

Abstract
The thymidine kinases (TK) of alphaherpesviruses phosphorylate nucleosides, allowing viral replication in non-dividing cells. They also phosphorylate acyclovir (ACV), a specific antiviral when modified. Despite encoding a TK homolog, Kaposi's sarcoma-associated herpesvirus (KSHV), a gammaherpesvirus, is relatively immune to the effects of ACV. In this issue, Gill et al (2015) show that rather than functioning as a thymidine kinase, the KSHV-TK homolog has evolved a unique function as a tyrosine kinase that is autophosphorylated. KSHV-TK autophosphorylation of three SH2 domains leads to Crk binding and likely sequestration of Crk from focal adhesions. KSHV-TK also binds to FAK with a concurrent loss of phosphorylation in the focal adhesions, leading to a loss of cell morphology and membrane blebbing. Rather than acting to create nucleotide pools for replication, the KSHV-TK homolog may play a pivotal role in viral pathogenesis by altering focal adhesions and cell detachment.
See also: MB Gill et al (February 2015)
Alphaherpesviruses encode thymidine kinases that allow the viruses to replicate in non-dividing cells that may not otherwise produce enough nucleotides for rapid viral replication. For example, herpes simplex virus 1 (HSV-1) goes latent in non-dividing neuronal cells and must reactivate from latency and replicate in these cells. In contrast, betaherpesviruses, like cytomegalovirus (CMV), replicate in dividing cells and appear to establish latency in dividing cells as well. Accordingly, the betaherpesviruses do not encode thymidine kinases for replication. The third herpesvirus family, the gammaherpesviruses, including Epstein–Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), encode a homolog of thymidine kinase with low homology to the alphaherpesvirus TKs.
Acyclovir (ACV) is a very effective and safe antiviral. It is phosphorylated by the thymidine kinase of alphaherpesviruses and then incorporated by the viral polymerase into the ongoing DNA chain, acting as a chain terminator. The ability of ACV to be activated by the viral thymidine kinase but not the host enzyme provides its tremendous specificity for infected cells. ACV is not effective in vivo against betaherpesviruses, likely due to their lack of thymidine kinases. However, the effect of acyclovir on gammaherpesviruses is more complex. EBV replication is inhibited by acyclovir in vitro, and there appears to be some decrease in viral shedding in saliva in vivo, though the mechanism of EBV activation of ACV remains unclear. In contrast, KSHV showed very little sensitivity to acyclovir in vitro. Only at high doses was ACV able to show significant inhibition of KSHV replication, despite the fact that KSHV encodes a thymidine kinase homolog (Kedes & Ganem, 1997).
The lack of KSHV sensitivity to ACV might be explained to some degree by the finding that the KSHV-TK has relatively weak thymidine kinase activity (Gustafson et al, 2000). Therefore, it was hypothesized that the KSHV-TK has evolved additional functions. In support of this hypothesis, KSHV-TK localizes to the cytoplasm, unlike the HSV-1 thymidine kinase, which is predominantly found in the nucleus. Overexpression of the KSHV-TK led to dramatic alterations in cell morphology including cell rounding and increased detachment (Gustafson et al, 2000). The HSV-1 and EBV thymidine kinases do not induce similar cellular morphology changes. Rhadinoviruses are one arm of the gammaherpesvirus family and include KSHV, rhesus rhadinovirus (RRV), and herpesvirus saimiri (HVS), among others, while EBV is in a distinct grouping. The KSHV-TK, as well as those of RRV and HVS, localizes to filamentous networks (Gill et al, 2005). Intriguingly, the KSHV-TK is tyrosine-phosphorylated, a property unique to the thymidine kinases of rhadinoviruses (Gill et al, 2005). Therefore, through tyrosine phosphorylation, the KSHV-TK has likely evolved additional functions that may play a role during lytic infection. This alternative role could explain why the KSHV-TK homolog was maintained during viral evolution despite its weak thymidine kinase activity.
In this issue, Gill and colleagues make the striking discovery that KSHV-TK has tyrosine kinase activity. Furthermore, KSHV-TK appears to be autophosphorylated. KSHV-TK with a triple glycine mutant in the active site of the KSHV-TK kinase, and is expected to be kinase dead, is no longer tyrosine-phosphorylated. Thus, the weak thymidine kinase domain might behave as a tyrosine kinase. KSHV-TK purified in E. coli is also tyrosine-phosphorylated, while the EBV and murine herpesvirus 4 thymidine kinase homologs are not. From this, they conclude that the KSHV-TK is a tyrosine kinase that is autophosphorylated. Using mass spectrometry and mutational analysis, they map the phosphorylation sites on KSHV-TK and find that three tyrosines are phosphorylated, Y-65, Y-85, and Y-120.
KSHV-TK induces cell contraction, and they find that there is also membrane blebbing in the absence of overt cell death. They also show that KSHV-TK is associated with actin filaments and induces central actin stress fibers in the cell. The stress fibers are inhibited by a dominant-negative RhoA, as well as a drug inhibitor of RhoA. RhoA is a GTPase that is involved in the remodeling of the actin cytoskeleton and associates with focal adhesions. RhoA is more strongly associated with GTP in KSHV-TK-expressing cells. KSHV-TK expression leads to a decrease in the phosphorylated form of focal adhesion kinase (FAK) and of the FAK-associated scaffold protein paxillin. The kinase-dead mutant of KSHV-TK does not induce dephosphorylation of FAK or paxillin. FAK immunoprecipitates with KSHV-TK and with a mutant that has all 3 phospho-tyrosine sites mutated to phenylalanines, but fails to immunoprecipitate with the kinase-dead mutant. Neither the kinase-dead mutant nor the triple tyrosine KSHV-TK mutant induce cell contraction or membrane blebbing. In FAK knockout cells and in cells where paxillin is knocked down, the wild-type KSHV-TK is unable to induce cellular contraction, indicating that FAK as well as RhoA is required for this effect.
Two of the phosphorylated tyrosines in KSHV-TK have SH2-like domains with proline at the +4 position (YxxP). This motif is known to be a Crk binding domain. The Crk family is a family of adapter proteins that bind to both SH2 and SH3 domains and are associated with FAK and paxillin in focal adhesions. Crk1, Crk2, and CrkL all bind to KSHV-TK but not to the version where Y-65 and Y-85, the two SH2 YxxP domains, are mutated to phenylalanines. Crk1 and CrkL are also tyrosine-phosphorylated in the presence of KSHV-TK but not in the presence of the tyrosine mutant, indicating that binding to the SH2-like domains of KSHV-TK appears to lead to Crk phosphorylation. Crk family members are known to promote cellular adhesion through binding to Rho-GTPase exchange factors and paxillin. This sets up a model where KSHV-TK is autophosphorylated allowing Crk family members to bind. This binding sequesters Crk family members away from Rho-GTPase exchange factors and paxillin allowing Rho-GTPase to be activated while at the same time KSHV-TK binds to FAK. Overall, this leads to dephosphorylation of FAK and paxillin causing disruption of the focal adhesions and, ultimately, cell contraction (see Fig1).
Figure 1.
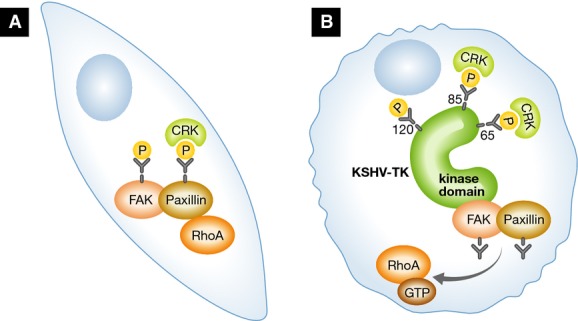
KSHV-TK is a tyrosine kinase that induces cell rounding and membrane blebbing
In untransduced cells (A), FAK and paxillin have normal tyrosine phosphorylation and the phosphorylation is protected, directly or indirectly, by Crk. In KSHV-TK transduced cells (B), KSHV-TK is autophosphorylated and Crk binds to the KSHV-TK SH2 domains and FAK binds to the KSHV-TK kinase domain. FAK and paxillin are no longer tyrosine-phosphorylated, leading to cell contraction and membrane blebbing through a RhoA/ROCK-mediated mechanism.
To provide relevance for these novel KSHV-TK functions during viral infection, the authors demonstrate that KSHV cells undergoing lytic infection induce similar cellular morphology changes as the cells expressing KSHV-TK alone. The same RhoA inhibitor that prevents cell contraction in KSHV-TK-expressing cells also blocks cell contraction in cells undergoing lytic replication. While this does not formally demonstrate that KSHV-TK is the viral gene responsible for this effect, the finding is certainly enticing. Additional work using a recently developed KSHV bacterial artificial chromosome to make mutants of the KSHV-TK kinase domain or tyrosine mutants will be necessary to demonstrate that KSHV-TK is solely responsible for the cellular contraction during lytic infection. However, the authors also show that when they replace the thymidine kinase gene of murine herpesvirus 4, which does not induce cellular contraction or membrane blebbing, with KSHV-TK the recombinant virus now induces cellular contraction, indicating that expression of KSHV-TK in the context of another herpesvirus is sufficient to induce this effect.
A remaining question is why has the KSHV-TK evolved to dephosphorylate FAK and paxillin and induce cell contraction and membrane blebbing? KSHV is the etiologic agent of Kaposi's sarcoma, an endothelial-based tumor. In the KS tumor cells, KSHV is predominantly latent, expressing only a small subset of genes, which does not include the KSHV-TK. During latent infection in endothelial cells, KSHV induces FAK phosphorylation through activation of integrin αVβ3 signaling (DiMaio et al, 2011). It is possible that activation of FAK and the alteration of focal adhesions during latent infection must be reversed for efficient lytic infection in specific cell settings. Alternatively, the cell contraction and blebbing could be helpful for replication or virus egress. Another hypothesis raised by the authors of the paper is that the detachment of the cells could aid in viral spread by allowing the cells undergoing lytic replication to migrate to distant sites. Further work with mutant viruses will be necessary to tease out the necessity for the KSHV-TK to have evolved tyrosine kinase activity.
References
- DiMaio TA, Gutierrez KD, Lagunoff M. Latent KSHV infection of endothelial cells induces integrin beta3 to activate angiogenic phenotypes. PLoS Pathog. 2011;7:e1002424. doi: 10.1371/journal.ppat.1002424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MB, Murphy JE, Fingeroth JD. Functional divergence of Kaposi's sarcoma-associated herpesvirus and related gamma-2 herpesvirus thymidine kinases: novel cytoplasmic phosphorylation that alter cellular morphology and disrupt adhesion. J Virol. 2005;79:14647–14659. doi: 10.1128/JVI.79.23.14647-14659.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MB, Turner R, Stevenson PG, Way M. KSHV-TK is a tyrosine kinase that disrupts focal adhesions and induces Rho-mediated cell contraction. EMBO J. 2015;34:448–465. doi: 10.15252/embj.201490358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson EA, Schinazi RF, Fingeroth JD. Human Herpesvirus 8 open reading frame 21 is a thymidine and thymidylate kinase of narrow substrate specificity that efficiently phosphorylates zidovudine but not ganciclovir. J Virol. 2000;74:684–692. doi: 10.1128/jvi.74.2.684-692.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedes DH, Ganem D. Sensitivity of Kaposi's sarcoma-associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J Clin Invest. 1997;99:2082–2086. doi: 10.1172/JCI119380. [DOI] [PMC free article] [PubMed] [Google Scholar]