the question of whether the same complexes form in other cell types that express RUNX2 as part of a pathological process. To address this, we examined RUNX2-related protein–protein interactions in PC3 cells, a metastatic prostate cancer cell line, and MDA-MB-231 cells, a metastatic breast cancer cell line, by co-immunoprecipitation analysis. First, we fractionated cells and conducted western blot analysis; we found that RUVBL2, INTS3 and BAZ1B are detected in the nuclear matrix of both PC3 and MDA-MB-231 cells (Fig. 4A). Endogenous interactions of RUNX2 with two of the three RUNX2-binding proteins were analyzed by co-immunoprecipitation using cell lysates from PC3 or MDA-MB-231 cells and N- or C-terminal-directed RUNX2 antibodies (Fig. 4B). The results suggest that RUNX2 complexes from both PC3 and MDA-MB-231 cells obtained using M70, the C-terminal-directed RUNX2 antibody, contain RUVBL2. In immunoprecipitate obtained using S19, the N-terminal-directed RUNX2 antibody, a strong RUNX2 band was seen and a band corresponding to BAZ1B was also present. Taken together, the co-immunoprecipitation data suggest that RUNX2 interacts with RUVBL2 and BAZ1B in PC3 and MDA-MB-231 cells. The interaction of RUNX2 with INTS3 appears to be more tenuous and requires elevated levels of RUVBL2 and/or RUNX2.
Fig. 4.
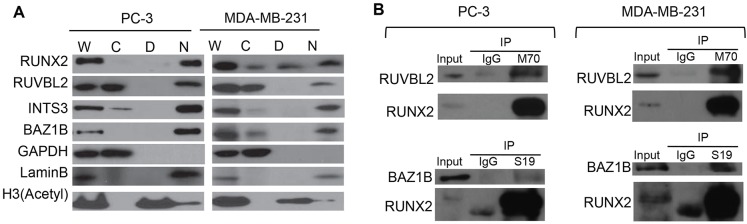
Interaction of RUNX2 with RUVBL2, INTS3 and BAZ1B in metastatic prostate PC3 or breast MDA-MB-231 cells. (A) Biochemical fractionation of PC3 and MDA-MB-231 cells. Proteins from whole-cell lysates (‘W’), cytoplasmic extracts, (‘C’), DNase I/salt extracts (‘D’) and nuclear matrix fraction (‘N’) from PC3 or MDA-MB-231 cells were resolved on 8% or 15% SDS-PAGE and analyzed by western blotting with the indicated primary antibodies. (B) Co-immunoprecipitation of RUNX2 with interacting proteins was analyzed by western blotting. To detect RUNX2–RUVBL2, RUNX2–INTS3 or RUNX2–BAZ1B endogenous interactions, 5 mg of whole-cell lysates from Saos2 or U2OS cells were immunoprecipitated (IP) with 5 µg of anti-RUNX2 antibodies or 5 µg of normal rabbit IgG as a negative control. Proteins were separated by SDS-PAGE and blotted, and immunoblots were probed with anti-RUVBL2, anti-INTS3 or anti-BAZ1B antibodies. Antibodies against GAPDH, histone H3 and lamin B were used as markers for cytoplasmic extracts, DNase I/salt extracts and the nuclear matrix fraction, respectively.
RUNX2 association with RUVBL2, INTS3 and BAZ1B in subnuclear domains points to a novel role for RUNX2 in the DNA damage response
Next, we examined the subcellular localization of RUNX2 in relation to each of the three proteins in Saos2 cells by confocal immunofluorescence microscopy. We observed colocalization of RUNX2 and any of the three proteins in only a few subnuclear domains (Fig. 5A). Because the interactions of RUVBL2, INTS3 or BAZ1B with RUNX2 appear to be, at least in part, interdependent, we performed gene ontology analysis to assess whether these proteins have common functions or participate in shared pathways (Fig. 5B,C). This analysis revealed that the three proteins are involved in cellular responses to DNA damage and are linked to histone protein H2AX (encoded by H2AFX) through either a RUVBL2–INTS3 network or a BAZ1B network (Fig. 5B). The finding from our studies that these two networks intersect is novel, and the implication is that RUNX2 is involved in H2AX-dependent genomic surveillance mechanisms. H2AX is a key sensor of genomic stress: it is phosphorylated on serine 139 (referred to as γ-H2AX) by DNA-dependent protein kinases, such as ataxia telangiectasia mutated (ATM) and ATM-related (ATR) (Fig. 5B,C). To further characterize this relationship, we directly investigated molecular and cellular responses involving this set of four proteins upon induction of DNA damage using UV irradiation, an accessible laboratory method for damaging DNA in a well-defined manner.
Fig. 5.

RUNX2 association with RUVBL2, INTS3 and BAZ1B in vivo. (A) Immunofluorescent staining of RUNX2 (Alexa Fluor 488, green) with RUVBL2, INTS3 or BAZ1B (Alexa Fluor 555, red) in Saos2 cells was analyzed by confocal microscopy (z-sections were 0.2 µm thickness; images were acquired using a 63x objective with oil immersion; 1.4 numerical aperture). Insets show differential interference contrast (DIC, lower right) and DAPI images (upper right). (B) The functional protein networks of RUVBL2, INTS3 and BAZ1B were analyzed using STRING (version 9.0). Boxes below the network diagrams show a simplified version of the model. (C) Reported biological functions of RUNX2, RUVBL2, INTS3 and BAZ1B, with references.
RUNX2 colocalizes with γ-H2AX at a limited number of distinct foci
To determine whether RUNX2 is involved in the nuclear response to DNA damage, cells transfected with RUNX2 or nontargeting siRNA were UV irradiated and analyzed by confocal immunofluorescence microscopy. In cells transfected with nontargeting siRNA, the number of RUNX2 foci with RUVBL2 or BAZ1B increased modestly 60 min after UV irradiation, whereas the RUNX2 foci that exhibited colocalization with INTS3 adjacent to one of the nucleoli were constitutively present regardless of UV (Fig. 6A, left). RUNX2 depletion by RNA interference did not change the subnuclear localization of any of the three proteins after UV irradiation, albeit that the localization of INTS3 in the nucleus was transiently altered 30 min after UV (Fig. 6A, right). The total protein level of γ-H2AX in irradiated cells increased after UV exposure; however, RUNX2, RUVBL2, INTS3 and BAZ1B levels did not change (Fig. 6C). This suggests that foci for RUVBL2, INTS3 and BAZ1B change in number or appearance upon UV exposure, but can form independently of RUNX2 (Fig. 6A). Because BAZ1B binds to both γ-H2AX (Barnes et al., 2010) and RUNX2 (Fig. 3), we examined whether RUNX2 associates with γ-H2AX in situ. We found that both large and small RUNX2 foci containing γ-H2AX were present at 30 min after UV irradiation (Fig. 6B). The partial colocalization of RUNX2 with γ-H2AX is consistent with our observation that the γ-H2AX-binding protein BAZ1B interacts with RUNX2.
Fig. 6.

UV effect on RUNX2 and interacting proteins. Saos2 cells were transfected with nontargeting (siNS) or RUNX2-targeting siRNA (siRUNX2), and were UV irradiated (300 J/m2) or not treated with radiation (NT). (A,B) Confocal microscopy analysis of co-immunofluorescent staining. Non-irradiated (NT) cells and 30 or 60 min post-UV-irradiation cells were permeabilized, fixed and stained with RUNX2-specific antibody and RUVBL2, INTS3, BAZ1B (A) or γ-H2AX (B) antibodies. Blue arrows point to RUNX2-associated RUVBL2 or BAZ1B foci. The thickness of z-sections is 0.2 µm. Scale bar: 5 µm. (C) Western blot analysis of H2AX, γ-H2AX, RUNX2, RUVBL2, INTS3, BAZ1B and GAPDH in whole-cell lysates from non-irradiated and UV-irradiated Saos2 cells. UV-irradiated cells were harvested at 5 or 30 min after UV irradiation (300 J/m2).
To investigate whether RUNX2 plays a role in the formation of γ-H2AX foci, we performed RUNX2 knockdown in Saos-2 cells (Figs 6, 7). Importantly, the total levels of γ-H2AX were increased in siRUNX2-transfected cells compared to the levels in cells transfected with negative-control siRNA (siNS) (Fig. 6C). This finding is consistent with similar observations in RUNX2-null cells (Zaidi et al., 2007b). As expected, UV irradiation increased the percentage of cells that exhibited nuclear γ-H2AX foci (Fig. 7B), as well as the number of foci per cell in nontargeting-siRNA-treated control cells (Fig. 7C). However, in RUNX2-depleted cells, there was a basal level of γ-H2AX foci in the majority of the cells (Fig. 7A,B), which increased after UV treatment (Fig. 7C,D). Taken together, our results suggest that RUNX2 depletion enhances the formation of γ-H2AX foci in the nucleus.
Fig. 7.

RUNX2 association with γ-H2AX by UV irradiation. Saos2 cells transfected with nontargeting siRNA (siNS) or RUNX2-targeting siRNA (siRUNX2) were not treated with radiation (NT) or were UV-irradiated (300 J/m2). (A) After UV irradiation, cells were incubated for 30 or 60 min, then permeabilized, fixed and co-stained with RUNX2 (green) and γ-H2AX (red) antibodies and analyzed by confocal microscopy. (B) Cells showing γ-H2AX foci in the nucleus were counted. The graph shows the ratio of the number of cells with nuclear γ-H2AX foci to the total cells in the field; 3–14 cells were counted per field, and at least 9 fields were counted per treatment. (C) The number of γ-H2AX foci per nucleus was counted and plotted; ≥36 cells were analyzed for each data point. The data show the mean±s.d. (D) Representative confocal images from 30 min after UV irradiation are shown. (E) Saos2 cells were transfected with the FLAG–H2AX expression construct, UV irradiated or left untreated, then FLAG–H2AX protein was immunoprecipitated (IP) and analyzed by western blotting (WB). Because the intensity of ECL signal from Ser139 phosphorylation on H2AX from input lanes was low, we included longer exposures of the blot for Ser139 phosphorylation (P-S139-H2AX) and total H2AX input lanes. The upper band from the P-S139-H2AX and H2AX blots corresponds to overexpressed FLAG–H2AX and the lower band is from endogenous H2AX. *Long indicates data from a longer exposure of blots for ECL detection. Bands corresponding to INTS3 and RUNX2 are indicated with red arrows.
Because RUNX2 interacts with the γ-H2AX-binding protein BAZ1B, we examined whether RUNX2 and γ-H2AX participate in protein–protein interactions within a larger complex. Saos2 cells were transfected with FLAG–H2AX expression construct, then FLAG–H2AX was immunoprecipitated from cell lysates. As expected, BAZ1B did co-immunoprecipitate with FLAG–H2AX (Fig. 7E). The immunoprecipitates also contained INTS3 and RUNX2, but signal above background was only seen in lysates from UV-irradiated cells. BAZ1B is a protein kinase that phosphorylates H2AX on tyrosine 142 (Y142) (Xiao et al., 2009). Indeed, phosphorylation of Y142 is observed in H2AX immunoprecipitates (Fig. 7E). These results suggest that RUNX2 binds to BAZ1B and γ-H2AX in response to UV.
RUNX2 decreases histone H3 acetylation on lysine 9 in response to UV irradiation
Because of the interrelationships between RUNX2, BAZ1B, γ-H2AX and the cellular response to UV, we further explored the biological function of RUNX2 in the UV-induced DNA damage response. For example, other post-translational modifications of histones occur in addition to phosphorylation of H2AX in response to DNA damage, including decreased acetylation of H3K9 and H3K56 by activation of HDACs or inhibition of acetyltransferases (e.g. GCN5) (Yu et al., 2011). In osteosarcoma cells transfected with nontargeting siRNA and exposed to UV irradiation, acetylation of H3K9 and H3K56 decreased, whereas phosphorylation of H2AX S139 increased (Fig. 8A,B). H2AX Y142 phosphorylation appeared to be slightly decreased when the signal intensity was normalized relative to total H2AX recovery (Fig. 8B).
Fig. 8.

RUNX2-dependent histone modification in response to UV. (A) Saos2 cells transfected with nontargeting siRNA (siNS) or RUNX2-targeting siRNA (siRUNX2) were non- irradiated (cont) or were UV irradiated (300 J/m2). Then, cells were further incubated for 60 min, and K56 and K9 acetylation of histone H3 and S139 and Y142 phosphorylation of histone H2AX were analyzed by western blotting. (B) The graph shows quantification of the western blot data shown in A performed using Image J software. Each band from acetylation or phosphorylation of H3 or H2AX was measured and normalized to total H3 or H2AX levels. The image represents the analysis of at least three independent western blots. Data show the mean±s.d. (C) Saos2 cells transfected with siNS or siRUNX2 were non-irradiated (Control) or UV irradiated (UV, 300 J/m2), and images of cells were analyzed by Nikon phase-contrast microscopy (10×). (D) Immunoprecipitation (IP) analysis of UV irradiated (UV, 60 min) or non-irradiated (–) Saos2 cells co-transfected with FLAG–H2AX expression plasmid and the indicated siRNA using anti-FLAG antibody. Immunoprecipitation with normal IgG was used as a control.
RUNX2 depletion by siRNA knockdown modestly attenuated the UV-dependent reductions in acetylation of both H3K9 and H3K56. The results of RUNX2 knockdown on UV-dependent phosphorylation of H2AX were more dramatic. We saw an increase in H2AX S139 phosphorylation after UV irradiation and the abolishment of the UV-related reduction in Y142 phosphorylation in cells transfected with RUNX2-specific siRNA (Fig. 8A,B). The latter finding supports the concept that RUNX2 modulates BAZ1B functions linked to H2AX phosphorylation and the response to UV irradiation. Because BAZ1B-mediated phosphorylation of Y142 in H2AX triggers apoptosis (Xiao et al., 2009), we examined cell survival in response to UV. Cells with decreased levels of RUNX2 appeared to be protected from UV-induced cell loss compared to nontargeting-siRNA-transfected control cells (Fig. 8C). Finally, immunoprecipitation analysis of Saos2 cells co-transfected with the FLAG–H2AX expression construct and RUNX2-targeting or nontargeting siRNA, and treated with UV irradiation indicated that the BAZ1B interaction with H2AX, which is normally increased by UV exposure, was actually diminished in the absence of RUNX2 (Fig. 8D). In conclusion, the combined results of this study are consistent with a model in which RUNX2 participates with BAZ1B and other associated proteins in cell survival in response to UV-induced DNA damage.
DISCUSSION
RUNX2 is a nuclear-matrix-associated transcription factor with biological activities in normal development or cancer that are determined by the dynamic association of interacting proteins to form different functional complexes depending on the physiological context. To understand this proteome of RUNX2-interacting proteins at the nuclear matrix, we employed a novel approach that combines the depletion of RUNX2 as a molecular scaffolding protein and high-resolution proteomic analysis of changes in the protein profile of the nuclear-matrix-related subnuclear fractions in cancer cell lines. Analysis of this subnuclear proteome in control cells containing RUNX2 revealed that, as expected, the nuclear matrix contains a large number of well-established structural molecules (e.g. lamin A/C and B, Numa, matrin-3) and many proteins involved in RNA metabolism. These observations are entirely consistent with results from a number of recent proteomic studies that characterize nuclear matrix proteins from embryonic stem cells, tumor cell types and plant cells (Oehr, 2003; Calikowski et al., 2003; Barboro et al., 2009; Albrethsen et al., 2010; Nasrabadi et al., 2010; Warters et al., 2010).
The main finding of the current study is the identification of three proteins that were shown to have shared RUNX2-dependent functions in UV-related DNA damage response. Interestingly, the nuclear matrix has been reported to provide a platform for DNA repair (Mullenders et al., 1990; Zaalishvili et al., 2000; Atanassov et al., 2005). One protein identified by our proteomic analysis is RUVBL2, a member of the AAA+ family (ATPase associated with diverse cellular activities) of DNA helicases (Izumi et al., 2010). Human RUVBL2 is the apparent homolog of the bacterial RUVB protein, which encodes a DNA helicase essential for homologous recombination and DNA double-strand break repair. Recently, RUVBL2 has also been identified in chromatin remodeling complexes including INO80, SRCAP, Uril and Tip60 (Gorynia et al., 2011). The second protein, INTS3, was initially characterized as an RNA polymerase II C-terminal domain binding factor involved in the 3′ processing of small nuclear RNAs (Inagaki et al., 2008). Recently, INTS3 has been identified as a key component of the DNA damage response (Huang et al., 2009; Skaar et al., 2009). BAZ1B, the third protein, is a novel tyrosine protein kinase related to the bromodomain family. It contains a structural motif characteristic of proteins that bind to acetylated lysines and are involved in the chromatin-dependent regulation of transcription (Xiao et al., 2009). BAZ1B phosphorylates H2AX at Tyr142, which plays a pivotal role in DNA repair and acts as a molecular marker that distinguishes between apoptotic and repair responses to genotoxic stress. The simultaneous identification of these three proteins as prominent RUNX2-dependent nuclear-matrix proteins suggested that they might share a RUNX2-related function.
RUVBL2, INTS3, BAZ1B and RUNX2 form a complex that regulates DNA repair following UV-induced DNA damage
In these experiments, we found that the association of endogenous RUNX2 with BAZ1B attenuated γ-H2AX foci formation, and resulted in higher rates of cell death after UV treatment than in RUNX2-depleted cells. The DNA-damage marker γ-H2AX relays signals from DNA lesions and recruits DNA damage repair machinery as well as chromatin remodelers to maintain genome stability. When DNA damage signal transduction is disrupted, however, cells fail to repair damaged DNA. We propose a model in which RUNX2 binds to the γ-H2AX complex with BAZ1B in response to DNA damage and inhibits BAZ1B. Although cellular DNA damage responses mediated by BAZ1B have not been well characterized, the molecular function of BAZ1B in response to DNA damage has been described (Xiao et al., 2009). According to the authors, phosphorylation of H2AX at Tyr142 by BAZ1B inhibits phosphorylation of H2AX at Ser139. So, the balance between survival and apoptosis can be maintained by two different but neighboring phosphorylation sites, Ser139 and Tyr142, of H2AX. However, our data indicated that in spite of increased phosphorylation at Tyr142, RUNX2-knockdown cells did not show higher levels of apoptosis as reported by others. Instead, these cells had higher survival rates after UV treatment than cells transfected with nontargeting siRNA. We attribute this result to the fact that the Saos2 cells used in our study do not express p53. The tumor suppressor p53 plays a key role in the DNA damage response, including arresting cells in G1 and G2 phases (Decraene et al., 2001) and activating wild-type p53-induced phosphatase 1 (WIP1, also known as protein phosphatase 1D), to reduce Ser139 phosphorylation of H2AX (Cha et al., 2010). For future studies, it would be interesting to evaluate the effects of treatments that induce double-strand DNA breaks, rather than the relatively benign single-strand breaks expected from the UV-irradiation levels used in the experiments described here.
Histone modification in the DNA damage response – histone acetylation
Eukaryotic cells protect their genome from UV-induced DNA damage through a sequence of molecular processes that include damage recognition, chromatin opening, DNA repair and chromatin sealing. Because chromatin is a multi-component complex of DNA and proteins, and the arrangement of nucleosomes throughout the genome is highly variable, this repair process has not been exhaustively characterized. Recently, however, histone acetylation was identified as a key player involved in opening chromatin to provide access for the DNA repair machinery. Histone H3 and H4 acetylation changes were observed after UV irradiation in yeast as well as human cells (Yu et al., 2011). Cells can modulate histone acetylation levels by recruiting either histone acetyltransferases (HATs) or histone deacetylases (HDACs), depending on the cellular context. H3K79 methylation is another important post-translational modification involved in opening and sealing chromatin following UV irradiation. DNA-damage-dependent histone modification happens not only in the main nucleosome components, but also in a variant histone H2AX with phosphorylation on S139 (γ-H2AX).
In conclusion, we have defined RUNX2-dependent protein–protein interactions that are associated with the nuclear matrix and discovered three proteins involved in the DNA damage response – BAZ1B, RUVBL2 and INTS3 – that require RUNX2 for scaffolding into a multi-functional complex. This complex supports histone displacement, DNA unwinding and stabilization of single-stranded DNA to mount an integrated response to DNA damage in breast, prostate and bone cancer cells.
MATERIALS AND METHODS
Cell culture and UV irradiation
Cells were grown at 37°C under 10% CO2. Saos2 and U2OS cells were cultured with McCoy's 5A medium with 15% FBS (Saos2) or 10% FBS (U2OS). MDA-MB-231 cells were cultured with α-MEM with 10% FBS. PC3 cells were cultured with T-medium with 10% FBS. L-glutamine and penicillin-streptomycin mixture (100 units/ml penicillin plus 100 µg/ml streptomycin) were added to the culture media for all cells (all cell culture reagents were from Thermo Fisher, Waltham, MA). For UV irradiation, cells were washed with PBS and irradiated in the UV-crosslinker model XL-1000 (Spectronics, Westbury, NY) with 300 J/m2. Culture media were added immediately after irradiation.
siRNAs, expression plasmids and transfection conditions
Nontargeting control siRNA (siNS) (D-001210–01-20) and RUNX2-targeting siRNA (siRUNX2) #4 were purchased from Thermo Fisher. The target sequence for RUNX2 (#4) is 5′-AAGGUUCAACGAUCUGAGAUUUU-3′. siRNAs (20 nmol) were diluted in Opti-MEM (0.5 ml, Thermo Fisher) and incubated with Oligofectamine (Thermo Fisher) for 30 min at room temperature. Cells were washed twice with PBS, and then washed in Opti-MEM just prior to transfection. Cells were incubated for 4 h at 37°C with the siRNA-Oligofectamine transfection mixture. 3× FBS-containing growth media were then added to cultures and siRNA-transfected cells were harvested 48 h later.
For transient transfection, appropriate plasmids were transfected into subconfluent Saos2 or U2OS cells using FuGENE6 or X-tremeGENE transfection reagent (Roche, Basel Switzerland). The expression plasmid for FLAG–H2AX was a gift from Dr Michael Rosenfeld (University of California, San Diego, CA). The expression plasmid for FLAG–RUVBL2 was purchased from Addgene. All transfections were equalized for total DNA by adding empty plasmid.
Biochemical fractionation
Cells were washed twice with cold PBS, harvested by scraping, then centrifuged for 10 min at 800 g, 4°C. After gentle aspiration of PBS from the pellet, the cells were extracted in cytoskeletal buffer [10 mM PIPES, pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, protease inhibitor cocktail (Roche), 1 mM AEBSF, 2 mM vanadyl ribonucleoside complexes (VRC), 25 nM MG132] containing 0.1% Triton X-100 for 5 min at 4°C to remove the soluble proteins. The mixture was centrifuged for 5 min at 800 g, 4°C, and the supernatant was collected as ‘cytoplasmic extract’. The pellet was resuspended in cytoskeletal buffer with 10,000 U/ml RNase-free DNase I (Sigma-Aldrich, St. Louis, MO) for 1 h at 30°C. Chromatin was then removed by salt extraction with 2 M ammonium sulfate to a final concentration of 250 mM for 10 min at room temperature and centrifuged for 5 min at 800 g, 4°C. The supernatant was collected as ‘DNase I/salt extract’. The remaining pellet was resuspended in protein sample buffer and collected as the ‘nuclear matrix fraction’. For proteomic analysis, equal volume fractions of each step of extraction and nuclear matrix fraction were loaded on SDS-polyacrylamide gels.
In-gel trypsin digestion
SDS-PAGE was performed with 4–15% linear-gradient polyacrylamide gels containing Tris-HCl (Bio-Rad, Hercules, CA). After electrophoresis, Coomassie-stained gel lanes were cut into 15 bands, each representing a unique molecular mass region, and processed for in-gel digestion. Briefly, control and siRUNX2 gel lanes were processed and transferred to 0.5-ml tubes. In some cases, a ‘short gel’ was run: nuclear matrix fractions were electrophoresed ∼2 cm by SDS-PAGE, Coomassie stained and then the entire protein band was processed for in-gel digestion. All gel pieces were de-stained twice with 200 µl of 25 mM ammonium bicarbonate in 50% acetonitrile for 30 min at 37°C, and reduced with 80 µl of 7.6 mg/ml dithiothreitol (DTT) for 10 min at 60°C. Excess solvent was removed and gel pieces were alkylated with 80 µl of 18.5 mg/ml iodoacetamide for 1 h at room temperature. The excess solvent was removed and gel pieces were washed twice with 200 µl of 25 mM ammonium bicarbonate in 50% acetonitrile for 15 min at 37°C and shrunk with 50 µl of acetonitrile for 10 min at room temperature. Excess acetonitrile was removed and the gel pieces were dried in a speedvac. Finally, 10 µl of 10 ng/µl trypsin (Promega, Madison, WI) and 10 µl of 25 mM ammonium bicarbonate were added to each tube. Additional 25 mM ammonium bicarbonate was added until the gel pieces were fully swollen (∼10–50 µl). Samples were then incubated for 4 h at 37°C and peptides were recovered by collecting excess solvent. Gel pieces were extracted twice more with 50 µl of 50% acetonitrile/5% formic acid, vortexing, centrifuging and incubating for 15 min. Solvent was removed and combined with the previously recovered solvent, for a total of three extractions. Combined extracts were dried on a speedvac.
Nanoflow LC-MS/MS and database search
Samples were reconstituted in 20 µl of 2% acetonitrile, 0.1% (v/v) formic acid, 0.01% (v/v) trifluoroacetic acid in water and analyzed using two different instrument platforms. Peptide samples from one full set of SDS gel pieces were injected (5 µl) using a NanoAcquity (Waters Corporation, Milford, MA) UPLC and loaded onto a Waters Symmetry C18 (180 µm i.d.×2 cm) trapping column at a flow rate of 5 µl/min for 3 min. Peptides were then separated by in-line gradient elution using a 75 µm i.d.×10 cm Waters BEH 130 (1.7 µm) analytical column, at a flow rate of 300 nl/min using a linear gradient from 3 to 90% B over 95 min (mobile phase A – 2% acetonitrile, 0.1% (v/v) formic acid, 0.01% (v/v) trifluoroacetic acid; mobile phase B – 98% acetonitrile, 0.1% formic acid, 0.01% trifluoroacetic acid). Peptides were eluted into an LTQ (Thermo Scientific) linear ion trap mass spectrometer operating in positive-ion electrospray and data-dependent acquisition modes. One mass spectrum was acquired over the m/z 400–2000 range, followed by serial tandem mass spectrometry (i.e. MS/MS) on the seven most abundant signals. Precursor ion isolation width was 2.0 Da, collision energy was 35%, ion population targets were 10,000 for MS and 5000 for MS/MS, and maximum ion fill times were 200 ms for both acquisition types. Precursor ions analyzed were subjected to dynamic exclusion for 30 s using a window of −0.5 to +1.5 m/z. The repeat count was 1 with a 30 s delay; ions at m/z 371 and 445 were also excluded from MS/MS.
Another set of similarly prepared samples were analyzed using a Proxeon Easy nanoLC (Thermo Scientific) system directly configured to an LTQ-Orbitrap Velos (Thermo Scientific) hybrid mass spectrometer. Peptide samples (2 µl) were loaded at 4 µl/min for 7 min onto a custom-made trap column (100 µm i.d. fused silica with Kasil frit) containing 2 cm of 200-Å, 5-µm Magic C18AQ particles (Michrom Bioresources, Auburn, CA). Peptides were then eluted using a custom-made analytical column (75 µm i.d. fused silica) with a gravity-pulled tip and packed with 25 cm of 100-Å, 5-µm Magic C18AQ particles (Michrom). Peptides were eluted with a linear gradient as described above. Mass spectrometry data were acquired using a data-dependent acquisition routine of acquiring one mass spectrum from m/z 350–2000 in the Orbitrap (resolution, 60,000; ion population, 1.0×106; maximum ion injection time, 500 ms) followed by tandem mass spectrometry in the linear ion trap (LTQ) of the ten most abundant precursor ions observed in the mass spectrum. MS/MS data were acquired using a precursor isolation width of 2.0 Da, a collision energy of 35%, an ion population of 5000 and a maximum ion fill time of 50 ms. Charge-state rejection of singly charged ions and dynamic exclusion was utilized [−0.1 to +1.1 Da window, repeat count 1 (30-s delay)] to minimize data redundancy and maximize peptide identification.
The raw data files were processed using Extract MSN software (Thermo Scientific) and searched against the human index of the SwissProt database (version 09/24/11) with Mascot (version 2.3.02; Matrix Science, London, UK) and X! Tandem [The GPM (www.thegpm.org); version Cyclone (2010.12.01.1)] software packages. LTQ Orbitrap Velos data were searched using a parent mass tolerance of 15 ppm and a fragment mass tolerance of 0.5 Da. LTQ data utilized a parent tolerance of 1.2 Da and a fragment tolerance of 1.0 Da. Full tryptic specificity with two missed cleavages was considered, and variable modifications of acetylation (protein N-term), cyclization of N-terminal S-carbamoylmethylcysteine (peptide N-term) and oxidation (methionine) and fixed modification of carbamidomethylation (cysteine) were considered. All search results were loaded into Scaffold software (Version 3.3.1; Proteome Software, Portland, OR) for comparative analyses using spectral counting of tandem mass spectra and full annotation of the data (Searle, 2010). Peptide identifications were accepted if they could be established at >95% probability by the Peptide Prophet algorithm (Keller et al., 2002) following Scaffold delta-mass correction. Protein identifications were accepted if they could be established at >99% probability and contained at least two identified peptides; protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al., 2003). Normalized spectral counts were calculated by dividing the spectral counts for an identified protein by the sum of the spectral counts per sample.
Quantitative gene expression analysis
RNA was isolated using Trizol reagent (Thermo Fisher Scientific) and trace DNA was removed using the DNA-free RNA kit (Zymo Research, Irvine, CA). cDNA was synthesized using Superscript III (Thermo Fisher Scientific) and amplified using gene-specific primers (supplementary material Table S2) and iTAQ SYBR green supermix (Bio-Rad Laboratories). Reactions were run and data collected on an ABI PRISM 700 system (Thermo Fisher Scientific). Primers for PCR are displayed in supplementary material Table S1.
Immunoprecipitation
Confluent cells on 100-mm plates were harvested and solubilized in lysis buffer (10 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.1% Nonidet P40, 1 mM DTT and 1 mM PMSF). Insoluble material was removed by centrifugation. The supernatants were incubated for 12 h at 4°C with 2 µg of anti-RUNX2 (M70 or S19; Santa Cruz Biotechnology, Santa Cruz, CA), anti-FLAG (M2; Sigma-Aldrich) and a mixture of anti-rabbit- and anti-goat-IgG (Santa Cruz Biotechnology). Following the addition of 30 µl of Protein-A/G–agarose beads (Santa Cruz Biotechnology), mixtures were incubated for 2 h at 4°C with rotation. Immune complexes were washed three times with lysis buffer; the agarose beads were then boiled for 10 min in sample buffer. Immunoprecipitates were run on 8% acrylamide SDS-PAGE or 4–20% gradient precast gels (Bio-Rad), followed by Western blot analysis with antibodies against RUVBL2, INTS3, BAZ1B, H2AX or phosphorylated Ser139-H2AX.
GST pulldown assays
The proteins containing N-terminal glutathione S-transferase (GST) fused in-frame to the RUNX homologs were obtained by expression in Escherichia coli BL21 strain as reported previously (Pande et al., 2013). A total of 1 µg of GST alone, GST-fused N-terminal RHD domain or C-terminal RUNX2 proteins was incubated with Glutathione Sepharose 4B (GE Healthcare) beads in 500 µl of binding buffer (20 mM Tris-HCl pH 8.0, 100 mM KCl, 0.5% NP-40, 10 mM EDTA, 0.05 mM PMSF, 1 mM DTT) for 30 min at 4°C with rotation. The beads were washed four times with 500 µl of binding buffer for 5 min at 4°C and incubated with 1 mg of whole-cell lysates from confluent Saos2 cells for 12 h at 4°C with rotation. After four washes with binding buffer, each for 5 min at 4°C, the beads were resuspended in 50 µl of sample buffer and boiled for 10 min at 95°C. The proteins that were retained with the beads were separated by 8% acrylamide SDS-PAGE and analyzed by western blotting with specific antibodies against RUVBL2 (Santa Cruz Biotechnology), INTS3 (Abcam, Cambridge, UK) and BAZ1B (Abcam).
Confocal microscopy
To prepare cells for immunofluorescence confocal microscopy analysis, Saos2 cells were washed in Hanks balanced salt solution (Thermo Fisher Scientific), and were incubated in 0.5% Triton X-100 in cytoskeletal buffer for 10 min. This step removes soluble proteins from both the cytoplasm and nucleus. Cells were later fixed in 4% formaldehyde in cytoskeletal buffer for 50 min on ice. For antibody staining, cells were stained with antibodies as described previously (Wagner et al., 2003). The primary antibodies were as follows: rabbit polyclonal antibody against RUNX2 (M70; 1∶100; Santa Cruz Biotechnology); mouse monoclonal antibody against RUVBL2 (B9; 1∶40; Santa Cruz Biotechnology); mouse monoclonal antibody against BAZ1B (1∶40; Santa Cruz Biotechnology); goat polyclonal antibody against INTS3 (1∶500; Abcam); mouse monoclonal antibody against phosphorylated S139 of H2AX (1∶100; Millipore). All antibodies were incubated overnight at 4°C. The following secondary antibodies (Thermo Fisher Scientific) were used: Alexa-Fluor-488-conjugated goat anti-rabbit-IgG (1∶2000); Alexa-Fluor-568-conjugated goat anti-mouse-IgG (1∶2000); Alexa-Fluor-594-conjugated donkey anti-goat-IgG (1∶2000); Alexa-Fluor-488-conjugated donkey anti-rabbit-IgG (1∶2000). Coverslips were mounted with ProlongGold (Thermo Fisher Scientific). Images were collected using a Leica TCS SP5 confocal microscope using a ×63 oil lens (numerical aperture 1.4) at optimum zoom, fixed pinhole size (100 µm) and optimum Z-plane interval (from 0.2–0.3-µm Z-stack), depending on the sample. The 405-nm, 488-nm and 561-nm excitation lasers were used to excite DAPI, Alexa Fluor 488, Alexa Fluor 568 and Alexa Fluor 594, respectively, and were activated sequentially during image collection. Images were analyzed using ImageJ and Photoshop 5.0.
Supplementary Material
Acknowledgments
We thank the members of our laboratory, especially Jason Dobson and Shirwin Pockwinse, for stimulating discussions and/or general support.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
S.Y., J.A.N., A.N.I., J.B.L., J.L.S., A.J.v.W. and G.S.S. planned and designed the study. S.Y. performed the experiments, with help from A.J.C.Q. and J.A.N. for confocal microscopy, and K.M.G. and S.A.S. for mass spectrometry. Data were analyzed and interpreted by S.Y., K.M.G., S.A.S., A.J.C.Q., J.A.N., A.N.I., J.B.L., J.L.S., A.J.v.W. and G.S.S. S.Y. drafted the manuscript, with help from J.A.N., A.N.I., J.B.L., J.L.S., A.J.v.W., L.A.M.-B., S.A.S. and G.S.S. All authors read and approved the final manuscript.
Funding
This work was supported, in whole or in part, by the National Institutes of Health [grant numbers P01 CA082834, P01 AR048818, R01 AR049069 and R01 AR039588]. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.160051/-/DC1
References
- Albrethsen J., Knol J. C., Piersma S. R., Pham T. V., de Wit M., Mongera S., Carvalho B., Verheul H. M. W., Fijneman R. J. A., Meijer G. A. et al. (2010). Subnuclear proteomics in colorectal cancer: identification of proteins enriched in the nuclear matrix fraction and regulation in adenoma to carcinoma progression. Mol. Cell Proteomics 9, 988–1005 10.1074/mcp.M900546-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanassov B., Gospodinov A., Stoimenov I., Mladenov E., Russev G., Tsaneva I., Anachkova B. (2005). Repair of DNA interstrand crosslinks may take place at the nuclear matrix. J. Cell. Biochem. 96, 126–136 10.1002/jcb.20518 [DOI] [PubMed] [Google Scholar]
- Barboro P., D'Arrigo C., Repaci E., Bagnasco L., Orecchia P., Carnemolla B., Patrone E., Balbi C. (2009). Proteomic analysis of the nuclear matrix in the early stages of rat liver carcinogenesis: identification of differentially expressed and MAR-binding proteins. Exp. Cell Res. 315, 226–239 10.1016/j.yexcr.2008.10.017 [DOI] [PubMed] [Google Scholar]
- Barnes L., Dumas M., Juan M., Noblesse E., Tesniere A., Schnebert S., Guillot B., Molès J. P. (2010). GammaH2AX, an accurate marker that analyzes UV genotoxic effects on human keratinocytes and on human skin. Photochem. Photobiol. 86, 933–941 10.1111/j.1751-1097.2010.00744.x [DOI] [PubMed] [Google Scholar]
- Barnett C., Krebs J. E. (2011). WSTF does it all: a multifunctional protein in transcription, repair, and replication. Biochem. Cell Biol. 89, 12–23 10.1139/O10-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezney R., Mortillaro M. J., Ma H., Wei X., Samarabandu J. (1995). The nuclear matrix: a structural milieu for genomic function. Int. Rev. Cytol. 162A, 1–65. [DOI] [PubMed] [Google Scholar]
- Bidwell J. P., Torrungruang K., Alvarez M., Rhodes S. J., Shah R., Jones D. R., Charoonpatrapong K., Hock J. M., Watt A. J. (2011). Involvement of the nuclear matrix in the control of skeletal genes: the NMP1 (YY1), NMP2 (Cbfa1), and NMP4 (Nmp4/CIZ) transcription factors. Crit. Rev. Eukaryot. Gene Expr. 11, 279–297 10.1615/CritRevEukarGeneExpr.v11.i4.20 [DOI] [PubMed] [Google Scholar]
- Bidwell J. P., Van Wijnen A. J., Fey E. G., Dworetzky S., Penman S., Stein J. L., Lian J. B., Stein G. S. (1993). Osteocalcin gene promoter-binding factors are tissue-specific nuclear matrix components. Proc. Natl. Acad. Sci. USA 90, 3162–3166 10.1073/pnas.90.8.3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calikowski T. T., Meulia T., Meier I. (2003). A proteomic study of the arabidopsis nuclear matrix. J. Cell. Biochem. 90, 361–378 10.1002/jcb.10624 [DOI] [PubMed] [Google Scholar]
- Cha H., Lowe J. M., Li H., Lee J. S., Belova G. I., Bulavin D. V., Fornace A. J., Jr (2010). Wip1 directly dephosphorylates gamma-H2AX and attenuates the DNA damage response. Cancer Res. 70, 4112–4122 10.1158/0008-5472.CAN-09-4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decraene D., Agostinis P., Pupe A., de Haes P., Garmyn M. (2001). Acute response of human skin to solar radiation: regulation and function of the p53 protein. J. Photochem. Photobiol. B 63, 78–83 10.1016/S1011-1344(01)00204-4 [DOI] [PubMed] [Google Scholar]
- Delcuve G. P., Rastegar M., Davie J. R. (2009). Epigenetic control. J. Cell. Physiol. 219, 243–250 10.1002/jcp.21678 [DOI] [PubMed] [Google Scholar]
- Dworetzky S. I., Fey E. G., Penman S., Lian J. B., Stein J. L., Stein G. S. (1990). Progressive changes in the protein composition of the nuclear matrix during rat osteoblast differentiation. Proc. Natl. Acad. Sci. USA 87, 4605–4609 10.1073/pnas.87.12.4605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzieciatkowska M., Hill R., Hansen K. C. (2014). GeLC-MS/MS analysis of complex protein mixtures. Methods Mol. Biol. 1156, 53–66 10.1007/978-1-4939-0685-7_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey E. G., Penman S. (1988). Nuclear matrix proteins reflect cell type of origin in cultured human cells. Proc. Natl. Acad. Sci. USA 85, 121–125 10.1073/pnas.85.1.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getzenberg R. H., Pienta K. J., Huang E. Y., Coffey D. S. (1991). Identification of nuclear matrix proteins in the cancer and normal rat prostate. Cancer Res. 51, 6514–6520. [PubMed] [Google Scholar]
- Gorynia S., Bandeiras T. M., Pinho F. G., McVey C. E., Vonrhein C., Round A., Svergun D. I., Donner P., Matias P. M., Carrondo M. A. (2011). Structural and functional insights into a dodecameric molecular machine – the RuvBL1/RuvBL2 complex. J. Struct. Biol. 176, 279–291 10.1016/j.jsb.2011.09.001 [DOI] [PubMed] [Google Scholar]
- Huang J., Gong Z., Ghosal G., Chen J. (2009). SOSS complexes participate in the maintenance of genomic stability. Mol. Cell 35, 384–393 10.1016/j.molcel.2009.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki Y., Yasui K., Endo M., Nakajima T., Zen K., Tsuji K., Minami M., Tanaka S., Taniwaki M., Itoh Y. et al. (2008). CREB3L4, INTS3, and SNAPAP are targets for the 1q21 amplicon frequently detected in hepatocellular carcinoma. Cancer Genet. Cytogenet. 180, 30–36 10.1016/j.cancergencyto.2007.09.013 [DOI] [PubMed] [Google Scholar]
- Izumi N., Yamashita A., Iwamatsu A., Kurata R., Nakamura H., Saari B., Hirano H., Anderson P., Ohno S. (2010). AAA+ proteins RUVBL1 and RUVBL2 coordinate PIKK activity and function in nonsense-mediated mRNA decay. Sci. Signal. 3, ra27 10.1126/scisignal.2000468 [DOI] [PubMed] [Google Scholar]
- Javed A., Barnes G. L., Pratap J., Antkowiak T., Gerstenfeld L. C., van Wijnen A. J., Stein J. L., Lian J. B., Stein G. S. (2005). Impaired intranuclear trafficking of Runx2 (AML3/CBFA1) transcription factors in breast cancer cells inhibits osteolysis in vivo. Proc. Natl. Acad. Sci. USA 102, 1454–1459 10.1073/pnas.0409121102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S., Dutta A. (2009). RVB1/RVB2: running rings around molecular biology. Mol. Cell 34, 521–33 10.1016/j.molcel.2009.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwaba S., Kitahashi K., Watanabe T., Onoda F., Ohtsu M., Murakami Y. (2010). The mammalian INO80 complex is recruited to DNA damage sites in an ARP8 dependent manner. Biochem. Biophys. Res. Commun. 402, 619–25 10.1016/j.bbrc.2010.10.066 [DOI] [PubMed] [Google Scholar]
- Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 10.1021/ac025747h [DOI] [PubMed] [Google Scholar]
- Kubben N., Voncken J. W., Misteli T. (2010). Mapping of protein- and chromatin-interactions at the nuclear lamina. Nucleus 1, 460–471 10.4161/nucl.1.6.13513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo Y. H., Zaidi S. K., Gornostaeva S., Komori T., Stein G. S., Castilla L. H. (2009). Runx2 induces acute myeloid leukemia in cooperation with Cbfbeta-SMMHC in mice. Blood 113, 3323–3332 10.1182/blood-2008-06-162248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian J. B., Javed A., Zaidi S. K., Lengner C., Montecino M., van Wijnen A. J., Stein J. L., Stein G. S. (2004). Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Crit. Rev. Eukaryot. Gene Expr. 14, 1–42 10.1615/CritRevEukaryotGeneExpr.v14.i12.10 [DOI] [PubMed] [Google Scholar]
- Lian J. B., Stein G. S., Javed A., van Wijnen A. J., Stein J. L., Montecino M., Hassan M. Q., Gaur T., Lengner C. J., Young D. W. (2006). Networks and hubs for the transcriptional control of osteoblastogenesis. Rev. Endocr. Metab. Disord. 7, 1–16 10.1007/s11154-006-9001-5 [DOI] [PubMed] [Google Scholar]
- Long F. (2012). Building strong bones: molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 13, 27–38 10.1038/nrm3254 [DOI] [PubMed] [Google Scholar]
- Lundgren D. H., Hwang S. I., Wu L., Han D. K. (2010). Role of spectral counting in quantitative proteomics. Expert Rev. Proteomics 7, 39–53 10.1586/epr.09.69 [DOI] [PubMed] [Google Scholar]
- Markaki Y., Gunkel M., Schermelleh L., Beichmanis S., Neumann J., Heidemann M., Leonhardt H., Eick D., Cremer C., Cremer T. (2010). Functional nuclear organization of transcription and DNA replication: a topographical marriage between chromatin domains and the interchromatin compartment. Cold Spring Harb. Symp. Quant. Biol. 75, 475–492 10.1101/sqb.2010.75.042 [DOI] [PubMed] [Google Scholar]
- Meldi L., Brickner J. H. (2011). Compartmentalization of the nucleus. Trends Cell Biol. 21, 701–708 10.1016/j.tcb.2011.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merriman H. L., van Wijnen A. J., Hiebert S., Bidwell J. P., Fey E., Lian J., Stein J., Stein G. S. (1995). The tissue-specific nuclear matrix protein, NMP-2, is a member of the AML/CBF/PEBP2/runt domain transcription factor family: interactions with the osteocalcin gene promoter. Biochemistry 34, 13125–13132 10.1021/bi00040a025 [DOI] [PubMed] [Google Scholar]
- Mullenders L. H., Venema J., van Hoffen A., Mayne L. V., Natarajan A. T., van Zeeland A. A. (1990). The role of the nuclear matrix in DNA repair. Prog. Clin. Biol. Res. 340A, 223–232. [PubMed] [Google Scholar]
- Nasrabadi D., Larijani M. R., Fathi A., Gourabi H., Dizaj A. V., Baharvand H., Salekdeh G. H. (2010). Nuclear proteome analysis of monkey embryonic stem cells during differentiation. Stem Cell Rev. 6, 50–61 10.1007/s12015-009-9109-6 [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 10.1021/ac0341261 [DOI] [PubMed] [Google Scholar]
- Nickerson J. A., Krockmalnic G., Wan K. M., Penman S. (1997). The nuclear matrix revealed by eluting chromatin from a cross-linked nucleus. Proc. Natl. Acad. Sci. USA 94, 4446–4450 10.1073/pnas.94.9.4446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehr P. (2003). Proteomics as a tool for detection of nuclear matrix proteins and new biomarkers for screening of early tumors stage. Anticancer Res. 23, 805–812. [PubMed] [Google Scholar]
- Pande S., Browne G., Padmanabhan S., Zaidi S. K., Lian J. B., van Wijnen A. J., Stein J. L., Stein G. S. (2013). Oncogenic cooperation between PI3K/Akt signaling and transcription factor Runx2 promotes the invasive properties of metastatic breast cancer cells. J. Cell. Physiol. 228, 1784–1792 10.1002/jcp.24339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap J., Lian J. B., Stein G. S. (2011). Metastatic bone disease: role of transcription factors and future targets. Bone 48, 30–36 10.1016/j.bone.2010.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A. J., Graybosch D. M., Huetsch J. C., Verdine G. L. (2005). A superhelical spiral in the Escherichia coli DNA gyrase A C-terminal domain imparts unidirectional supercoiling bias. J. Biol. Chem. 280, 26177–26184 10.1074/jbc.M502838200 [DOI] [PubMed] [Google Scholar]
- Searle B. C. (2010). Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 10, 1265–1269 10.1002/pmic.200900437 [DOI] [PubMed] [Google Scholar]
- Simon D. N., Wilson K. L. (2011). The nucleoskeleton as a genome-associated dynamic ‘network of networks’. Nat. Rev. Mol. Cell Biol. 12, 695–708 10.1038/nrm3207 [DOI] [PubMed] [Google Scholar]
- Skaar J. R., Richard D. J., Saraf A., Toschi A., Bolderson E., Florens L., Washburn M. P., Khanna K. K., Pagano M. (2009). INTS3 controls the hSSB1-mediated DNA damage response. J. Cell Biol. 187, 25–32 10.1083/jcb.200907026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien D., Sharp Z. D., Smith C. L., Mancini M. A. (1998). Functional subnuclear partitioning of transcription factors. J. Cell. Biochem. 70, 213–221 [DOI] [PubMed] [Google Scholar]
- van Driel R., Wansink D. G., van Steensel B., Grande M. A., Schul W., de Jong L. (1995). Nuclear domains and the nuclear matrix. Int. Rev. Cytol. 162A, 151–189. [DOI] [PubMed] [Google Scholar]
- Wagner S., Chiosea S., Nickerson J. A. (2003). The spatial targeting and nuclear matrix binding domains of SRm160. Proc. Natl. Acad. Sci. USA 100, 3269–3274 10.1073/pnas.0438055100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warters R. L., Cassidy P. B., Sunseri J. A., Parsawar K., Zhuplatov S. B., Kramer G. F., Leachman S. A. (2010). The nuclear matrix shell proteome of human epidermis. J. Dermatol. Sci. 58, 113–122 10.1016/j.jdermsci.2010.03.001 [DOI] [PubMed] [Google Scholar]
- Xiao A., Li H., Shechter D., Ahn S. H., Fabrizio L. A., Erdjument-Bromage H., Ishibe-Murakami S., Wang B., Tempst P., Hofmann K. et al. (2009). WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 457, 57–62 10.1038/nature07668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S., Teng Y., Waters R., Reed S. H. (2011). How chromatin is remodelled during DNA repair of UV-induced DNA damage in Saccharomyces cerevisiae. PLoS Genet. 7, e1002124 10.1371/journal.pgen.1002124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaalishvili T. M., Gabriadze I. Y., Margiani D. O., Philauri V. R., Surguladze N. M. (2000). Participation of Poly(ADP-ribose)-polymerase of nuclear matrix in DNA repair. Biokhimiia 65, 659–661. [PubMed] [Google Scholar]
- Zaidi S. K., Javed A., Choi J. Y., van Wijnen A. J., Stein J. L., Lian J. B., Stein G. S. (2001). A specific targeting signal directs Runx2/Cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J. Cell Sci. 114, 3093–3102. [DOI] [PubMed] [Google Scholar]
- Zaidi S. K., Sullivan A. J., van Wijnen A. J., Stein J. L., Stein G. S., Lian J. B. (2002). Integration of Runx and Smad regulatory signals at transcriptionally active subnuclear sites. Proc. Natl. Acad. Sci. USA 99, 8048–8053 10.1073/pnas.112664499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S. K., Sullivan A. J., Medina R., Ito Y., van Wijnen A. J., Stein J. L., Lian J. B., Stein G. S. (2004). Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 23, 790–799 10.1038/sj.emboj.7600073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S. K., Javed A., Pratap J., Schroeder T. M., J Westendorf J., Lian J. B., van Wijnen A. J., Stein G. S., Stein J. L. (2006). Alterations in intranuclear localization of Runx2 affect biological activity. J. Cell. Physiol. 209, 935–942 10.1002/jcp.20791 [DOI] [PubMed] [Google Scholar]
- Zaidi S. K., Young D. W., Javed A., Pratap J., Montecino M., van Wijnen A., Lian J. B., Stein J. L., Stein G. S. (2007a). Nuclear microenvironments in biological control and cancer. Nat. Rev. Cancer 7, 454–463 10.1038/nrc2149 [DOI] [PubMed] [Google Scholar]
- Zaidi S. K., Pande S., Pratap J., Gaur T., Grigoriu S., Ali S. A., Stein J. L., Lian J. B., van Wijnen A. J., Stein G. S. (2007b). Runx2 deficiency and defective subnuclear targeting bypass senescence to promote immortalization and tumorigenic potential. Proc. Natl. Acad. Sci. USA 104, 19861–19866 10.1073/pnas.0709650104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C., van Wijnen A. J., Stein J. L., Meyers S., Sun W., Shopland L., Lawrence J. B., Penman S., Lian J. B., Stein G. S. et al. (1997). Identification of a nuclear matrix targeting signal in the leukemia and bone-related AML/CBF-alpha transcription factors. Proc. Natl. Acad. Sci. USA 94, 6746–6751 10.1073/pnas.94.13.6746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C., McNeil S., Pockwinse S., Nickerson J., Shopland L., Lawrence J. B., Penman S., Hiebert S., Lian J. B., van Wijnen A. J. et al. (1998). Intranuclear targeting of AML/CBFalpha regulatory factors to nuclear matrix-associated transcriptional domains. Proc. Natl. Acad. Sci. USA 95, 1585–1589 10.1073/pnas.95.4.1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink D., Fischer A. H., Nickerson J. A. (2004). Nuclear structure in cancer cells. Nat. Rev. Cancer 4, 677–687 10.1038/nrc1430 [DOI] [PubMed] [Google Scholar]
- Zybailov B., Coleman M. K., Florens L., Washburn M. P. (2005). Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal. Chem. 77, 6218–6224 10.1021/ac050846r [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.