

Abstract
Locating ligand binding sites and finding the functionally important residues from protein sequences as well as structures became one of the challenges in understanding their function. Hence a Naïve Bayes classifier has been trained to predict whether a given amino acid residue in membrane protein sequence is a ligand binding residue or not using only sequence based information. The input to the classifier consists of the features of the target residue and two sequence neighbors on each side of the target residue. The classifier is trained and evaluated on a nonredundant set of 42 sequences (chains with at least one transmembrane domain) from 31 alpha-helical membrane proteins. The classifier achieves an overall accuracy of 70.7% with 72.5% specificity and 61.1% sensitivity in identifying ligand binding residues from sequence. The classifier performs better when the sequence is encoded by psi-blast generated PSSM profiles. Assessment of the predictions in the context of three-dimensional structures of proteins reveals the effectiveness of this method in identifying ligand binding sites from sequence information. In 83.3% (35 out of 42) of the proteins, the classifier identifies the ligand binding sites by correctly recognizing more than half of the binding residues. This will be useful to protein engineers in exploiting potential residues for functional assessment.
1. Introduction
Membrane proteins are an important class of molecules which play key roles in various biologically important functions such as the maintenance of ionic and proton balance, transport of substrates, ions, energy, and information across the membrane, light harvesting, photosynthesis, and other biological processes [1]. Membrane proteins are classified mainly into two types: (i) formed by bundles of apolar transmembrane α-helices (TMH) and (ii) β-barrels (TMS). It has been estimated that nearly 45% of the drugs on the market target membrane protein receptors [2]. Advancement of high throughput technologies enable the whole genome sequencing of a number of organisms. It has been estimated that, in many genomes, TM proteins comprise 20–35% of all proteins [3, 4] and hence significant progress has been made in recent years in the determination of the structures of membrane proteins. Attempts have been made to determine the complete structures or domains of membrane proteins by crystallographic and solution or solid-state NMR spectroscopy methods [5, 6]. This in turn increases the entries significantly in various structural databases.
The vast majority of signal transduction events begin with the interactions of extracellular signaling molecules (ligands) to their respective membrane-bound receptors. However, identifying functional residues in proteins is a complex issue, even when atomic detailed structures are available [7]. Various approaches are evolved to study the functional residues [8, 9]. Zhang and Grigorov, 2006, studied a hierarchically organized structural relationship among protein binding sites using similarity networks [10]. Further the prediction of function from sequence and structural data has been extensively reviewed by Watson et al. [11]. These studies showed that almost all the functions of membrane proteins are mediated by interactions, which have a pivotal role in biological processes essential to life, and hence understanding protein-ligand interactions is of prime importance. Traditionally protein-ligand interactions are studied through laboratory experiments, which are often time consuming and costly [12, 13]. Accordingly computational methods have evolved and become increasingly dominant in understanding protein-ligand interactions.
Protein-ligand interactions have been extensively studied in recent years for various reasons [14–18] such as carbohydrate recognition, drug interaction, and DNA binding. Moreover, the prediction of ligand binding sites is an essential part of the drug discovery process. Knowing the location of binding sites greatly facilitates the search for hits, the lead optimization process, the design of site-directed mutagenesis experiments, and the hunt for structural features that influence the selectivity of binding in order to minimize the drug's adverse effects. Several reports throw light on the prediction as well as design of ligands and ligand binding sites [19–21] using amino acid residue features and various algorithms of machine learning [22–28]. Recently Xie and Hwang, 2015, reviewed the underlying concepts of the methods used by various tools for predicting protein-ligand binding sites [29]. Prediction of protein functional residues using sequence conservation and multiple sequence alignments have also been reported [30–32]. However, our understanding about the interaction of ligands with membrane proteins is very limited when compared to the other class of proteins known as globular proteins.
The recent explosion in the availability of complete genome sequences has led to the cataloging of tens of thousands of new proteins and putative proteins. Previous research focused mainly on prediction of membrane proteins and their types [33–36]. Due to the absence of intricacies of structural information the problem of ligand binding prediction in membrane proteins is ignored for a long time; however, the growth of the databases and construction of well-defined dataset paved ways to this study. Hence, in this work, we started from analyzing a set of nonredundant membrane protein-ligand complexes and derived several important sequence descriptors and trained a Naïve Bayes classifier. Bayesian classifiers are probabilistic models, based on Bayesian theorem, robust to real data noise and missing values [37]. The Naïve Bayes classifier is one of the most effective and efficient classification algorithms in the literature [38] showing good performance. With the help of the machine learning technique we tried to predict the ligand binding residues in membrane proteins from sequence information alone.
2. Materials and Methods
2.1. Data Sets
A data set of ligand-binding membrane proteins was extracted from structures of known membrane protein-ligand complexes in the Protein Data Bank [39]. The dataset was culled using the list of membrane proteins obtained from PDBTM [40], TMPDB [41], MPDB [42], and a large collection of membrane protein structures [43]. The resulting dataset consists of 31 membrane proteins from which 42 sequences (nonhomologous chains are taken into account) were considered in the present study with mutual sequence identity ≤ 30% using BLASTCLUST program from NCBI and each protein has at least 50 amino acid residues. All the structures have resolution better than 3.0 Å and R factor less than 0.3.
2.2. Ligand Information
Several reports in the literature used all nonprotein and nonwater molecules as ligands [44]. In this study, ligand is considered as a molecule that binds with the proteins that have structural and/or functional role and will be present within a cut-off distance of 4.5 Å from any of the protein atoms.
2.3. Definition of Ligand Binding Residues
Any of the atoms of the ligand is in contact with the any of the atoms of a particular residue, which is said to be in binding if the distance between them is lower than the cut-off value 4.5 Å. This definition has also been used in our previous studies. The 42 proteins sequences (chains with at least one transmembrane domain from 31 membrane proteins) in the dataset consist of 10657 residues in total and 1431 of them (13.43%) are identified as ligand binding residues.
2.4. Description of Naïve Bayes Classifier
We used the Naïve Bayes implementation in the Weka package from the University of Waikato, New Zealand [45, 46], for predicting the ligand binding residues in membrane proteins. For each input target residue, the classifier produces a Boolean output (with 1 denoting a binding residue and 0 denoting a nonbinding residue). The Naïve Bayes classifier assumes independence of the attributes given the class. For an input X = x 1, x 2,…, x n, a Naïve Bayes classifier assigns it a class label c by optimizing the posterior:
| (1) |
In the case of two-class classification (c ∈ {0,1}), this is equivalent to determining c by comparing the ratio likelihood with a parameter θ as in
| (2) |
c is predicted to be 1 if the ratio likelihood is greater than θ, and 0 otherwise. θ takes the value of 1. When a target residue and its neighbors were encoded using numeric features such as binding propensity and hydrophobicity, the numerical values were normalized using the normalization filter of Weka. We used leave-one-protein-out cross-validation to validate the classifier. In each round of experiment, all proteins except one were used as the training set and the remaining protein was used to test the classifier.
2.5. Naïve Bayes Classifier Using Sequence Based Parameters as Input
The input to the Naïve Bayes classifier contains the identities of 2n + 1 residues in the form of X = (x t−n, x t−n+1,…, x t−1, x t, x t−1,…, x t+n−1, x t+n), where x t is the property of target residue and x t−n, x t−n+1,…, x t−1 and x t+1, x t+n−1, x t+n are the identities of n residues on each side of the target residue. Different values of n from 1 to 7 were tried and the best performance was obtained when n = 2 (corresponding to a window size of 5). A training example is an ordered pair (X, c), where c ∈ {0,1}. 1 indicates that the target residue (the residue in the center of the input window) is a binding residue and 0 indicates that target residue is not a binding residue. For a test example X, the classifier outputs 1 (i.e., X is predicted to be a binding residue) or 0 (i.e., X is predicted to be a nonbinding residue) as the class label of X.
2.6. Naïve Bayes Classifier Using PSSM Profiles as Inputs
In the present study, a reference database with known nonredundant membrane protein sequences constructed separately was used for the purpose of generating PSSM profiles. We set parameters of PSI-BLAST [47] using BLOSUM62 substitution matrix, three iteration runs, and exception value 0.001. The other parameters are set using default values. The PSI-BLAST program by querying each protein chain against the nonredundant database is used to generate PSSM profiles which are in the form of 20N matrix, where N is the total number of amino acid residues in the queried protein sequence. Let the residue i be represented by a i = (a i,1,…, a i,20) where 1 ≤ i ≤ N. Each query residue is represented by a vector of 20 attributes. The input pattern to the Naïve Bayes classifier using the PSSM profile features for the residue i is x i = (ai k −,…, ai,…, ai k +) where k is the number of neighborhood residues on either side. We construct a matrix with window size s = 2k + 1 centered on the target residue i. The used profile x i is the form of a 20 × s matrix. These profiles are normalized into the range (0, 1) using the normalization option of Weka. Another set of attributes was also generated in such a way that it utilizes the values of BLOSUM62 matrix as features.
2.7. Performance Measures
We utilized the following parameters to evaluate the performance of our prediction method because no single performance measure provides a complete picture of performance of the classifier: accuracy, correlation coefficient (MCC), specificity, and sensitivity. These measures are defined as
| (3) |
where TP is the number of true positives (residues predicted to be binding residues that are in fact binding residues); FP is the number of false positives (residues predicted to be binding residues that are in fact not binding residues); TN is the number of true negatives (residues predicted to be nonligand binding residues that are in fact not ligand binding residues); FN is the number of false negatives (residues predicted to be nonligand binding residues that are in fact ligand binding residues); N is the total number of residues (TP + TN + FP + FN).
3. Results and Discussion
In this work, we trained a Naïve Bayes classifier to predict whether a given amino acid residue in a membrane protein sequence is ligand binding or not based on its sequence information. The Naïve Bayes classifier algorithm as implemented in Weka, a machine learning package, is adopted. The Naïve Bayes classifier is adopted for several reasons. The prime advantage of the Bayesian classifiers is that they are probabilistic models, based on Bayesian theorem, robust to real data noise and missing values [37]. The Naïve Bayes classifier assumes independence of the attributes used in classification but it has been tested on several artificial and real datasets, showing good performance even when strong attribute dependence is present. It is one of the most effective and efficient classification algorithms in the literature and is simple to implement and use [38].
3.1. Prediction Results
We used a dataset of 42 nonredundant transmembrane protein sequences (chains with at least one transmembrane domain from 31 membrane proteins) to train the Naïve Bayes classifier. Three methods were used to encode the protein sequence. They are amino acid properties, BLOSUM62 and PSSM profiles based encodings. In the sequence based method 48 important amino acid properties (for more details see [48, 49]) such as hydrophobicity, polarity, molecular weight, and charge, are used as features to encode the protein sequence. It will be noted that some of the parameters are related. In addition it may be advisable to keep a larger input in order to avoid losing useful parameters. And hence we used all the parameters. The best prediction performance measures were obtained for a window size of 5 (k = 2), keeping the central residue as the target residue. The overall prediction accuracy obtained by this method was 64% and the other prediction measures are shown in Table 1.
Table 1.
The prediction performance of the Naïve Bayes classifier using different encodings of sequence.
| Number | Type of encoding | Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|
| (1) | Sequence | 63.8% | 60.5% | 64.4% |
| (2) | Blosum62 | 67.9% | 59.0% | 69.4% |
| (3) | PSSM | 70.7% | 61.1% | 72.2% |
BLOSUM matrices are based on observed alignments. Though BLOSUM62 is tailored for comparisons of moderately distant proteins, it has been used in detecting closer relationships between proteins since they best represent the physiochemical characteristics of the amino acid substitutions. And hence we used this as a feature set for training the classifier. The prediction performance of classifier trained using BLOSUM62 elements as input was relatively better than that of the sequence based classifier. The performance measures are shown in Table 1.
Further we have incorporated the evolutionary information in the form of PSSM profiles based encoding. Position specific iterative BLAST (PSI BLAST) is a strong measure of residue conservation in a given location. When a residue is important for biological function it is conserved through cycles of PSI BLAST. The performance measures of the PSSM profiles based classifier are given in Table 1. Interestingly, the prediction accuracy (71%) is higher than the other two classifiers trained with the same dataset. The large predictive power of the evolutionary information as measured in this work may be due to the reason that residue conservation in protein families is directly related to its contribution to protein stability or function. It has been established by several researchers that the prediction of structural properties is significantly enhanced by the use of PSSM profiles compared to predictions based on unique representations of amino acid sequence and its environment. In addition the ligand binding in membrane proteins is largely influenced by their particular structural architecture.
We analyzed the predicted binding residues by the highest performance classifier to understand the reliability of the method. Interestingly, in 83.3% (35 out of 42) of the proteins, the classifier identifies the ligand binding sites by correctly recognizing more than half of the binding residues. In more than 90% of the proteins, the classifier correctly identifies at least 20% of the binding residues suggesting the possibility of using such classifiers to identify potential ligand-binding membrane proteins. The per protein prediction accuracy is given in Table 2. Moreover, those nonbinding residues predicted as binding residues will be in contact if we just increase the cutoff distance about 6–8 Å. Most of the false positive residues are either sequence neighbors or structural neighbors that can influence ligand binding.
Table 2.
Per protein prediction accuracy.
| Pdb ID | Number of residues | Sensitivity | Specificity | Accuracy |
|---|---|---|---|---|
| 1ar1_b | 252 | 50 | 83.2 | 79.44 |
| 1be3_e | 196 | 80 | 89.6 | 89.06 |
| 1ds8_h | 246 | 40 | 91.6 | 90.50 |
| 1ds8_l | 281 | 54.8 | 65.2 | 62.45 |
| 1ehk_a | 544 | 43.5 | 71 | 66.30 |
| 1ehk_b | 166 | 30 | 77 | 74.07 |
| 1eys_m | 318 | 52.6 | 75.5 | 67.20 |
| 1f88_a | 338 | 25.5 | 55.9 | 50.90 |
| 1fx8_a | 254 | 29.5 | 68 | 61.2 |
| 1iwo_a | 994 | 10 | 82.3 | 80.81 |
| 1j4n_a | 249 | 50 | 68.2 | 67.34 |
| 1j95_a | 98 | 60 | 62.9 | 62.77 |
| 1kb9_c | 385 | 66.7 | 79.4 | 74.80 |
| 1kb9_h | 93 | 20 | 73.8 | 70.78 |
| 1kb9_i | 55 | 100 | 38.6 | 47.06 |
| 1kf6_c | 130 | 100 | 19.8 | 23.02 |
| 1kf6_d | 119 | 36.8 | 62.5 | 58.26 |
| 1kpl_a | 430 | 23.8 | 72.8 | 70.43 |
| 1kqf_b | 289 | 47.8 | 88.4 | 78.60 |
| 1lgh_a | 56 | 79.4 | 16.7 | 57.69 |
| 1lnq_a | 301 | 0 | 79.8 | 78.45 |
| 1m0k_a | 222 | 59.5 | 68.3 | 65.13 |
| 1m56_a | 547 | 50.4 | 69.1 | 65.19 |
| 1m56_b | 260 | 33.3 | 79.9 | 77.73 |
| 1nek_c | 129 | 92.3 | 27.9 | 48 |
| 1nek_d | 113 | 92.6 | 3.7 | 25.69 |
| 1okc_a | 292 | 27.5 | 88.5 | 71.53 |
| 1oy9_a | 1006 | 100 | 84 | 84.03 |
| 1p49_a | 549 | 44.4 | 73.1 | 70.11 |
| 1p7b_a | 258 | 60 | 76.7 | 76.38 |
| 1ppj_g | 75 | 14.3 | 64.9 | 54.93 |
| 1pv7_a | 417 | 37.5 | 65.7 | 65.13 |
| 1q16_c | 224 | 57.1 | 42.9 | 38.89 |
| 1q90_a | 292 | 35.1 | 89.6 | 82.64 |
| 1q90_b | 212 | 50 | 78.3 | 66.35 |
| 1qle_c | 273 | 35.5 | 65.5 | 62.08 |
| 1rc2_b | 231 | 40 | 63.2 | 61.67 |
| 1v54_d | 144 | 84.2 | 42.1 | 47.86 |
| 1v54_g | 84 | 60 | 20 | 40 |
| 1v54_j | 58 | 30 | 81.8 | 72.22 |
| 1vf5_b | 138 | 42.4 | 83.2 | 73.13 |
| 1vf5_d | 168 | 61.1 | 82.9 | 80.49 |
|
| ||||
| Average | 50.18 | 66.04 | 65.25 | |
3.2. ROC Curve
The receiver operating characteristic curve (ROC curve) is a plot of the “sensitivity” (TP/(TP + FN)) versus the “1-specificity” (FP/(TN + FP)) [50]. It shows the tradeoff between true positive rate and false positive rate when different threshold values are used for the classifier. Figure 1 shows such a plot for the predictor with sequence, BLOSUM62, and PSSM profiles based encoding obtained using Weka. It could be noted from the figure that there is slight improvement while using PSSM profiles as the input features for the classifier.
Figure 1.
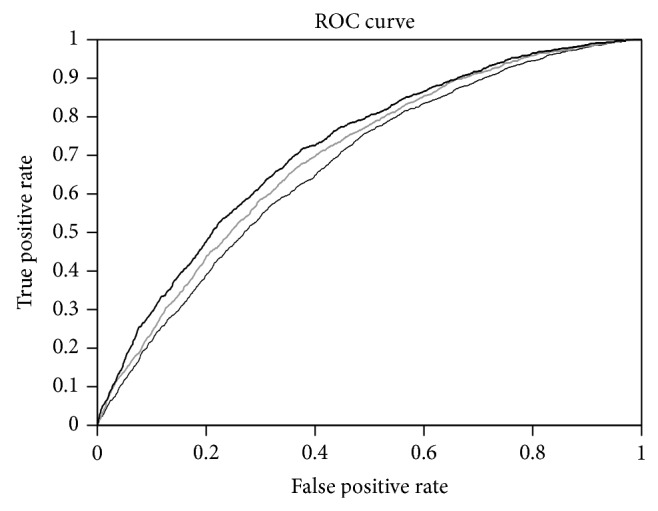
Showing ROC plots of classifiers trained with sequence (thin), BLOSUM62 (grey), and PSSM (dark) based encoding of the amino acid sequence of membrane proteins.
3.3. Comparison with Other Algorithms
Though several methods address the issue of protein-ligand interactions [22–27, 30–32], the method reported here is particularly for membrane proteins. Since the features derived are from the dataset of membrane protein sequences, its performance is very poor for globular proteins. However, for comparison of performance, few other algorithms implemented in Weka, for example, SMO, RBF network, Multilayer perceptron, IBk, ADTree, and J48, were also tested with the same data set, among which the analysis shows that the cross-validation sensitivity and net prediction accuracy are good for the current Naïve Bayes classifier (Table 3). Sequence based methods employing only sequence information presented in this work are new and will have a much wider application as no structure information will be required for prediction. We expect that this will trigger interest in the prediction of ligand binding sites in membrane proteins using machine learning methods and the performance will improve with the availability of more data.
Table 3.
The prediction performance of different machine learning algorithms.
| Algorithm | Accuracy % |
Sensitivity % |
Specificity % |
Net prediction % |
|---|---|---|---|---|
| Naïve Bayes | 70.9 | 59.5 | 72.2 | 65.9 |
| SMO | 86.5 | 0 | 100 | 50 |
| RBF network | 86.5 | 0 | 100 | 50 |
| Multilayer perceptron | 84.6 | 29.6 | 93.1 | 61.4 |
| IBk | 83.3 | 31.3 | 91.4 | 61.4 |
| ADTree | 86.6 | 4.5 | 99.3 | 51.9 |
| J48 | 87.4 | 28.1 | 93.5 | 60.8 |
3.4. WEB Based Tool
With the optimized parameters during cross-validation the current PSSM profiles based Naïve Bayes predictor has also been implemented as a web based tool which will be freely accessed from following url: http://tmbeta-genome.cbrc.jp/tm-lig/tm-lig.html. The only input to this predictor is the membrane protein sequence. The web server will automatically generate PSSMs of the given sequence against a reference data and use them as the input to the Naïve Bayes classifier trained for predictions of 42 membrane proteins. It requires less than a minute. The results presented include the raw probability scores and annotation of the residues whether ligand binding or not.
3.5. Identification of Binding Residues in Cytochrome BD Oxidase
We used the trained classifier to identify the binding residues for an unknown membrane protein sequence randomly selected from swissprot, a sequence database. The protein is cytochrome bd oxidase from E. coli, important for anaerobic oxidation [51]; its structure has not yet been determined. The predicted results were compared with the functional information available from the literature [51–54]. Interestingly, 50% of the residues predicted to be ligand binding are involved in interaction. In addition, few of the predicted residues belong to the segments which were experimentally determined to be functionally important [52]. Since the structure of this protein has not been determined, a homology model has been built (data not shown) and comparative analysis of binding sites with the related structures revealed that nearly 80% of the predicted residues are found to be along the lining of the proposed binding sites. This indeed increases the confidence level of using this predictor prior to planning for any mutagenesis or any functional assessment related experiment with more confidence rather than a random start.
4. Conclusion
Using a well constructed dataset, a Naïve Bayes predictor is trained and tested to predict the ligand binding residues in membrane proteins from amino acid sequence. Several encodings were used to test the performance of the predictor and PSSM profiles based predictor was shown to have better prediction accuracy (71%). With the level of success achieved in this study, putative ligand-binding sites predicted by the classifiers trained using a machine learning approach should be useful for guiding experimental investigations into the role of specific residues of a protein in its interaction with ligand, for example, by localizing candidate residues for mutagenesis. This paves ways for further improvements and predictions based on sequence methods for membrane protein-ligand interactions. Currently, we are investigating other machine learning classification methods to improve the accuracy of the prediction, which warrants further exploration.
Acknowledgments
The computational facilities at CBRC and the financial assistance from JSPS are gratefully acknowledged. The authors thank the anonymous reviewers for their valuable suggestions.
Conflict of Interests
The authors declare that they have no conflict of interests related to this paper.
References
- 1.Fleming K. G. Riding the wave: structural and energetic principles of helical membrane proteins. Current Opinion in Biotechnology. 2000;11(1):67–71. doi: 10.1016/s0958-1669(99)00056-7. [DOI] [PubMed] [Google Scholar]
- 2.Drews J. Drug discovery: a historical perspective. Science. 2000;287(5460):1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 3.Wallin E., von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Science. 1998;7(4):1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens T. J., Arkin I. T. Do more complex organisms have a greater proportion of membrane proteins in their genomes? Proteins: Structure, Function, and Genetics. 2000;39(4):417–420. doi: 10.1002/(sici)1097-0134(20000601)39:4x003C;417::aid-prot140x0003e;3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 5.Opella S. J., Nevzorov A., Mesleh M. F., Marassi F. M. Structure determination of membrane proteins by NMR spectroscopy. Biochemistry and Cell Biology. 2002;80(5):597–604. doi: 10.1139/o02-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyogoku Y., Fujiyoshi Y., Shimada I., et al. Structural genomics of membrane proteins. Accounts of Chemical Research. 2003;36(3):199–206. doi: 10.1021/ar0101279. [DOI] [PubMed] [Google Scholar]
- 7.Jones S., Thornton J. M. Searching for functional sites in protein structures. Current Opinion in Chemical Biology. 2004;8(1):3–7. doi: 10.1016/j.cbpa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Folkertsma S., van Noort P., van Durme J., et al. A family-based approach reveals the function of residues in the nuclear receptor ligand-binding domain. Journal of Molecular Biology. 2004;341(2):321–335. doi: 10.1016/j.jmb.2004.05.075. [DOI] [PubMed] [Google Scholar]
- 9.Pogozheva I. D., Przydzial M. J., Mosberg H. I. Homology modeling of opioid receptor-ligand complexes using experimental constraints. The AAPS Journal. 2005;7(2):E434–E448. doi: 10.1208/aapsj070243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z., Grigorov M. G. Similarity networks of protein binding sites. Proteins: Structure, Function, and Bioinformatics. 2006;62(2):470–478. doi: 10.1002/prot.20752. [DOI] [PubMed] [Google Scholar]
- 11.Watson J. D., Laskowski R. A., Thornton J. M. Predicting protein function from sequence and structural data. Current Opinion in Structural Biology. 2005;15(3):275–284. doi: 10.1016/j.sbi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Palmer R. A., Niwa H. X-ray crystallographic studies of protein-ligand interactions. Biochemical Society Transactions. 2003;31(5):973–979. doi: 10.1042/BST0310973. [DOI] [PubMed] [Google Scholar]
- 13.Clarkson J., Campbell I. D. Studies of protein-ligand interactions by NMR. Biochemical Society Transactions. 2003;31(5):1006–1009. doi: 10.1042/BST0311006. [DOI] [PubMed] [Google Scholar]
- 14.Simonson T., Archontis G., Karplus M. Free energy simulations come of age: protein-ligand recognition. Accounts of Chemical Research. 2002;35(6):430–437. doi: 10.1021/ar010030m. [DOI] [PubMed] [Google Scholar]
- 15.Hinz H. J. Thermodynamics of protein-ligand interactions: calorimetric approaches. Annual Review of Biophysics and Bioengineering. 1983;12:285–317. doi: 10.1146/annurev.bb.12.060183.001441. [DOI] [PubMed] [Google Scholar]
- 16.Perozzo R., Folkers G., Scapozza L. Thermodynamics of protein-ligand interactions: history, presence, and future aspects. Journal of Receptors and Signal Transduction. 2004;24(1-2):1–52. doi: 10.1081/rrs-120037896. [DOI] [PubMed] [Google Scholar]
- 17.Lian L. Y., Barsukov I. L., Sutcliffe M. J., Sze K. H., Roberts G. C. K. Protein-ligand interactions: exchange processes and determination of ligand conformation and protein-ligand contacts. Methods in Enzymology. 1994;239:657–700. doi: 10.1016/s0076-6879(94)39025-8. [DOI] [PubMed] [Google Scholar]
- 18.Hendlich M., Bergner A., Günther J., Klebe G. Relibase: design and development of a database for comprehensive analysis of protein-ligand interactions. Journal of Molecular Biology. 2003;326(2):607–620. doi: 10.1016/s0022-2836(02)01408-0. [DOI] [PubMed] [Google Scholar]
- 19.Taroni C., Jones S., Thornton J. M. Analysis and prediction of carbohydrate binding sites. Protein Engineering. 2000;13(2):89–98. doi: 10.1093/protein/13.2.89. [DOI] [PubMed] [Google Scholar]
- 20.Zvelebil M. J. J. M., Sternberg M. J. E. Analysis and prediction of the location of catalytic residues in enzymes. Protein Engineering Design & Selection. 1988;2(2):127–138. doi: 10.1093/protein/2.2.127. [DOI] [PubMed] [Google Scholar]
- 21.Bartlett G. J., Porter C. T., Borkakoti N., Thornton J. M. Analysis of catalytic residues in enzyme active sites. Journal of Molecular Biology. 2002;324(1):105–121. doi: 10.1016/S0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- 22.Mirceva G., Kulakov A. Improvement of protein binding sites prediction by selecting amino acid residues’ features. Journal of Structural Biology. 2015;189(1):9–19. doi: 10.1016/j.jsb.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 23.Chen P., Huang J. Z., Gao X. LigandRFs: random forest ensemble to identify ligand-binding residues from sequence information alone. BMC Bioinformatics. 2014;15(supplement 15):p. S4. doi: 10.1186/1471-2105-15-s15-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yan C., Wang Y. A graph kernel method for DNA-binding site prediction. BMC Systems Biology. 2014;8(supplement 4, article S10) doi: 10.1186/1752-0509-8-s4-s10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liou Y.-F., Charoenkwan P., Srinivasulu Y., et al. SCMHBP: prediction and analysis of heme binding proteins using propensity scores of dipeptides. BMC Bioinformatics. 2014;15(supplement 16, article S4) doi: 10.1186/1471-2105-15-S16-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Izidoro S. C., de Melo-Minardi R. C., Pappa G. L. GASS: identifying enzyme active sites with genetic algorithms. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu746. [DOI] [PubMed] [Google Scholar]
- 27.Liu B., Liu B., Liu F., Wang X. Protein Binding Site Prediction by Combining Hidden Markov Support Vector Machine and Profile-Based Propensities. The Scientific World Journal. 2014;2014:6. doi: 10.1155/2014/464093.464093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park B., Im J., Tuvshinjargal N., Lee W., Han K. Sequence-based prediction of protein-binding sites in DNA: comparative study of two SVM models. Computer Methods and Programs in Biomedicine. 2014;117(2):158–167. doi: 10.1016/j.cmpb.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 29.Xie Z.-R., Hwang M.-J. Molecular Modeling of Proteins. Vol. 1215. New York, NY, USA: Springer; 2015. Methods for predicting protein—ligand binding sites; pp. 383–398. (Methods in Molecular Biology). [DOI] [PubMed] [Google Scholar]
- 30.Capra J. A., Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007;23(15):1875–1882. doi: 10.1093/bioinformatics/btm270. [DOI] [PubMed] [Google Scholar]
- 31.Fischer J. D., Mayer C. E., Söding J. Prediction of protein functional residues from sequence by probability density estimation. Bioinformatics. 2008;24(5):613–620. doi: 10.1093/bioinformatics/btm626. [DOI] [PubMed] [Google Scholar]
- 32.Janda J.-O., Busch M., Kück F., Porfenenko M., Merkl R. CLIPS-1D: analysis of multiple sequence alignments to deduce for residue-positions a role in catalysis, ligand-binding, or protein structure. BMC Bioinformatics. 2012;13(1, article 55) doi: 10.1186/1471-2105-13-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gromiha M. M., Suwa M. Influence of amino acid properties for discriminating outer membrane proteins at better accuracy. Biochimica et Biophysica Acta. 2006;1764(9):1493–1497. doi: 10.1016/j.bbapap.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Gromiha M. M., Suwa M. Discrimination of outer membrane proteins using machine learning algorithms. Proteins: Structure, Function and Genetics. 2006;63(4):1031–1037. doi: 10.1002/prot.20929. [DOI] [PubMed] [Google Scholar]
- 35.Suresh M. X., Gromiha M. M., Suwa M. Membrane protein-ligand interactions: new insights from statistical analysis and molecular modeling studies. Seibutsu Butsuri. 2006;46:p. S408. [Google Scholar]
- 36.Suresh M. X., Gromiha M. M., Suwa M. Analysis and discrimination of ligand binding membrane proteins using a simple statistical approach. Proceedings of the 17th International Conference on Genome Informatics (GIW '06); 2006; Yokohama, Japan. p. p. 120. [Google Scholar]
- 37.Elkan C. CS97-557. San Diego, Calif, USA: Department of Computer Science and Engineering, University of California; 1997. Boosting and naive Bayesian learning. [Google Scholar]
- 38.Zhang H., Su J. Naive Bayesian classifiers for ranking. Proceedings of the 15th European Conference on Machine Learning (ECML '04); 2004; Pisa, Italy. [Google Scholar]
- 39.Berman H. M., Westbrook J., Feng Z., et al. The protein data bank. Nucleic Acids Research. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tusnády G. E., Dosztányi Z., Simon I. PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Research. 2005;33(supplement 1):D275–D278. doi: 10.1093/nar/gki002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ikeda M., Arai M., Okuno T., Shimizu T. TMPDB: a database of experimentally-characterized transmembrane topoligies. Nucleic Acids Research. 2003;31(1):406–409. doi: 10.1093/nar/gkg020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raman P., Cherezov V., Caffrey M. The membrane protein data bank. Cellular and Molecular Life Sciences. 2006;63(1):36–51. doi: 10.1007/s00018-005-5350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White S. H., Wimley W. C. Membrane protein folding and stability: physical principles. Annual Review of Biophysics and Biomolecular Structure. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 44.McCammon M. G., Robinson C. V. Structural change in response to ligand binding. Current Opinion in Chemical Biology. 2004;8(1):60–65. doi: 10.1016/j.cbpa.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 45.Witten I. H., Frank E. Data Mining: Practical Machine Learning Tools and Techniques. San Francisco, Calif, USA: Morgan Kaufmann; 2005. [Google Scholar]
- 46.Hall M., Frank E., Holmes G., Pfahringer B., Reutemann P., Witten I. H. The WEKA data mining software: an update. ACM SIGKDD Explorations Newsletter. 2009;11(1):10–18. doi: 10.1145/1656274.1656278. [DOI] [Google Scholar]
- 47.Altschul S. F., Madden T. L., Schäffer A. A., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gromiha M. M., Oobatake M., Sarai A. Important amino acid properties for enhanced thermostability from mesophilic to thermophilic proteins. Biophysical Chemistry. 1999;82(1):51–67. doi: 10.1016/s0301-4622(99)00103-9. [DOI] [PubMed] [Google Scholar]
- 49.Gromiha M. M., Suresh M. X. Discrimination of mesophilic and thermophilic proteins using machine learning algorithms. Proteins: Structure, Function and Genetics. 2008;70(4):1274–1279. doi: 10.1002/prot.21616. [DOI] [PubMed] [Google Scholar]
- 50.Baldi P., Brunak S., Chauvin Y., Andersen C. A. F., Nielsen H. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 2000;16(5):412–424. doi: 10.1093/bioinformatics/16.5.412. [DOI] [PubMed] [Google Scholar]
- 51.Zhang J., Hellwig P., Osborne J. P., et al. Site-directed mutation of the highly conserved region near the Q-loop of the cytochrome bd quinol oxidase from Escherichia coli specifically perturbs heme b 595 . Biochemistry. 2001;40(29):8548–8556. doi: 10.1021/bi010469m. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J., Hellwig P., Osborne J. P., Gennis R. B. Arginine 391 in subunit I of the cytochrome bd quinol oxidase from Escherichia coli stabilizes the reduced form of the hemes and is essential for quinol oxidase activity. The Journal of Biological Chemistry. 2004;279(52):53980–53987. doi: 10.1074/jbc.m408626200. [DOI] [PubMed] [Google Scholar]
- 53.Yang K., Zhang J., Vakkasoglu A. S., et al. Glutamate 107 in subunit I of the cytochrome bd quinol oxidase from Escherichia coli is protonated and near the heme d/heme b 595 binuclear center. Biochemistry. 2007;46(11):3270–3278. doi: 10.1021/bi061946. [DOI] [PubMed] [Google Scholar]
- 54.Gromiha M. M., Yabuki Y., Suresh M. X., Thangakani A. M., Suwa M., Fukui K. TMFunction: database for functional residues in membrane proteins. Nucleic Acids Research. 2009;37(1):D201–D204. doi: 10.1093/nar/gkn672. [DOI] [PMC free article] [PubMed] [Google Scholar]