
Figure 1.
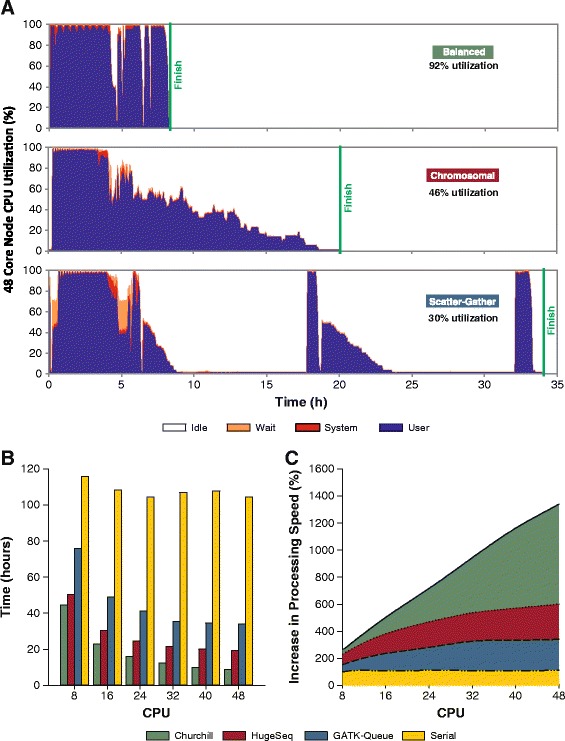
Churchill optimizes load balancing, resulting in improved resource utilization and faster run times. Three different strategies for parallelization of whole genome sequencing secondary data analysis were compared: balanced (utilized by Churchill), chromosomal (utilized by HugeSeq) and scatter-gather (utilized by GATK-Queue). The resource utilization, timing and scalability of the three pipelines were assessed using sequence data for a single human genome sequence dataset (30× coverage). (A) CPU utilization was monitored throughout the analysis process and demonstrated that Churchill improved resource utilization (92%) when compared with HugeSeq (46%) and GATK-Queue (30%). (B) Analysis timing metrics generated with 8 to 48 cores demonstrated that Churchill (green) is twice as fast as HugeSeq (red), four times faster than GATK-Queue (blue), and 10 times faster than a naïve serial implementation (yellow) with in-built multithreading enabled. (C) Churchill scales much better than the alternatives; the speed differential between Churchill and alternatives increases as more cores in a given compute node are used.