

Abstract
ASCL1 is an important regulatory transcription factor in pulmonary neuroendocrine (NE) cell development, but its value as a biomarker of NE differentiation in lung adenocarcinoma (AD) and as a potential prognostic biomarker remains unclear. We examined ASCL1 expression in lung cancer samples of varied histologic subtype, clinical outcome and smoking status and compared with expression of traditional NE markers. ASCL1 mRNA expression was found almost exclusively in smokers with AD, in contrast to non-smokers and other lung cancer subtypes. ASCL1 protein expression by immunohistochemical (IHC) analysis correlated best with synaptophysin compared with chromogranin and CD56/NCAM. Analysis of a compendium of 367 microarray-based gene expression profiles in stage I lung adenocarcinomas identified significantly higher expression levels of the RET oncogene in ASCL1-positive tumors (ASCL1+) compared with ASCL1− tumors (q-value <10−9). High levels of RET expression in ASCL1+ but not in ASCL1− tumors was associated with significantly shorter overall survival (OS) in stage 1 (P = 0.007) and in all AD (P = 0.037). RET protein expression by IHC had an association with OS in the context of ASCL1 expression. In silico gene set analysis and in vitro experiments by ASCL1 shRNA in AD cells with high endogenous expression of ASCL1 and RET implicated ASCL1 as a potential upstream regulator of the RET oncogene. Also, silencing ASCL1 in AD cells markedly reduced cell growth and motility. These results suggest that ASCL1 and RET expression defines a clinically relevant subgroup of ∼10% of AD characterized by NE differentiation.
Keywords: prognostic biomarker, MASH1, lung cancer, neuroendocrine, microarray
Introduction
The clinical significance of neuroendocrine (NE) differentiation in lung adenocarcinoma (AD), and the most appropriate biomarkers for this assessment, has long been debated. In the absence of a gold standard, investigators have most commonly used immunohistochemical (IHC) staining of one or a combination of NE markers, such as chromogranin (CHGA), synaptophysin (SYP), neuron-specific enolase or neural cell adhesion molecule (CD56/NCAM) to assess the role of NE differentiation in lung cancer survival. Notably, previous reports have not included ASCL1, despite the pivotal role this gene has in the development of NE cells in the lung.
ASCL1 is a member of a family of basic helix-loop-helix (bHLH) transcriptional factors that have a major role in cell fate determination during neurogenesis.1 ASCL1 also has an essential role in the development of NE cells specifically in the lung. This is evidenced by ASCL1-null mice that lack pulmonary NE cells but form enteric neurons and NE cells in pancreatic islets and gut with positive staining for CHGA, SYP, calcitonin (CGRP/CALCA) and other NE markers.2 In addition, animal studies have suggested an important role for ASCL1 in the development of tumors with NE differentiation. Double-transgenic mice expressing ASCL1 and SV40 large T antigen develop tumors resembling non-small cell lung cancer (NSCLC) with NE features.3 Suppressing ASCL1 expression in lung cancer cell lines with RNA interference methods induces cell cycle arrest and apoptosis in vitro and inhibits xenograft growth in vivo.3 Collectively, these reports underscore the importance of ASCL1 in normal lung and in lung carcinogenesis.
This study examines the role of ASCL1 in the NE differentiation of lung adenocarcinoma (AD). We find expression of the RET oncogene to be highly associated with expression of ASCL1, and expression of both ASCL1 and RET identifies a subgroup of AD with aggressive behavior and poor patient outcome. ShRNA in vitro experiments support an important link between ASCL1 and RET in pulmonary AD and suggest that ASCL1 may influence the motility of AD cells.
Results
Comparative IHC analysis of NE markers (ASCL1, CHGA, SYP and CD56/NCAM)
Immunohistochemical staining quality of ASCL1, CHGA, SYP and CD56/NCAM was comparable, and all slides were interpretable. Scattered immunoreactive bronchiolar basal-located NE cells were considered as positive internal controls for the IHC reaction. Labeling indices and immunoreactivity for ASCL1, CHGA, SYP and CD56/NCAM for AD, small cell carcinoma (SCLC), large cell neuroendocrine carcinoma (LCNEC) and large cell carcinoma (LCC), respectively, are shown in Supplementary Table S1.
The pattern of ASCL1 immunoreactivity varied according to the tumor histological subtype. In AD, 15 of 83 cases (18%) showed ASCL immunoreactivity (ASCL1+AD), and ASCL1+ cells were focal and admixed with ASCL1− cells (Figures 1a and b), resulting in low-to-moderate labeling indices (Supplementary Table S1). ASCL1 immunoreactivity was more frequently identified than staining for CHGA (Figure 1c) and CD56/NCAM (Figure 1e); however, SYP (Figure 1d) was the most common IHC NE marker expressed in this group as shown in Supplementary Table S2 and illustrated in Figure 1p. One ASCL1+ AD was not immunoreactive for any of the other IHC NE markers, including SYP, whereas six ADs showing reactivity for at least one of the other NE markers were ASCL1− AD (Supplementary Table S3). In SCLC (Figures 1f and g) and LCNEC (Figures 1k and l), ASCL1 immunostaining was diffuse, resulting in moderate-to-high labeling indices (Supplementary Table S1). Among SCLC (Figures 1h–j) and LCNEC (Figures 1m–o), all cases were immunoreactive for ASCL1 as well as for most other IHC NE markers (CHGA, SYP and CD56/NCAM); whereas the four LCC were not immunoreactive for any IHC NE marker, including ASCL1 (Figure 1p).
Figure 1.
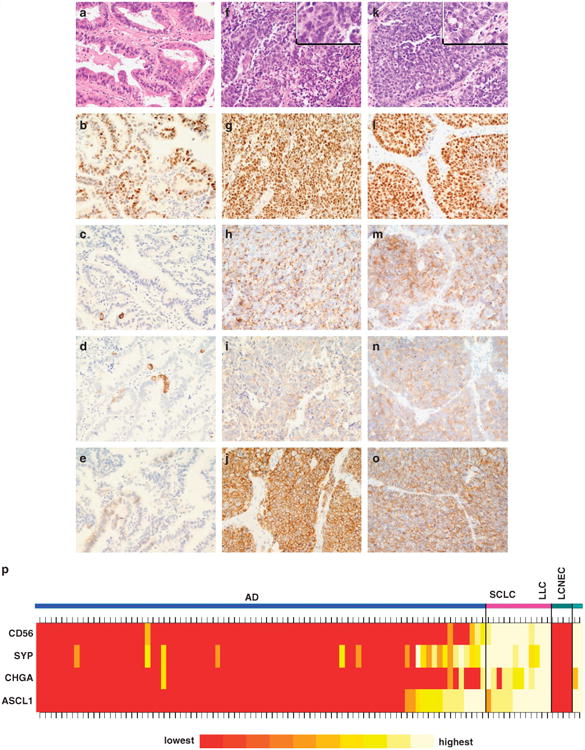
Immunohistochemical analysis. (a–e) AD characterized by ASCL1 mRNA expression (ASCL1 ± AD): (a) Adenocarcinoma with an acinar pattern. H&E, × 400. (b) ASCL1 protein expression (nuclear pattern, 70%, 2+), × 400. (c) CHGA (cytoplasmic pattern, 5%, 3+), × 400. (d) SYP (cytoplasmic pattern, 5%, 2+), × 400. (e) CD56/NCAM (membranous pattern, 5%, 2+), × 400. (f–j) SCLC: (f) High-grade tumor characterized by extensive areas of necrosis and cells with high nuclear to cytoplasmic ratio, delicate nuclear chromatin and inconspicuous nucleoli. H&E, × 400. (g) ASCL1 protein expression (nuclear pattern, 95%, 3+), × 400. (h) CHGA (cytoplasmic pattern, 100%, 2+), × 400. (i) SYP (cytoplasmic pattern, 100%, 1 +), × 400. (j) CD56/NCAM (membranous pattern, 100%, 2+), × 400. (k–o) LCNEC: (k) poorly differentiated tumor with high mitotic activity (> 10 mitotic figures/2mm2) and organoid nesting composed by cells with vesicular nuclei, evident nucleoli and moderate amount of cytoplasm. H&E, × 400. (l) ASCL1 protein expression (nuclear pattern, 95%, 3+). × 400 (m) CHGA (cytoplasmic pattern, 90%, 3+), × 400. (n) SYP (cytoplasmic pattern, 95%, 2+), × 400. (o) CD56/NCAM (membranous pattern, 90%, 3+), × 400. (p) Heat map of IHC protein expression of ASCL1, CHGA, SYP and CD56/NCAM.
ASCL1 mRNA expression is more prevalent in AD than in SQCC
The expressions of ASCL1 and other known NE markers in Data set 1 (see Supplemetary Figure S1 and details in the Supplementary Information) consisting of AD (n = 232), squamous cell carcinoma (SQCC) (n=100), SCLC (n = 15), adjacent non-neoplastic lung (n = 12), and LCC and LCNEC (n = 9) are shown as a heatmap in Figure 2. The heatmap includes the genes associated with squamous and glandular differentiation, DSG3 and NKX2.1/TTF1, respectively, and RET. ASCL1 expression had the highest correlation with calcitonin (CPRG/CALCA) (correlation coefficient = 0.65). However, in general, there was not a high correlation between the mRNA expression levels of the NE markers (Supplementary Table S4). Most NE markers had similar frequency of expression in the AD and SQCC. In contrast, ASCL1 was much more specific to AD than to SQCC. For a quantitative analysis, we selected a threshold for ASCL1 mRNA expression that corresponded to a positive IHC stain (Supplementary Figure S2). Microarray signal intensity levels above this threshold had excellent correlation with the IHC staining (correlation coefficient = 0.89, Supplementary Figure S2). In AD, 44 of 232 cases (19.0%) were ASCL1+. On the other hand, in 100 SQCC, only one of 100 cases (1.0%) was ASCL1+. Of the nine LCC, two had strong expression of ASCL1 and the other NE markers and were thus re-classified as LCNEC. Importantly, ASCL1 was also highly expressed in other NE lung tumors, including SCLC and carcinoid tumors (CT). Six of 10 (60%) and 14 of 15 (93%) CT and SCLC, respectively, were ASCL1+.
Figure 2.
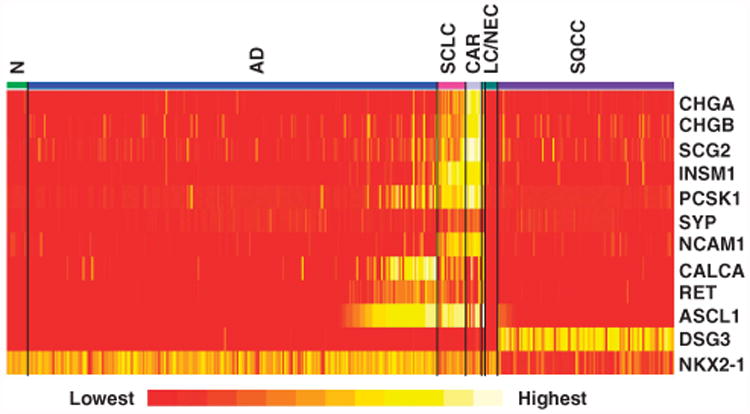
Heat map of the microarray Log2 expression values using known NE genes and RET. The map also includes SQCC and AD specific genes (DSG3 and NKX2.1, respectively). ASCL1 expression is much more frequent in AD than in SQCC. Each gene is represented by the most variable probeset with the highest s.d.
NE differentiation is rare in non-smoker adenocarcinomas
Expression levels of known NE markers were examined in the Mayo Clinic lung cancer samples from non-smokers, which included 75 AD, 32 CT, 8 adenosquamous carcinomas and SQCC and 125 adjacent non-neoplastic samples. Compared with non-neoplastic adjacent tissue, all NE markers were overexpressed in a majority of CT as expected (Supplementary Figure S3). In contrast, the expression levels of NE markers in AD were within the range of non-neoplastic tissue. We did not identify a subset of AD with marked overexpression of any of the NE markers. These data suggest that NE differentiation in lung AD is largely restricted to smokers.
Survival analysis of lung AD in relation to ASCL1 mRNA expression
Given that ASCL1 is expressed in about 20% of AD, to obtain sufficient statistical power for survival analysis, we combined our AD microarray data with four publicly available AD data sets where patient survival data were available (see Supplementary Information). We did not identify an association between the ASCL1 expression and survival or time to progression in stage I tumors nor in combined stage II-IV tumors (P≥0.28). However, we noted that the Kaplan-Meir (KM) survival curves for ASCL1+ and ASCL1− stage I tumors had different drop off profiles (Supplementary Figure S4). ASCL1+ patients who died had significantly shorter survival times causing a sharp drop in the survival curve. Upon further investigation, we noted that this pattern was consistent in all five data sets (Supplementary Table S5). In the combined data sets, ASCL1+ patients who died had statistically shorter survival times than ASCL1− patients who died (P<5 × 10−6). This trend did not change after excluding samples suspected of LCNEC in the Director's Challenge data set4 (see Supplementary Information). A statistical test indicated non-proportional hazards in ASCL1−/ASCL1+ tumors when we used censoring survival times of 6.5 or more years (P<0.05). These observations suggested that ASCL1+ status reflects a different underlying biology for these tumors. We assessed the role of traditional prognostic markers such as age, gender, race, smoking status (former or current), and tumor grade by Cox analysis. The only significant parameters were age (P= 10−6) and gender (P = 0.045). When we stratified stage I AD by ASCL1 status, we did not identify any differences in age and gender between the ASCL1+ and ASCL1− patients (group t-test and χ2-test, P≥0.09).
RET mRNA expression in ASCL1 ± AD is predictive of overall survival
To gain further insight into the biology of ASCL1+ tumors, we compared gene expression data for ASCL1+ and ASCL1− tumors. Gene expression analysis used Data set 2 (see Supplementary Figure S1 and details in Supplementary Information), which was a compendium of four sets of microarray data with follow-up information and more than 22 000 common Affymetrix probesets from 593 AD including 367 stage I AD. Genes (probesets) overexpressed in ASCL1+ compared with ASCL1− tumors were identified by signal to noise ratio (SNR). The top 12 genes (16 probesets) are listed in Supplementary Table S6 in the supplementary material. All genes in the list were significantly overexpressed in ASCL1+ tumors after correcting for multiple comparisons (q-value< 10−6).5 Interestingly, RET was the fourth most overexpressed gene (Supplementary Table S6). Among these genes, only RET was predictive of overall survival (OS) (see below). Furthermore, RET is a well-characterized oncogene in other tumors, such as thyroid cancers, and is a ‘druggable’ target. Therefore, we pursued this gene further. Figure 3a illustrates the expression of RET in ASCL1− and ASCL1+ stage I AD. The overexpression of RET in ASCL1+ tumors was consistent across all four data sets (Figure 3a). RET expression was more consistent with ASCL1 than other NE markers (Figure 2). Depending on the microarray signal threshold for calling a transcript present (Log2 signal intensity of 3.5 or 4.5), 91–95% of samples that expressed RET also expressed ASCL1. In contrast, only up to 55% of samples that expressed RET also expressed CHGA, CHGB, SCG2, SYP, INSM1, PCSK1 or NCAM1. Also noted was a small portion of samples with high levels of RET in the absence of ASCL1 (black circles in Figure 3a), indicating that in rare cases RET is expressed independently of ASCL1 (see Discussion).
Figure 3.
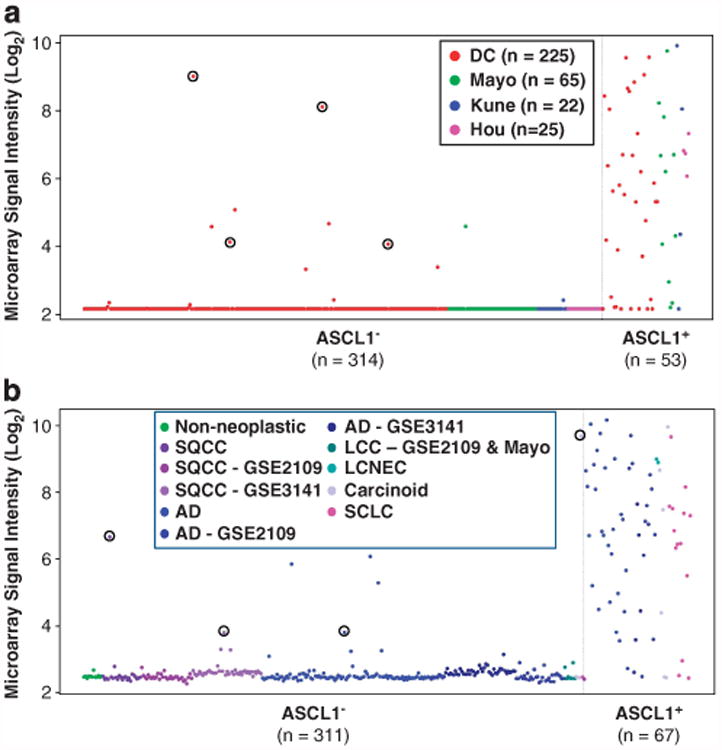
Overrepresentation of tumors expressing RET (211421_s_at probe set) in ASCL1+ compared with ASCL1− in (a) stage I ADs (Data set 2) and in (b) all lung cancers (Data set 1). This overrepresentation was consistent in all data sets. Few samples (shown in red circles) expressed RET, while ASCL1 was below noise level (arbitrary Log2 signal intensity of 3.5, see supplementary information). Data points correspond to the samples from the Mayo Clinic unless stated in the figure legends.
RET and ASCL1 co-expression was not limited to stage I AD. We observed a similar ASCL1/RET co-expression in all stages of AD and other lung cancer subtypes. Figure 3b illustrates the expression of RET in Data set 1. In SQCC where ASCL1 was largely absent, RET was also rarely expressed. In SCLC, CT and LCC, RET expression was largely restricted to ASCL1+ tumors.
As in Figure 3a, RET mRNA was detectable in a limited number of lung cancers that did not express ASCL1 (black circles in Figure 3b).
Two probesets corresponding to RET were significant in predicting the OS in stage I ASCL1+ tumors by Cox analysis (P-values of 0.029 and 0.006). High expression of RET was associated with shorter survival. In contrast, we did not identify an association between the OS and RET expression level in ASCL1− tumors. For illustration, we selected a threshold for ‘low’ and ‘high’ expression of RET in a KM plot as shown in Figure 4a (see Materials and Methods). The results did not appreciably change after excluding samples where an alternative diagnosis of LCNEC was possible.4 Using the same threshold, RET mRNA was also significant in predicting OS in all AD (Figure 4b).
Figure 4.
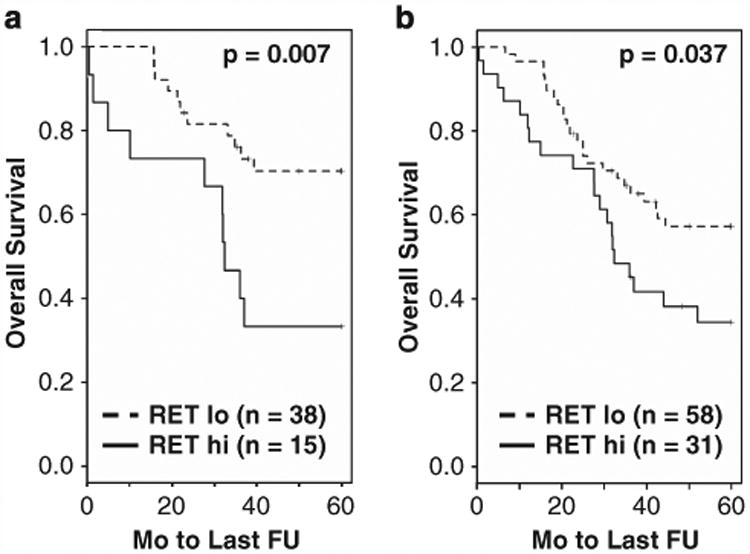
Overall survival in ASCL1+ stage I (a) and all (b) AD as a function of the RET mRNA expression level by microarrays (see Materials and methods).
RET protein expression analysis by IHC
A select set of our AD samples with expression data by the microarray analysis were immunostained for RET (see Supplementary Information). RET protein expression by IHC was more prevalent than expected from the microarray data, perhaps owing to the sensitivity of antibody to multiple variants of RET. We observed a blush staining in some ASCL1− cases with RET mRNA expression below detection levels by microarray (Figure 5a). However, ASCL1+ cases from aggressive tumors (patients that died of AD) usually had intensely stained areas (Figure 5b). There was a significant correlation between RET IHC scores and microarray signal intensity by the RET probeset used in OS analysis (rho = 0.43, P<0.02, data not shown). We also developed dual ASCL1/RET IHC stain and frequently found areas in tumors with positive staining for both proteins (see Figure 5c). However, because of the discrepancies in sensitivity and specificity of RET and ASCL1 antibodies or because of ASCL1 independent activation of RET (see Discussion), we also encountered areas with positive RET staining without ASCL1 expression.
Figure 5.
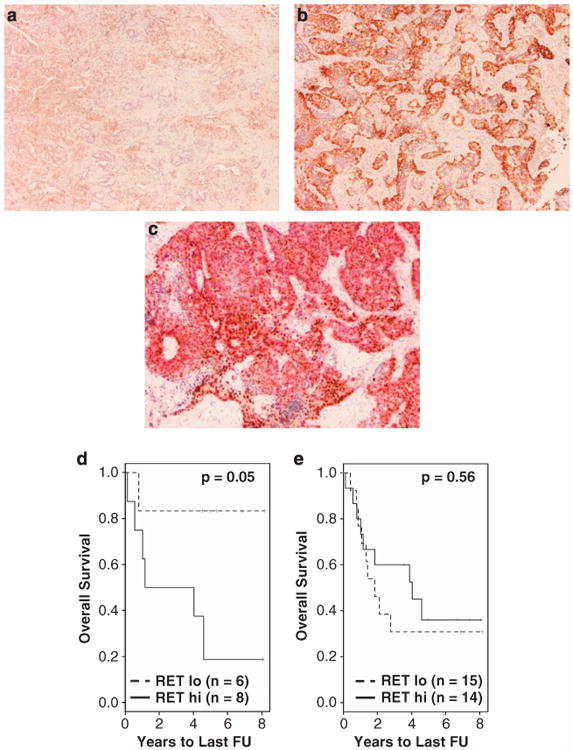
RET protein expression by IHC. RET staining in fatal adenocarcinoma was typically much less intense in ASCL1− (a) than in ASCL1+ (b) tumors. (c) Co-IHC of ASCL1 (nuclear brown staining) and RET (cytoplasmic red staining) identified areas with overlapping expression of the two proteins. (d) KM plot of 14 ASCL1+ AD samples indicates a significant association with OS (P = 0.05). (e) When samples were not stratified by the ASCL1 expression, RET IHC was not significant in predicting OS.
In the presence of ASCL1 IHC positivity, RET protein level was predictive of OS in AD (log-rank test P-value = 0.05, Figure 5d). When cases were not stratified by the ASCL1 expression, RET IHC was not predictive of OS (Figure 5e). Median survival times of tumors with ‘low’ levels of RET was slightly less than that of tumors with ‘high’ levels of RET, but this difference was statistically insignificant. These observations indicate that RET protein is predictive of OS survival only in the context of ASCL1 expression.
Gene set analysis of ASCL1± tumors
To gain further insight into the biology of ASCL1+ tumors, we performed gene set enrichment analysis by the GSA program and MSigDB version 3 with close to 7000 gene sets (see Supplementary Information). The results are shown in Supplementary Table S7. Notably, positively and negatively associated gene sets included OSADA_ASCL1_TARGETS_UP and _DN, respectively. These sets contained genes that were up and down regulated by ASCL1 in a study of ASCL1-transduced A549 lung AD cells.6 Importantly, RET was among the target genes upregulated by ASCL1 in the OSADA_ASCL1_TARGETS_UP set corroborating our observations in patient data. In the module corresponding to human chromosome and cytogenetic bands, 12q22 and 8p22 were enriched. Ten (of 37) genes (including ASCL1) on chr12q22 and 12 (of 41) genes on chr 8p22 were significantly overexpressed in ASCL1+ tumors. The high concentration of overexpressed genes in these regions suggested potential epigenetic or genetic changes.
We also examined mRNA correlates of aggressive behavior in stage I ASCL1+ AD. Tumors from patients who died in less than 3.5 years following surgery (n = 21) and from patients who survived more than 6 years following surgery (n = 20) were designated as aggressive and non-aggressive tumors, respectively. When we ranked probesets by SNR in aggressive versus non-aggressive tumors, two probesets for RET were among the list of top 10 probesets (data not shown). GSA analysis in these tumors identified six gene sets (Supplementary Table S8). Most notably, KANG-CISPLATIN-RESISTANCE-UP was positively associated with aggressive tumors. This set includes genes that were upregulated in gastric cancer cell lines resistant to cisplatin.7 Further experimental work is needed to determine the significance of these pathways in the outcome of ASCL1+ tumors.
Knockdown of ASCL1 reduces level of RET and inhibits cell growth in vitro
To examine the influence of ASCL1 in regulating RET, we knocked down ASCL1 by stable transfection of shRNAs in HCC1833 lung AD cell line, which has high endogenous levels of ASCL1 and RET. As shown by western blots in Figure 6a, RET protein was markedly reduced in cells transfected with ASCL1 shRNA (ASCL1-sh) compared with cells transfected with the empty vector (VEC-sh). These results are in line with previous data suggesting that ASCL1 acts upstream of RET. Also, consistent with the activating role of RET in the JAK/STAT3 pathway,8 the level of STAT3, a downstream target of RET was reduced in ASCL1-sh compared with VEC-sh (Figure 6a). We further examined the influence of ASCL1 in proliferation. As shown in Figure 6b, ASCL1-sh cells grew considerably slower than VEC-sh cells as measured by BrdU incorporation. ASCL1-sh doubling times were about twice longer than VEC-sh doubling times (data not shown). Furthermore, silencing ASCL1 expression significantly reduced cell motility as measured using a scratch assay (Supplementary Figure S5). These results indicate that lowering ASCL1 and RET reduces the invasive properties of the AD cells.
Figure 6.
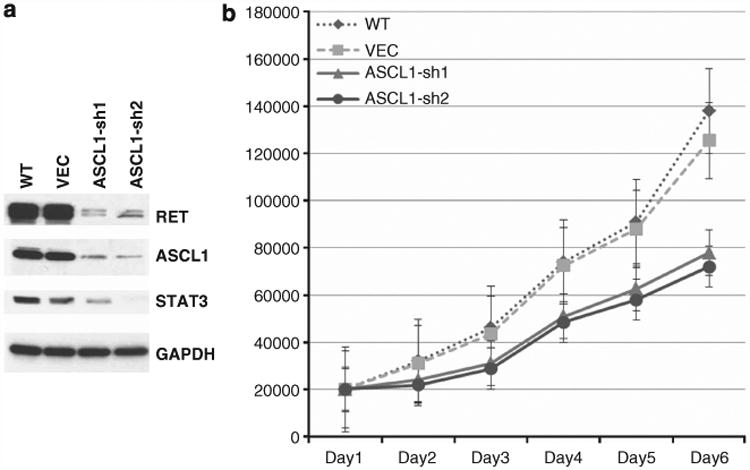
In vitro analysis of ASCL1 and RET in HCC1833 AD cells. (a) Wild-type cells (WT) express high levels of ASCL1 and RET. Knocking down of ASCL1 by stable shRNA transfection (ASCL1-sh1 and ACSL1-sh2) resulted in significant reduction of RET protein levels in these cells compared with cells transfected with empty vector (VEC), suggesting that ASCL1 acts upstream of RET. Also, STAT3 levels were reduced in ASCL1-sh cells, suggesting potential activation of the JAK/STAT3 pathway. (b) ASCL1-sh cells had markedly reduced proliferation rates compared with VEC and WT cells.
Discussion
Our results suggest that ASCL1 is a relevant NE biomarker of lung AD. Although other NE markers such as CHGA and SYP were commonly expressed in AD, only ASCL1 expression in conjunction with RET demonstrated significant differences in clinical outcome, suggesting a biologically and clinically relevant pathway of NE differentiation.
The prognostic significance of NE differentiation in AD has been controversial because of studies of varying sample size using different NE markers. Although we did not find a direct association between ASCL and survival, the unusual appearance of the KM curve for stage I AD ASCL1+ tumors, with a significant drop off profile compared with ASCL1− tumors led us to hypothesize that these tumors may be biologically different. Stage I AD in patients with ASCL1+ tumors who died had a significantly shorter survival than patients with ASCL1− tumors. This finding is supported by the study by Lonescu et al.9 that reported a similar survival profile in pulmonary AD using SYP, NCAM and CHGA as NE markers.9 Although Lonescu et al.9 did not specify the stage of AD, the similarity of the survival profile in that study with our findings in stage I AD (Supplementary Figure S4) is notable. Further analysis investigating the significance of this survival profile led us to identify increased expression of RET in ASCL1+ compared with ASCL1− lung AD. Importantly, in ASCL1+ AD, high expression of RET was significantly associated with shorter OS, an association not seen in ASCL1− tumors. Experimental and in silico gene set analyses identified RET as a gene driven by ASCL1 in human AD cells. These findings suggest that NE differentiation in lung AD as assessed by ASCL1 is clinically relevant.
A recent report by Fujiwara et al.10 examined the expression of NE markers including ASCL1 in lung AD. In their study, 100 genes that were correlated with ASCL1 expression were used to categorize ADs into alveolar cell, squamoid and NE subtypes. Compared with alveolar subtype, the NE subtype was significantly associated with poor survival in a univariate analysis (P = 0.0075) but not in a multivariate analysis that included stage. Importantly, RET expression was not examined, and analysis was not restricted to early-stage lung AD.
Both ASCL1 and RET are intimately involved in the control of neuronal fate in early development and are among the earliest genes to be expressed by undifferentiated neural crest cells. Homozygous knockout mice for each of these genes die soon after birth from respiratory abnormalities, manifesting similar effects on neuronal precursor cell differentiation fates.11–15 Gain-of-function mutations of RET are associated with the development of various human cancers, including medullary thyroid carcinoma (MTC), multiple endocrine neoplasias, phaeochromocytoma and parathyroid hyperplasia.16–18
It is unclear whether ASCL1 directly or indirectly regulates RET. A recent study by Rheinbay et al.19 examined direct regulatory targets of ASCL1 in human glioblastoma stem cells MGG8 by ChIP-seq analysis. In these cells, ASCL1 binding to the RET promoter was weak (data not shown). However, RET transcript levels were also low and undetectable. Therefore, it is unclear if the weak binding of ASCL1 to the RET promoter was due to weak interactions or due to the RET promoter being inaccessible. Previous mechanistic studies indicate that ASCL1 acts upstream of RET through induction of the homeobox gene transcription factor PHOX2A.13,20 Transduction of ASCL1 in A549 lung AD cells significantly increased the expression levels of a set of 46 genes including RET.6 Conversely, suppression of ASCL1 is linked to a decrease in the expression of RET. Fujiwara et al.10 partially knocked down ASCL1 by siRNA in DMS79 SCLC cell line, which has high endogenous ASCL1 expression, and analysis of their microarray expression data (GSE21216 on the GEO website) shows a more than two-fold decrease in the RET expression by the ASCL1 siRNA (data not shown). Similarly, silencing of ASCL1 in MTC cells in culture resulted in decreased RET expression and development of a mature thyroid c-cell differentiation pattern.21 We have observed similar effects in lung cancer cell line HCC1833 (Figure 6a). Silencing ASCL1 led to decrease in the level of RET and a decrease in cell proliferation and motility.
Notably, transcription factors other than ASCL1 also influence the regulation of RET expression.22 Specifically, although homozygous deletion of ASCL1 in sympathetic ganglia arrests the development of neuronal precursor cells due to the lack of PHOX2A, RET is still expressed due to the independent action of other transcription factors.20 Also, chromosomal rearrangement resulting in KIF5B–RET fusion protein is another potential ASCL1 independent mechanism of the overexpression of the RET oncogene.23 Interestingly, the RET fusion events are frequent in AD from non-smokers.24 However, we believe these fusions will be much less frequent in AD with NE differentiation as our study shows that these tumors occur in smokers (Supplementary Figure S3). We also noted a few instances of RET expression in the absence of ASCL1 (Figure 3).
To our knowledge, this is the first report of NE differentiation in AD being significantly more common in smokers than in never smokers (NS). Smoking is a well-established risk factor for lung carcinogenesis25,26 and results in different affected pathways.27–29 For AD, KRAS mutations are more frequently found in smokers, whereas EGFR mutations30 and EML4-ALK fusions31,32 are more common in NS. Further studies are needed to determine whether ASCL1+ tumors with RET expression is yet another distinct molecular subtype of pulmonary AD.
Using ASCL1 to identify a specific subtype of ADs could potentially lead to new therapeutic options. RNA interference33 and chimeric promoter constructs34 targeting ASCL1 indicated that this gene may be an effective molecular therapeutic target for lung cancer. Additionally, at least three of the genes that are the most highly associated with ASCL1 (RET, SCN3A and TUBB2B) can be targeted by currently available drugs (see Supplementary Table S6 in the Supplementary Material). Additional functional studies are needed to determine if any of these genes drive tumor progression. If results of such studies are positive, these targets may provide promising new treatment strategies.
In summary, we have provided evidence that ASCL1 expression identifies a distinct group of lung AD in smokers in which the RET oncogene is overexpressed. RET expression in these tumors is associated with shorter OS. We also demonstrated that ASCL1 is a potential upstream regulator of the RET oncogene.
Material and Methods
Mayo patient sample population for microarray expression profiling
Using the Mayo Clinic frozen tumor bank, lung specimens resected from 303 patients between 1997 and 2007 were selected. Neoadjuvant therapy was not given to any patient included in this study. Formalin-fixed paraffin-embedded H&E sections from the corresponding surgical specimens were reviewed, and the diagnoses were confirmed according to the 2004 World Health Organization classification of tumors.35 Bronchioloalveolar carcinoma variant of lung AD was excluded; hence, all ADs analyzed were clearly and predominantly invasive tumors. NS were characterized by less than 100 cigarettes per lifetime. Samples exclusively from NS (n = 130) were analyzed on the Illumina platform, and samples from former and current S patients (n = 186) and NS (n = 18) were analyzed on the Affymetrix platform. Supplementary Table S9 describes the clinicopathologic features of all Mayo samples. This study was conducted under Mayo Clinic Institutional Review Board approved protocols.
Immunohistochemical analysis
IHC procedures for ASCL1, CHGA, SYP, CD56/NCAM and RET including antibody clones and dilutions, and IHC score generations are in the Supplementary Material.
Associations between RET expression and survival in ASCL1 ± tumors in the microarray data
Details describing sample preparation for microarray analysis, selection of ASCL1 +/− tumors, and ASCL1 survival, and gene set analysis are in the Supplementary Material. By Cox proportional hazards regression analysis in R (coxph), two probesets corresponding to the RET oncogene (215771_x_at, 205879_x_at) had significant associations with OS in stage I AD after we censored follow-up data at 5 years (see Results). To visualize this association by a KM plot, we examined varying the threshold for ‘low’ and ‘high’ expression levels of RET (215771_x_at). Values in 3.0–6.5 were significant with P-values ranging 0.0005-0.029. Excluding AD samples where an alternative diagnosis of LCNEC was possible did not appreciably change these results.4 The reported KM plot used a threshold of 3.5, as signal intensities below this threshold are usually not detected by RT–PCR. If we did not censor at 5 years, P-values ranged from 0.00053 to 0.037 as the threshold changed from 3.0 to 6.5. Same probeset and threshold was used in the KM plot of all AD stages. Also, a KM plot for RET stains was generated by using the mean of all RET IHC scores (see above) as the threshold for selecting ‘low’ and ‘high’ levels.
Stable ASCL1 shRNA transfection and western blotting
HCC1833 cells were transfected with 15 μg of either empty vector or ASCL1 shRNA containing 5′-GCCAACAAGAAGATGAGTAAG-3′ target sequence (Sigma-Aldrich, St Louis, MO, USA; Cat# NM_004316.3-1019s21c1) using Fugene 6 HD (Roche Applied Science, Indianapolis, IN, USA). Clones were selected by puromycin (1 μg/ml) for 3 weeks. Single clones were expanded and tested by western blotting, using anti-ASCL1 (BD Biosciences, San Jose, CA, USA; cat# 556604), anti-RET (EPITOMICS, Burlingame, CA, USA; cat# EPR2871), anti-STAT3 (Cell Signaling Technology, Danvers, MA, USA; cat# 9132), and anti-GAPDH (Santa Cruz Biotechnology, Inc., Dallas, TX, USA; cat# sc-365062) antibodies.
Cell Proliferation and migration Assay
Cells were seated at 2.0 × 104 cells in 12-well plates. On the next day (Day 1), three wells containing wild-type cells (WT), cells stably transfected with empty vector (VEC) or ASCL1 shRNA (ASCL1-sh) were collected, and the numbers of living and dead cells were counted by a Countess automated cell counter (Life Technologies, Grand Island, NY, USA). Cell counting was continued daily until Day 6, and the results were plotted. Cell migration was measured using a scratch assay. WT, VEC or ASCL1-sh cells were seeded at 1.0 × 105 cells in an eight-well chamber slide and cultured for 48 h. The cell monolayer was wounded with a p10 tip. The slides were then washed and incubated at 37 °C in growth medium for 3 days. Phase contrast images were taken on Day 1 and Day 3, and the distance moved toward the center for each sample was compared.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported by a Waterman Biomarker Discovery grant and by the Mayo Clinic Center for Individualized Medicine.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Kageyama R, Nakanishi S. Helix-loop-helix factors in growth and differentiation of the vertebrate nervous system. Curr Opin Genet Dev. 1997;7:659–665. doi: 10.1016/s0959-437x(97)80014-7. [DOI] [PubMed] [Google Scholar]
- 2.Borges M, Linnoila RI, van de Velde HJ, Chen H, Nelkin BD, Mabry M, et al. An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature. 1997;386:852–855. doi: 10.1038/386852a0. [DOI] [PubMed] [Google Scholar]
- 3.Linnoila RI, Zhao B, DeMayo JL, Nelkin BD, Baylin SB, DeMayo FJ, et al. Constitutive achaete-scute homologue-1 promotes airway dysplasia and lung neuroendocrine tumors in transgenic mice. Cancer Res. 2000;60:4005–4009. [PubMed] [Google Scholar]
- 4.Bryant CM, Albertus DL, Kim S, Chen G, Brambilla C, Guedj M, et al. Clinically relevant characterization of lung adenocarcinoma subtypes based on cellular pathways: an international validation study. PLoS ONE. 2010;5:e11712. doi: 10.1371/journal.pone.0011712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osada H, Tomida S, Yatabe Y, Tatematsu Y, Takeuchi T, Murakami H, et al. Roles of achaete-scute homologue 1 in DKK1 and E-cadherin repression and neuroendocrine differentiation in lung cancer. Cancer Res. 2008;68:1647–1655. doi: 10.1158/0008-5472.CAN-07-5039. [DOI] [PubMed] [Google Scholar]
- 7.Kang HC, Kim IJ, Park JH, Shin Y, Ku JL, Jung MS, et al. Identification of genes with differential expression in acquired drug-resistant gastric cancer cells using high-density oligonucleotide microarrays. Clin Cancer Res. 2004;10(1 Pt 1):272–284. doi: 10.1158/1078-0432.ccr-1025-3. [DOI] [PubMed] [Google Scholar]
- 8.Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005;16:441–467. doi: 10.1016/j.cytogfr.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 9.Ionescu DN, Treaba D, Gilks CB, Leung S, Renouf D, Laskin J, et al. Nonsmall cell lung carcinoma with neuroendocrine differentiation-an entity of no clinical or prognostic significance. Am J Surg Pathol. 2007;31:26–32. doi: 10.1097/01.pas.0000213319.04919.97. [DOI] [PubMed] [Google Scholar]
- 10.Fujiwara T, Hiramatsu M, Isagawa T, Ninomiya H, Inamura K, Ishikawa S, et al. ASCL1-coexpression profiling but not single gene expression profiling defines lung adenocarcinomas of neuroendocrine nature with poor prognosis. Lung Cancer. 2012;75:119–125. doi: 10.1016/j.lungcan.2011.05.028. [DOI] [PubMed] [Google Scholar]
- 11.Dauger S, Renolleau S, Vardon G, Nepote V, Mas C, Simonneau M, et al. Ventilatory responses to hypercapnia and hypoxia in Mash-1 heterozygous newborn and adult mice. Pediatr Res. 1999;46:535–542. doi: 10.1203/00006450-199911000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Burton MD, Kawashima A, Brayer JA, Kazemi H, Shannon DC, Schuchardt A, et al. RET proto-oncogene is important for the development of respiratory CO2 sensitivity. J Auton Nerv Syst. 1997;63:137–143. doi: 10.1016/s0165-1838(97)00002-7. [DOI] [PubMed] [Google Scholar]
- 13.Dauger S, Guimiot F, Renolleau S, Levacher B, Boda B, Mas C, et al. MASH-1/RET pathway involvement in development of brain stem control of respiratory frequency in newborn mice. Physiol Genomics. 2001;7:149–157. doi: 10.1152/physiolgenomics.00056.2001. [DOI] [PubMed] [Google Scholar]
- 14.Huber K, Bruhl B, Guillemot F, Olson EN, Ernsberger U, Unsicker K. Development of chromaffin cells depends on MASH1 function. Development. 2002;129:4729–4738. doi: 10.1242/dev.129.20.4729. [DOI] [PubMed] [Google Scholar]
- 15.Shoba T, Dheen ST, Tay SS. Retinoic acid influences the expression of the neuronal regulatory genes Mash-1 and c-ret in the developing rat heart. Neurosci Lett. 2002;318:129–132. doi: 10.1016/s0304-3940(01)02491-0. [DOI] [PubMed] [Google Scholar]
- 16.Lodish MB, Stratakis CA. RET oncogene in MEN2, MEN2B, MTC and other forms of thyroid cancer. Expert Rev Anticancer Ther. 2008;8:625–632. doi: 10.1586/14737140.8.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marsh DJ, Gimm O. Multiple endocrine neoplasia: types 1 and 2. Adv Otorhinolaryngol. 2011;70:84–90. doi: 10.1159/000322479. [DOI] [PubMed] [Google Scholar]
- 18.Kouvaraki MA, Shapiro SE, Perrier ND, Cote GJ, Gagel RF, Hoff AO, et al. RET proto-oncogene: a review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid. 2005;15:531–544. doi: 10.1089/thy.2005.15.531. [DOI] [PubMed] [Google Scholar]
- 19.Rheinbay E, Suva ML, Gillespie SM, Wakimoto H, Patel AP, Shahid M, et al. An aberrant transcription factor network essential for wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013;3:1567–1579. doi: 10.1016/j.celrep.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo L, Tiveron MC, Anderson DJ. MASH1 activates expression of the paired homeodomain transcription factor Phox2a, and couples pan-neuronal and subtype-specific components of autonomic neuronal identity. Development. 1998;125:609–620. doi: 10.1242/dev.125.4.609. [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Carson-Walter EB, Baylin SB, Nelkin BD, Ball DW. Differentiation of medullary thyroid cancer by C-Raf-1 silences expression of the neural transcription factor human achaete-scute homolog-1. Surgery. 1996;120:168–172. doi: 10.1016/s0039-6060(96)80284-4. discussion 73. [DOI] [PubMed] [Google Scholar]
- 22.Huber K, Karch N, Ernsberger U, Goridis C, Unsicker K. The role of Phox2B in chromaffin cell development. Dev Biol. 2005;279:501–508. doi: 10.1016/j.ydbio.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 23.Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. 2012;30:4352–4359. doi: 10.1200/JCO.2012.44.1477. [DOI] [PubMed] [Google Scholar]
- 25.Freedman ND, Leitzmann MF, Hollenbeck AR, Schatzkin A, Abnet CC. Cigarette smoking and subsequent risk of lung cancer in men and women: analysis of a prospective cohort study. Lancet Oncol. 2008;9:649–656. doi: 10.1016/S1470-2045(08)70154-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khuder SA. Effect of cigarette smoking on major histological types of lung cancer: a meta-analysis. Lung Cancer. 2001;31:139–148. doi: 10.1016/s0169-5002(00)00181-1. [DOI] [PubMed] [Google Scholar]
- 27.Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE. 2008;3:e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Massion PP, Zou Y, Chen H, Jiang A, Coulson P, Amos CI, et al. Smoking-related genomic signatures in non-small cell lung cancer. Am J Respir Crit Care Med. 2008;178:1164–1172. doi: 10.1164/rccm.200801-142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powell CA, Spira A, Derti A, DeLisi C, Liu G, Borczuk A, et al. Gene expression in lung adenocarcinomas of smokers and nonsmokers. Am J Respir Cell Mol Biol. 2003;29:157–162. doi: 10.1165/rcmb.2002-0183RC. [DOI] [PubMed] [Google Scholar]
- 30.Boldrini L, Ali G, Gisfredi S, Ursino S, Baldini E, Melfi F, et al. Epidermal growth factor receptor and K-RAS mutations in 411 lung adenocarcinoma: a population-based prospective study. Oncol Rep. 2009;22:683–691. doi: 10.3892/or_00000488. [DOI] [PubMed] [Google Scholar]
- 31.Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi T, Sonobe M, Kobayashi M, Yoshizawa A, Menju T, Nakayama E, et al. Clinicopathologic features of non-small-cell lung cancer with EML4-ALK fusion gene. Ann Surg Oncol. 2010;17:889–897. doi: 10.1245/s10434-009-0808-7. [DOI] [PubMed] [Google Scholar]
- 33.Osada H, Tatematsu Y, Yatabe Y, Horio Y, Takahashi T. ASH1 gene is a specific therapeutic target for lung cancers with neuroendocrine features. Cancer Res. 2005;65:10680–10685. doi: 10.1158/0008-5472.CAN-05-1404. [DOI] [PubMed] [Google Scholar]
- 34.Poulsen TT, Pedersen N, Juel H, Poulsen HS. A chimeric fusion of the hASH1 and EZH2 promoters mediates high and specific reporter and suicide gene expression and cytotoxicity in small cell lung cancer cells. Cancer Gene Ther. 2008;15:563–575. doi: 10.1038/cgt.2008.24. [DOI] [PubMed] [Google Scholar]
- 35.Travis WD World Health Organization. International Agency for Research on Cancer., International Association for the Study of Lung Cancer., International Academy of Pathology. Pathology and Genetics of Tumours of The Lung, Pleura, Thymus and Heart. Lyon Oxford: IARC Press Oxford University Press; 2004. distributor. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.