

ABSTRACT
In kidney transplant patients with BK polyomavirus (BKPyV) nephropathy, viral variants arise bearing rearranged noncoding control regions (rr-NCCRs) that increase viral early gene expression, replicative fitness, and cytopathology. rr-NCCRs result from various deletions and duplications of archetype NCCR (ww-NCCR) sequences, which alter transcription factor binding sites (TFBS). However, the role of specific TFBS is unclear. We inactivated 28 TFBS in the archetype NCCR by selective point mutations and examined viral gene expression in bidirectional reporter constructs. Compared to the archetype, group 1 mutations increased viral early gene expression similar to rr-NCCR and resulted from inactivating one Sp1 or one Ets1 TFBS near the late transcription start site (TSS). Group 2 mutations conferred intermediate early gene activation and affected NF1, YY1, and p53 sites between early and late TSS. Group 3 mutations decreased early and late gene expression and included two other Sp1 sites near the early TSS. Recombinant viruses bearing group 1 NCCRs showed increased replication in human renal epithelial cells similar to clinical rr-NCCR variants. Group 2 and 3 viruses showed intermediate or no replication, respectively. A literature search revealed unnoticed group 1 mutations in BKPyV nephropathy, hemorrhagic cystitis, and disseminated disease.
IMPORTANCE The NCCRs of polyomaviruses mediate silent persistence of the viral genome as well as the appropriately timed (re)activation of the viral life cycle. This study indicates that the basal BKPyV NCCR is critically controlled by a hierarchy of single TFBS in the archetype NCCR that direct, modulate, and execute the bidirectional early and late viral gene expression. The results provide new insights into how BKPyV NCCR functions as a viral sensor of host cell signals and shed new light on how transcription factors like Sp1 control bidirectional viral gene expression and contribute to replication and pathology.
INTRODUCTION
BK polyomavirus (BKPyV) is one of now more than 12 human PyVs (1) and is known to infect more than 90% of the human population during early childhood (2–6). Following primary infection, BKPyV persists latently in the renourinary tract, with intermittent periods of asymptomatic shedding into urine (5, 7). BKPyV disease typically occurs in individuals with altered immune functions, to which viral determinants are thought to contribute (3, 8). Thus, BKPyV causes nephropathy in 1 to 14% of kidney transplant patients and hemorrhagic cystitis in 5 to 20% of allogeneic hematopoietic stem cell transplant recipients (2, 9).
BKPyV strains excreted in urine of healthy immunocompetent individuals typically bear a noncoding control region (NCCR) of linear archetype (ww) architecture (ww-NCCR) (5, 10, 11) and grow slowly in primary human urothelial and renal tubular epithelial cells (8, 12). In immunosuppressed kidney transplant patients with early stages of BKPyV-associated nephropathy, the virus contains mostly archetype ww-NCCR (8, 13). However, when immunosuppression is not readily reduced to permit specific T cells to resume control over BKPyV replication (14–17), viral variants with rearranged NCCRs (rr-NCCRs) emerge that are associated with higher plasma viral loads and more severe renal allograft pathology (8). Similar rr-NCCRs of JC polyomavirus have been linked with the emergence of progressive multifocal leukoencephalopathy, a debilitating, often fatal brain disease in immunocompromised patients with HIV/AIDS or receiving transplantation or therapies for cancer and immune diseases (18–22). The archetype NCCR of polyomaviruses is approximately 400 bp in length and harbors the origin of replication as well as bidirectional promoter/enhancer functions that, in concert with the host cell signals, control the timing and sequential activation of early viral gene region (EVGR) expression, viral DNA genome replication, and late viral gene region (LVGR) expression. Notably, EVGR and LVGR are encoded and transcribed in opposite directions from the NCCR (23). The ∼2.3-kb EVGR encodes the small T antigen, the large T antigen (LTag), and several splicing variants, whereas the ∼2.5-kb LVGR encodes the capsid proteins VP1, VP2, and VP3 and, in the case of BKPyV and JCPyV, the small nonstructural agnoprotein (2).
Sequencing of naturally occurring BKPyV rr-NCCRs variants in kidney transplant patients revealed that the rearrangements alter the number and composition of transcription factor binding sites (TFBS) as a result of partial duplications and deletions of the archetype sequence (8, 13, 24, 25). Phenotypic analysis using bidirectional reporter constructs mimicking the polyomavirus genome organization indicated that the clinical rr-NCCR variants constitutively activate EVGR expression compared to archetype NCCR (8, 26). However, no common sequence or TFBS pattern could be identified. To address the role of specific TFBS in rr-NCCR of BKPyV, we chose a minimally disruptive approach by introducing selective point mutations predicted to inactivate TFBS without affecting the overall length or architecture of the archetype ww-NCCR. The results indicate for the first time a hierarchy of TFBS with at least three phenotypic groups. Group 1 mutants showed high early and low late gene expression, similar to rr-NCCR emerging in vivo, and resulted from inactivating single Sp1 and Ets1 sites close to the late transcription start site (TSS). Group 2 and group 3 mutants showed intermediate and low gene expression, respectively. In a database search, we identified corresponding mutations in the BKPyV NCCRs from patients with significant BKPyV pathology, such as nephropathy, hemorrhagic cystitis, and disseminated infection, that had not been identified as viral pathology determinants (27–29). Our results provide new insights into how polyomavirus NCCRs function through specific TFBS and shed new light on how Sp1 controls bidirectional BKPyV gene expression and its role in BKPyV pathology.
MATERIALS AND METHODS
Prediction and mutation of potential TFBS.
The DNA genome of the archetype BKPyV ww(1.4) strain is 5,142 bp and contains the ww-NCCR of 376 bp flanked by the EVGR start codon of LTag at 5142 bp and the LVGR start codon of agnoprotein at 377 bp. The programs PROMO and Alibaba2.1 were used to determine the locations of potential TFBS in the ww-NCCR and in the rr-NCCR sequences of different variants with intermediate stringency to get a good compromise between false and missing predictions. The predicted TFBS were graphically depicted within the OPQRS architecture (Fig. 1). Empirically, minimal transitions and transversions were introduced into the predicted TFBS, and the potential effect was analyzed using both programs. Mutations abrogating TFBS without affecting neighboring TFBS were selected (Fig. 2A; also, see Table S1 in the supplemental material), and the corresponding NCCRs were chemically synthesized (Eurogentec, Belgium). Similarly, a selection of TFBS mutants was placed into the del(R8–18) rr-NCCR background described previously (8), wherein an 11-bp sequence has been deleted from the R block by site-directed mutagenesis, causing a small increase and decrease in EVGR and LVGR, respectively (Fig. 2B). All final plasmid constructs were verified by standard dideoxy sequencing.
FIG 1.


Schematic representation of BKPyV genome. (A) The early viral gene region (EVGR), encoding large and small T antigen (Tag), the late viral gene region (LVGR), encoding capsid proteins (VP1, -2, and -3) and agnoprotein (agno), and the noncoding control region (NCCR). The NCCR of the archetype BKPyV ww(1.4) and the rearranged rr-NCCRs found in Dunlop, in natural ins(7.3), del(5.3), del(15.10) variants, and in the del(13.8) variant from kidney transplant patients with nephropathy are divided into sequence blocks O, P, Q, R, and S (subscript numerals are numbers of base pairs; total base pairs are on the right). The colored symbols indicate predicted transcription factor binding sites (TFBS) according to the legend. (B) Flow cytometry and quantification of indicated NCCRs cloned into the bidirectional reporter vector pHRG. x axis, GFP fluorescence; y axis, RFP fluorescence. For each measurement, 5,000 transfected (fluorescent) cells were gated except for mock transfection, where 10,000 untransfected cells were measured. Numbers in the quadrants (Q) are absolute numbers of detected cells and percentages with respect to the gated cells. Ex., excitation wavelength; Em., emission wavelength. (C) Summary plot. Red bars, sum of red cells (Q1+Q2); green bars, sum of green cells (Q2+Q4); yellow bars, double-positive cells (Q2). (D) Normalized mean fluorescence intensity (MFI). For each measurement, the weighted MFI was calculated (see Materials and Methods); late expression was normalized to the ww(1.4) value (green MFI = 100%), and early expression was normalized to the Dunlop value (red MFI = 100%). Quantification results are from 3 independent replicates.
FIG 2.

Mutant BKPyV NCCRs. The mutant NCCR sequences (see Table S1 in the supplemental material) are named according to mutant site (italics) and represented by colorless symbols with dashed lines, and they were tested for early (EVGR) and late (LVGR) gene expression in the bidirectional reporter pHRG. (A) Mutant NCCRs introduced into archetype ww(1.4)-NCRR. (B) Mutant NCCRs introduced into the rr-NCCR carrying a 11-bp deletion in the R block (R8–18). (C) Dual TFBS mutants.
Cell culture.
Primary renal proximal tubule epithelial cells (RPTECs; PCS-400-010; ATCC, Manassas, VA, USA) were grown in epithelial cell medium (EpiCM; no. 4101; ScienceCell Research Laboratory, Carlsbad, CA, USA) supplemented with epithelial cell growth supplement (EpiCGS, no. 4152, ScienceCell Research Laboratory, Carlsbad, USA) and 2% fetal bovine serum (FBS; no. 0010; ScienceCell Research Laboratory). HEK293 cells (CRL1573; ATCC) were propagated in Dulbecco's modified Eagle's medium, high-glucose formulation (DMEM-H; D5671; Sigma-Aldrich, St. Louis, MO, USA), containing 10% FBS (S0113; Biochrome AG, Berlin, Germany). COS-7 cells (CRL1651; ATCC, Manassas, VA, USA) were grown in DMEM-H containing 5% FBS. All cultures were supplemented with 2 mM l-glutamine (K0302; Biochrome AG, Berlin, Germany).
FACS-based, bidirectional reporter assay.
For the bidirectional reporter assay, HEK293 cells were seeded in 12-well plates and transfected at 70 to 80% confluence with Lipofectamine 2000 (11668-019; Invitrogen, Carlsbad, CA) at a ratio of 3:1 (3 μl reagent and 1 μg plasmid DNA) in Opti-MEM (Gibco, Grand Island, NY, USA) according to the manufacturers' instructions. Medium was replaced with DMEM-H–10% FBS the next morning. At 48 h posttransfection, cells were rinsed once with PBS–2.5 mM EDTA and then detached, suspended, and transferred to 5-ml polystyrene round-bottom fluorescence-activated-cell-sorting (FACS) tubes (BD, Franklin Lakes, NJ, USA) with 1 ml PBS–2.5 mM EDTA. Directly before each measurement, DAPI (D8417; Sigma-Aldrich, St. Louis, MO, USA) was added (final concentration, 1 ng/ml) as a dead-cell marker, and cells were resuspended. FACS measurements were carried out on a Fortessa Cytometer (BD, Franklin Lakes, NJ, USA) at medium flow with the following settings: forward scatter (FSC) at 220 V, side scatter (SSC) at 220 V; GFP, excitation at 488 nm (blue laser) and emission at 530/30 nm at a detector voltage of 373 V; RFP, excitation at 561 nm (yellow-green laser) and emission at 586/15 nm at a detector voltage of 500 V; DAPI, excitation at 405 nm (violet laser) and emission at 450/50 nm at a detector voltage of 302 V. In order to calculate the weighted mean fluorescence intensity (MFI) for red (early) and green (late) expression, the cell number (N) and mean fluorescence (I) of quadrant 1 (Q1) (red cells), Q2 (red and green cells), and Q4 (green cells) were inserted into the following formulas:
EVGR expression of all constructs was normalized to that of the Dunlop strain (set as 100%), and LVGR expression was normalized to that of the archetype ww(1.4) (set as 100%). The mean values and standard deviations were calculated by Graph Pad Prism 6 (Mac OS).
Transfection of BKPyV genome into COS-7 and RPTECs and infection of RPTECs.
Transfection of QIAamp DNA kit-purified BKPyV genomes into cells was initiated by cutting BKPyV plasmid DNA with BamHI and religating the diluted DNA as described previously (18). Transfection of religated BKPyV genomic DNA into COS-7 cells was performed at 90 to 95% confluence in 6-well plates using Lipofectamine 2000 (11668-019; Invitrogen, Carlsbad, CA) or GeneExpresso 8000 (EG-1074; Excellgen Inc., Gaithersburg, MD) at a reagent/DNA ratio of 3:1 according to the manufacturers' instructions. At 4 h after transfection, medium was replaced with DMEM-H containing 2% fetal calf serum (FCS). At 14 days posttransfection, COS-7 cells were harvested by scraping off cells in 1/10 of the cell culture supernatant. Virus was released by 3 cycles of freeze-thawing of the cells and centrifugation at 800 × g for 5 min. Transfection of religated BKPyV genomic DNA into RPTECs was performed at 90 to 95% confluence in 6-well plates using ViaFect transfection reagent (E4982; Promega, Madison, WI, USA) at a reagent/DNA ratio of 3:1 according to the manufacturers' instructions. At 24 h after transfection, medium was replaced with supplemented EpiCM medium (ScienceCell Research Laboratory, Carlsbad, CA, USA). At 1, 2, 3, 5, and 7 days posttransfection, 1 ml of supernatant for quantification of viral load was taken and RPTECs were harvested by scraping off cells for Western blotting. For the reinfection of RPTECs with COS-7 supernatants, cells were seeded at 75,000 cells per well of a 24-well plate in 0.5 ml of supplemented EpiCM medium. After 24 h at a confluence of approximately 50%, RPTECs were exposed to 200 μl of the corresponding virus preparations at 37°C for 2 h followed by removal, washing, and replacement with supplemented EpiCM medium.
Determination of BKPyV viral loads by real-time PCR.
BKPyV loads were quantified after DNA extraction from 100 μl cell culture supernatants with the Corbett X-tractor Gene and the Corbett VX reagents (Qiagen, Hombrechtikon, Switzerland). The real-time PCR protocol for detection of BKPyV DNA samples targets the BKPyV large T-coding sequence and has been described elsewhere (30).
Immunofluorescence and antibodies.
Five, seven, and 14 days postinfection, RPTECs were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min and permeabilized with 0.2% Triton in PBS for 10 min at room temperature (8). After incubation with monoclonal mouse anti-simian virus 40 (SV40) large T antigen (1:50; DP02; Merck, Darmstadt, Germany), monoclonal mouse anti-VP1 (1:300; 10309-5E6; Abnova, Taipei City, Taiwan), and polyclonal rabbit anti-agnoprotein (1:800) (31), diluted in 3% skim milk-PBS at 37°C for 1 h, cells were washed twice with PBS at room temperature for 5 min and incubated with the secondary antibodies anti-mouse IgG1–Alexa Fluor 647 (1:800; A-21240; Life Technologies, Carlsbad, CA, USA), anti-mouse IgG2a–Alexa Fluor 568 (1:300; A-21134; Life Technologies), and anti-rabbit IgG–Alexa Fluor 488 (1:1,000; A-21441; Life Technologies) and Hoechst 33342 dye (0.5 μg/ml; H21492; Life Technologies) as described for primary antibodies. After two washes, specimens were mounted in 90% glycerol (1.04095; Merck) in PBS containing 1% N-propylgallate (P-3130; Sigma) as an antifading agent or ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI; P36935; Life Technologies).
Microscopy and digital image processing.
Microscopy was performed using an epifluorescence microscope (model TE200; Nikon) equipped with suitable filters and a digital camera (Hamamatsu). DAPI-, LTag-, VP1-, and agnoprotein-positive cells were counted in a minimum of three microscopic fields (10× objective). Images were taken at the times indicated in the figure legends. Digital images were recorded with Openlab 2.2 software, and raw images were processed in Fiji (version 1.49e).
Western blotting.
Cells were lysed in NP-40-buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8.0], 1% NP-40, and protease inhibitors [Roche]). Cell lysates were separated by SDS-PAGE and electrotransferred onto 0.45-μm Immobilon-FL polyvinylidene difluoride (PVDF) membrane (IPFL00010; Millipore/Merck, Darmstadt, Germany). Membranes were blocked with Odyssey blocking buffer (927-40000; Licor, Lincoln, NE, USA) diluted 1:2 in Tris-buffered saline (TBS). The membrane was incubated with the following primary antibodies: polyclonal rabbit anti-LTag (1:5,000), polyclonal rabbit anti-VP1 (1:10,000), polyclonal rabbit antiagnoprotein (1:10,000), and monoclonal mouse anti-actin (1:5,000; Abcam, Cambridge, England) in Odyssey blocking buffer–TBS–0.1% Tween 20. Washing was performed with TBS–0.1% Tween 20. Secondary antibodies were donkey anti-mouse Alexa Fluor 680 (A10038; Invitrogen, Life Technologies, Carlsbad, CA, USA) and goat anti-rabbit antibody–IRDye 800CW (926-32211; Licor); both were used at a 1:10,000 dilution. Detection and quantification were done with the Licor Odyssey CLx system.
EMSAs.
Electrophoretic mobility shift assay (EMSA) analysis of TFBS was performed as described previously (32). Briefly, 10 μg of nuclear extract from RPTEC cells was mixed with binding buffer [20 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), pH 6.8; 50 mM NaCl; 1 mM dithiothreitol (DTT); 0.25 mg/ml bovine serum albumin (BSA); 100 μM ZnSO4; 0.05% NP-40; 4% Ficoll] and approximately 5 fmol of 32P-labeled, duplexed oligonucleotide in a final volume of 20 μl (Table 1). Nuclear extracts were prepared as described by Schreiber et al. (33) by scraping cells with ice-cold PBS off 10-cm dishes and collecting them in Eppendorf tubes. After centrifugation, they were swollen in hypotonic buffer (10 mM HEPES, pH 7.9; 10 mM KCl; 0.1 mM EDTA; 2.5 mM DTT) and lysed by addition of NP-40 (final concentration, 0.5%). After centrifugation of the nuclei, proteins were extracted in nuclear extract buffer (20 mM HEPES, pH 7.9; 25% glycerol; 400 mM NaCl; 1 mM EDTA; 2.5 mM DTT). Competing, unlabeled oligonucleotides were used in a 200-fold excess. Transcription factors were identified by supershifts. Specific antibodies to Sp1 (200 ng; SC-59; Santa Cruz Biotechnology), to NF-1 (200 ng; SC-5567; Santa Cruz Biotechnology), to Ets1 (200 ng; SC-22802; Santa Cruz Biotechnology), or to PEA3 (200 ng; SC-166629; Santa Cruz Biotechnology) were added to the binding buffer. After incubation of the mixture on ice for 30 min, samples were separated on a native 4% polyacrylamide gel with 0.25× Tris-borate-EDTA (TBE) as the running buffer. The detection of β-decay was carried out with a Fujifilm FLA-7000 Image plate reader.
TABLE 1.
Oligonucleotides used for electrophoretic mobility shift assays
| Name | Sequence (5′→3′) |
|---|---|
| Sp1 Promega fw | ATT CGA TCG GGG CGG GGC GAG C |
| Sp1 Promega rev | G CTC GCC CCG CCC CGA TCG AAT |
| Sp1-1 fw | GGA GGC AGA GGC GGC CTC GGC CTC |
| Sp1-1 rev | GAG GCC GAG GCC GCC TCT GCC TCC |
| Sp1-1 mut fw | GGA AAC ATA TCC ATC CTA GGC ATC |
| Sp1-1 mut rev | GAT GCC TAG GAT GGA TAT GTT TCC |
| Sp1-2 fw | AGG CCA CAG GGA GGA GCT GCT TA |
| Sp1-2 rev | TA AGC AGC TCC TCC CTG TGG CCT |
| Sp1-2 mut fw | AGG CCA CAG TTA TGA GCT GCT TA |
| Sp1-2 mut rev | TA AGC AGC TCA TAA CTG TGG CCT |
| Sp1-4 fw | ATA GTG AAA CCC CGC CCC TGA AA |
| Sp1-4 rev | TT TCA GGG GCG GGG TTT CAC TAT |
| Sp1-4 mut fw | ATA GTG AAA CCG ATA CCC TGA AA |
| Sp1-4 mut rev | TT TCA GGG TAT CGG TTT CAC TAT |
| Sp1-3/NF1-2 fw | GCA TGA CTG GGC AGC CAG CCA GT |
| Sp1-3/NF1-2 rev | AC TGG CTG GCT GCC CAG TCA TGC |
| Sp1-3/NF1-2 mut fw | GCA TGA ACA TGC AAC CAG CCA GT |
| Sp1-3/NF1-2 mut rev | AC TGG CTG GTT GCA TGT TCA TGC |
| Sp1-4 5CG fw | ATA GTG AAA CCC GGC CCC TGA AA |
| Sp1-4 5CG rev | TT TCA GGG GCC GGG TTT CAC TAT |
| Sp1-4 5CG/10CT fw | ATA GTG AAA CCC GGC CCT TGA AA |
| Sp1-4 5CG/10CT rev | TT TCA AGG GCC GGG TTT CAC TAT |
| Sp1-4 10CT fw | ATA GTG AAA CCC CGC CCT TGA AA |
| Sp1-4 10CT rev | TT TCA AGG GCG GGG TTT CAC TAT |
| Sp1-4 7CG/10CT fw | ATA GTG AAA CCC CGG CCC TGA AA |
| Sp1-4 7CG/10CT rev | TT TCA GGG CCG GGG TTT CAC TAT |
| Ets1-2 fw | ACA CAA GAG GAA GTG GAA ACT G |
| Ets1-2 rev | C AGT TTC CAC TTC CTC TTG TGT |
| Ets1-2 HI-u6/CAP-m5 fw | ACA CAA GAG GGA GTG GAA ACT G |
| Ets1-2 HI-u6/CAP-m5 rev | C AGT TTC CAC TCC CTC TTG TGT |
| Ets1-2 15.10 fw | ACA CAA CAG GAA GTG GAA ACT G |
| Ets1-2 15.10 rev | C AGT TTC CAC TTC CTG TTG TGT |
| ets1-2 fw | ACA CAA TAT TAA GTG GAA ACT G |
| ets1-2 rev | C AGT TTC CAC TTA ATA TTG TGT |
RESULTS
To investigate the role of TFBS in archetype and rearranged BKPyV NCCRs, two different computer prediction programs were used with moderate stringency to locate major TFBS in silico. In the archetype (ww) NCCR, approximately 30 TFBS were consistently predicted by both programs and included multiple sites for some factors like Sp1 or NF1 (Fig. 1). In rearranged (rr)-NCCRs like the one found in clinical BKPyV variants from kidney transplant patients with nephropathy (8, 24) or in the laboratory-adapted Dunlop strain BKPyV-DUN (34), various TFBS were duplicated, deleted, and/or replaced (Fig. 1). Typically, the continuity of the O143-P68 segment bearing the viral origin of replication and the early TSS remained intact in the rr-NCCRs, but complete or partial duplications of the P68 block were observed [ins(7.3) and DUN] (Fig. 1A). Conversely, deletions in clinical rr-NCCRs typically affected the Q39-R63-S63 sequence blocks around the late TSS [del(5.3), del(15.10), del(13.8), and DUN] (Fig. 1A). To experimentally quantify the effect on viral gene expression, the rr-NCCR variants and the ww-NCCR were cloned into the reporter gene vector pHRG, which, as reported earlier (8), recapitulates the bidirectional organization of the polyomavirus DNA genome and allows monitoring of NCCR-directed EVGR and LVGR expression by the red fluorescent protein (RFP) and the enhanced green fluorescent protein (eGFP), respectively. The corresponding NCCR reporter constructs were transfected into the embryonic kidney cell line HEK293. As shown in Fig. 1B, the archetype ww-NCCR conferred strong LVGR and weak EVGR expression, as indicated by the respective red and green signals detected by flow cytometry. In contrast, the rr-NCCR of BKPyV-DUN and all of the clinical rr-NCCR variants showed an opposite pattern, with strong EVGR expression in red but weak LVGR expression in green (Fig. 1B). Quantification showed that, compared to ww-NCCR, rr-NCCR increased both the number of cells expressing the EVGR as well as the mean fluorescence intensity (MFI) of EVGR expression in the cells (Fig. 1C and D). Despite the respective dominance of LVGR or EVGR expression, both the archetype and the rearranged NCCR pattern showed limited expression in the respective opposite direction within the same single cells (Fig. 1C, yellow bars). Moreover, although increased EVGR expression was clearly the common hallmark of the clinical rr-NCCRs, the quantitative results indicated that there were differences between the various clinical rr-NCCR variants with respect to EVGR and LVGR levels (Fig. 1D). However, the diversity in TFBS content, the altered OPQRS architecture, and the overall length of the clinical rr-NCCR variants (Fig. 1A) did not point to an obvious hierarchy of TFBS or their role in BKPyV EVGR and LVGR expression.
To dissect the potential role of various TFBS, we introduced point mutations into the archetype ww-NCCR that were predicted to abrogate binding of respective proteins. Importantly, the overall architecture and length of 376 bp of the archetype ww-NCCR remained unaltered (Fig. 2A; for sequences, see the supplemental material). Additionally, some TFBS mutants were either combined with other TFBS mutations (Fig. 2A and C) or placed in the background of the experimentally designed rr-NCCR del(R8–18) deletion mutant described previously (8) (Fig. 2B) to investigate their effect in a defined deletion background known to activate EVGR expression.
The resulting mutant ww-NCCRs were placed in the bidirectional reporter gene vector pHRG, and the reporter constructs were verified by sequencing, and transfected into HEK293 cells. RFP and GFP expression was detectable at 1 day posttransfection (dpt) and persisted for at least 5 days (not shown). Analysis of the EVGR and LVGR reporter gene expression revealed that the TFBS mutant ww-NCCRs could be grouped into three phenotypic groups (Fig. 3).
FIG 3.

Phenotypic characterization of NCCR point mutations. (A) The indicated constructs were transfected into HEK293 cells, and fluorescence images (20× objective) were taken 2 days posttransfection (dpt). The archetype ww(1.4) strain and the Dunlop strain served as references. The group 1 prototype is sp1-4a (high early and low late expression), the group 2 prototype is nf1-4 (intermediate to high early and intermediate late expression), and the group 3 prototype is sp1-2 (low early and low to intermediate late expression). (B) Plots of representative FACS measurements of indicated NCCRs in HEK293 at 2 dpt as described in Fig. 1. (C) Cell counts of red, green, and double-positive cells. (D) Weighted mean fluorescence intensity (MFI) (Fig. 1).
Group 1 mutants were exemplified by the prototype mutant with a sp1-4a mutation of the SP1-4 site and characterized by stronger EVGR than LVGR expression (Fig. 3). Thus, the group 1 mutants resembled the clinical rr-NCCR BKPyV variants from kidney transplant patients with nephropathy (Fig. 1). Although EVGR expression was dominant, some LVGR expression still occurred, as shown by the cells that were positive for both RFP and GFP (Fig. 3C, yellow bars).
Group 2 mutations exemplified by the prototype nf1-4 mutant also showed an increased EVGR signal but a lesser reduction of LVGR expression of the archetype signal compared to group 1 mutations (Fig. 3). This was reflected in a higher number of LVGR-expressing cells (Fig. 3C) and a higher green MFI (Fig. 3D).
Group 3 mutations were exemplified by the prototype sp1-2 mutant and characterized by reduced green LVGR expression compared to that of the parent archetype ww-NCCR, but, unlike in group 1 and group 2 mutants, the red EVGR expression was not increased (Fig. 3).
The group 1 phenotype was also observed by a different mutation similarly predicted to inactivate the SP1-4 (called sp1-4b) in the Q39 block and by an inactivating mutation of the ETS1-2 site (called ets1-2) in the R63 block (Fig. 4A; also, see the sequences in Table S1 in the supplemental material). Comparison with the clinical rr-NCCR variants suggested that these point mutations at least partly explained the phenotype seen in the clinical rr-NCCR variants bearing deletions: in the del(15.10) NCCR, SP1-4 was missing, in the del(13.8) NCCR, ETS1-2 was missing, and both sites were missing in the del(5.3) NCCR.
FIG 4.
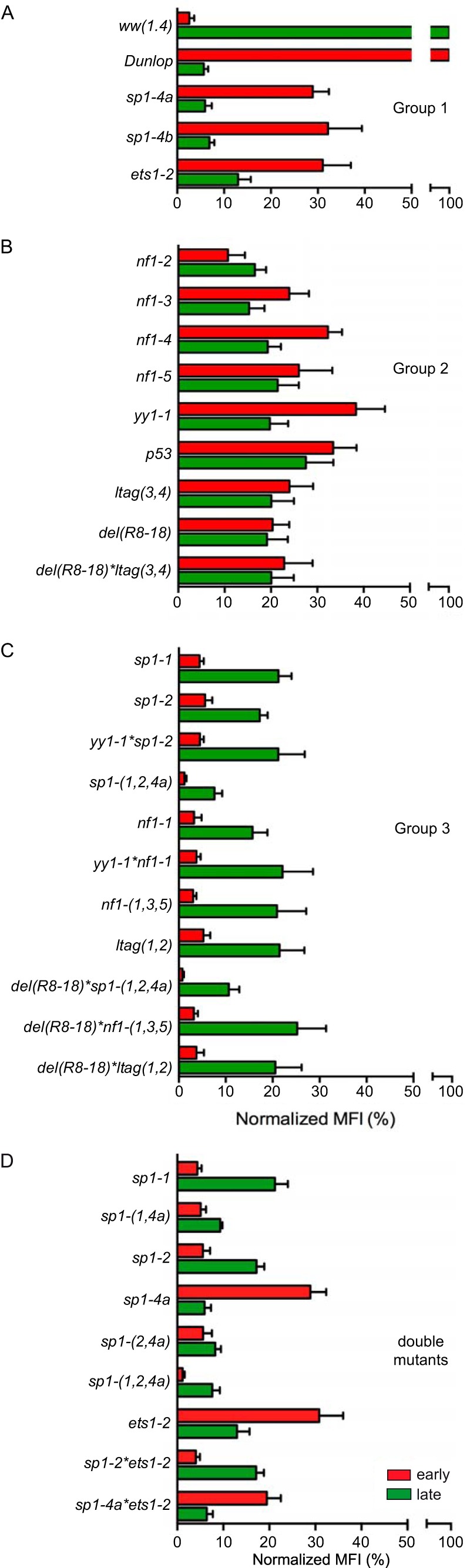
Summary of mutant NCCR phenotype groups. The indicated mutant NCCR bidirectional reporter constructs were transfected into HEK293 cells and grouped according to normalized MFI (Fig. 1D) (see Materials and Methods). (A) Group 1 NCCR mutants (high early expression and low late expression). (B) Group 2 NCCR mutants (intermediate early and late expression). (C) Group 3 NCCR mutants (low early and low intermediate late expression). (D) MFI comparison of single-site mutants with selected double and triple mutants.
The group 2 phenotype was seen to result from single TFBS mutations targeting p53, yy1, and different NF1 sites (called nf1-2, nf1-3, nf1-4, and nf1-5, from early to late TSS) and from the dual mutation ltag(3,4) (Fig. 4B). Group 2 mutations showed an increased EVGR expression relative to the archetype ww(1.4), but LVGR expression appeared to be less impaired than observed for the group 1 mutations (Fig. 4B). The intermediate group 2 phenotype suggested that the group 1 TFBS targets SP1-4, and ETS1-2 TFBS were essential for LVGR expression, whereas group 2 TFBS had a modulatory role. Interestingly, the nf1 mutations seemed to have subtle differences with increasing EVGR expression, as the mutated sites were located further from the early TSS and closer to the late TSS (Fig. 4B).
Group 3 mutations resulted in an overall reduced LVGR and EVGR expression and included other Sp1 TFBS, called sp1-1 and sp1-2, located close to the early TSS, nf1-1, and ltag(1,2), as well as their yy1*sp1-2, sp1-(1,2,4a), nf1-(1,3,5), del(R8–18)*sp1-(1,2,4a), del(R8–18)*nf1-(1,3,5), and del(R8–18)*ltag(1,2) combination mutants (Fig. 4C). Thus, with the exception of the sp1-1, sp1-2, and nf1-1 mutants, all other group 3 mutants targeted more than one site in the NCCR. Accordingly, some of these combinations shifted the expression from a group 2 to a group 3 pattern, e.g., for yy1 when combined with sp1-2, as in the yy1*sp1-2 mutant, or with nf1-1, as in the yy1*nf1-1 mutant (Fig. 4C). Most prominently, the EVGR activation of the group 1 mutation sp1-4a was reversed as was the sp1-(1,2,4a) triple mutation (Fig. 4C). Also, the activating background of the del(R8–18) mutant was reversed, when combined with the mutation sp1-(1,2,4a), nf1-(1,3,5), or ltag(1,2). This suggested that some TFBS, such as SP1-1 or SP1-2, were of particular importance to the overall bidirectional promoter/enhancer function of the BKPyV NCCR.
To investigate the role of Sp1 in more detail, several additional TFBS mutants were generated and analyzed (Fig. 4D). Combining the inactivation of SP1-4 and SP1-1 in the sp1-(1,4a) NCCR showed a reduction of LVGR expression (characteristic of sp1-4a) and a reduction of EVGR expression (characteristic of sp1-1), indicating that either site had an independent contribution to viral gene expression (Fig. 4D). The same was observed for the combination of sp1-2 and sp1-4a in sp1-(2,4a). The independent contribution of each of these Sp1 sites was supported by the further decrease observed for the triple combination sp1-(1,2,4a). Similarly, sp1-2 also decreased EVGR expression when combined with ets1-2, as in the sp1-2*ets1-2 double mutant. Taken together, the data suggested that the different Sp1 sites played an important independent role in the control of BKPyV EVGR and LVGR expression. The pronounced inactivating effect of the group 3 mutations sp1-1, sp1-2, nf1-1, and ltag(1,2) also suggested an explanation for why deletions have not been observed in the O143-P68 block of prominent clinical rr-NCCR variants, wherein these sites are located (Fig. 1).
Given the substantial effects of the ww-NCCR point mutations on EVGR and LVGR reporter expression, it was of interest to study their consequences for BKPyV replication. To that end, group 1 sp1-4a and ets1-2 mutant NCCRs were placed in the BKPyV backbone and transfected into primary human renal proximal tubule epithelial cells (RPTECs). Compared to the archetype (ww) NCCR BKPy, a rapid increase in supernatant viral loads was seen for the del(5.3) NCCR BKPyV (Fig. 5A). Both group 1 mutants, the sp1-4a- and ets1-2 NCCR BKPyV recombinants, also replicated significantly faster than the archetype but more slowly than the natural variant del(5.3) NCCR BKPyV over 7 days posttransfection (dpt) (Fig. 5A). Group 1 mutants showed enhanced early and late viral gene expression compared to the archetype, although this was less than in the clinical variant del(5.3)-bearing virus, as immunofluorescence (Fig. 5B) or immunoblots (Fig. 5C) showed LTag and VP1 expression being highest for del(5.3), followed by sp1-4a, ets-2, and then the ww-NCCR virus.
FIG 5.

Replication and protein expression of recombinant NCCR BKPyV. (A) RPTECs were transfected with the indicated recombinant BKPyV NCCR genomes. Supernatant viral loads were quantified by quantitative PCR after 1, 2, 3, 5, and 7 days posttransfection (dpt). Increases in BKPyV supernatant viral loads are expressed on a log scale. (B) Immunofluorescence was performed for large T antigen (LTag; red), agnoprotein (agno; green), and viral protein 1 (VP1; cyan) at 7 dpt. The nuclei are marked with DAPI (blue). The graph shows expression of the indicated viral proteins relative to that of the archetype ww(1.4) protein. (C) Western blot of indicated cell extracts showing LTag (triangles) and VP1 in green (triangles); actin (red) served as a loading control. Actin-normalized band fluorescence intensity (bottom diagrams) was quantified using Licor Image Studio, with ww(1.4) as a reference.
To compare the replication of different recombinant NCCR mutant viruses over time, supernatant BKPyV loads were determined over 7 dpt and 13 dpt (Fig. 6A). As expected, supernatant viral loads of the del(5.3) and DUN NCCR had increased by more than 1,000-fold by 7 dpt. The group 1 mutations sp1-4a and ets1-2 replicated slightly less efficiently than the natural del(5.3) variant but better than the archetype virus, a difference that became more evident by 13 dpt. In contrast, the group 3 mutations sp1-2 and sp1-1 as well as sp1-(1,2,4a) permitted only little or no replication of the respective recombinant BKPyV mutant viruses (Fig. 6A).
FIG 6.

Replication and infectivity of BKPyV NCCR mutant strains. (A) RPTECs were transfected using the indicated recombinant BKPyV NCCR variants. Supernatant viral loads quantified by quantitative PCR after 1, 7, and 13 days posttransfection (dpt). The increase in supernatant viral loads was normalized to the value at 1 dpt. (B) Supernatant taken from COS-7 cells at 14 dpt were used to infect RPTECs. Immunofluorescence was performed to detect large T antigen (LTag; red) and agnoprotein (agno; green) at 7 days postinfection; cell nuclei are in blue.
To investigate the infectivity of the mutant ww-NCCR viruses, COS-7 cells were transfected, and the respective supernatants were used to infect primary human RPTECs. At 7 days postinfection (dpi), the archetype ww(1.4) NCCR virus displayed only a few cells that were positive for LTag in the nuclei and agnoprotein in the cytoplasm (Fig. 6B). Instead, the del(5.3) and DUN NCCR viruses abundantly expressed the early LTag and the late agnoprotein, as reported previously (8). Group 1 mutant sp1-4a and sp1-4b NCCR viruses showed increased infectivity compared to the archetype, whereas the group 2 and group 3 mutations resulted in few and no infected cells, respectively. In particular, sp1-1 and sp1-(1,2,4a) mutations seemed to result in virtually undetectable infectivity. Overall, infection and replication matched the bidirectional phenotypic reporter assay rather well.
Given the prominent effect of mutating the predicted Sp1 sites on EVGR and LVGR expression, Sp1 binding to the archetype and the mutant sequences was examined by electrophoretic mobility shift assay (EMSA) (Fig. 7). For the archetype-derived oligonucleotides bearing the wild type SP1-1, SP1-2, and SP1-4 TFBS (Table 1), a similar band shift pattern was observed, that was reduced in the presence of unlabeled oligonucleotides bearing the Sp1 consensus sequence (Fig. 7A). Addition of an Sp1-specific monoclonal antibody (Sp1 MAb) caused a supershift, confirming the identity of the bound factor. For oligonucleotides bearing the putative SP1-3 site, however, a different EMSA pattern was obtained, which could neither be competed with a consensus SP1-oligonucleotide sequence nor be shifted with the Sp1 MAb. Instead, binding of NF1 was documented through competition by an unlabeled oligonucleotide containing the NF1 consensus sequence and a supershift induced by NF1-specific antibody (Fig. 7A). This site corresponded to nf1-2. Thus, all point mutations in fact led to a significant reduction or loss of the respective transcription factor binding activity in the EMSA (Fig. 7A), which is an important finding for the interpretation of the reporter assay results.
FIG 7.

Binding of Sp1 and Ets1 to archetype and mutant NCCR sequences and reporter gene expression. (A) Electrophoretic mobility shift assays (EMSAs) were performed using labeled and duplexed oligonucleotides carrying the indicated NCCR sequences with nuclear extracts (NUX) alone or in the presence of unlabeled competitor (comp) or monoclonal antibody (MAb), where indicated. Names of oligonucleotides (top row) refer to putative binding sites; wt, wild type sequence; mut, mutated binding site; NUX, 10 μg of RPTEC nuclear extract; comp, competition with 1 pmol unlabeled oligonucleotide with Sp1 or NF1 consensus sequence; MAb, 200 ng of monoclonal Sp1 or NF1 antibody for supershift; FP, free probe. (B) (Top) Alignment of naturally occurring SP1-4 mutants as described by Priftakis et al. (27). Names refer to the position relative to the SP1-4 binding site start and type of exchange. (Bottom) Binding analysis of SP1-4 mutants. (C) Oligonucleotides bearing the indicated archetype or mutant NCCR sequences were incubated with nuclear extracts as described in Materials and Methods. Archetype, ETS1-2 sequence from the BKPyV ww(1.4) strain; HI-u6/CAP-m5, sequence derived from a natural variant found in a patient with systemic BKPyV replication and capillary leak pathology (29); 15.10, sequence derived from a natural variant del(15.10) from kidney transplant patients with nephropathy (8); NUX, 10 μg of RPTEC nuclear extract; MAb, 200 ng monoclonal Ets1, PEA3, or Sp1 antibody for supershift; FP, free probe. (D) The indicated mutant NCCR bidirectional reporter constructs were transfected into HEK293 cells and grouped according to normalized MFI (Fig. 1D) (see Materials and Methods).
Since the sp1-4 mutations significantly increased EVGR expression and BKPyV replication similarly to natural rr-NCCR variants found in kidney transplant patients, database entries were searched for sequence alterations affecting this specific site. Indeed, in a study of bone marrow transplant patients with hemorrhagic cystitis, overrepresentation of sp1-4 mutations in clinical BKPyV variants had been described (27) (Fig. 7B, top). Testing the effect of these mutations on Sp1 binding by EMSA revealed a reduced Sp1 binding for the mutations 5CG, 5CG/10CT, and 7CG/10CT (Fig. 7B). The data lend support to the notion that sp1-4 mutations reducing Sp1 binding are found in clinically relevant BKPyV diseases.
EMSA was also performed for the ETS1-2 site. The labeled archetype sequence showed binding by mobility shift (Fig. 7C), whereas no binding to the group 1 mutant ets1-2 sequence was seen (Fig. 7C, rightmost lanes). Addition of a monoclonal Ets1 antibody induced a supershift, while antibodies to Sp1 or to PEA3, another Ets family member, had no effect (Fig. 7C). This indicated that ETS1-2 contained an Ets1-binding site that was abrogated by the respective group 1 point mutations. As indicated above (Fig. 1A), ETS1-2 was found to be deleted in the natural rr-NCCR BKPyV variants from kidney transplant patients with nephropathy, e.g., together with the SP1-4 site in the del(5.3) mutant and in the del(13.8) mutant (8). In another natural rr-NCCR variant, the del(15.10) variant, (8), a deletion of the SP1-4 site occurred together with a point mutation that was predicted to change the archetype BKPyV ETS1-2 site to a perfect Ets1 consensus. Indeed, EMSA revealed strong binding to the mutated Ets1 site found in the del(15.10) NCCR sequence that could be supershifted by Ets1 antibody but not by PEA3 antibody, and the unlabeled del(15.10) sequence was an effective competitor for the archetype ww-NCCR sequence (Fig. 7C). Interestingly, the del(15.10) NCCR showed a group 2 rather than a group 1 phenotype (8). This is in line with the notion that both the SP1-4 and the ETS1-2 TFBS conferred independent regulation on EVGR and LVGR and that loss of the SP1-4 was partially counteracted by stronger Ets1 binding (Fig. 7D). A database search revealed that the ETS1-2 site was altered in two clinical cases with significant BKPyV disease. The first case was an HIV-1 infected patient with BKPyV-associated hemorrhagic cystitis (29). Here, the mutant ets1-2 sequence was predicted to abrogate Ets1 binding, while a novel Sp1 site was potentially created. Indeed, the corresponding sequences showed a changed mobility pattern, as determined by EMSA, that could no longer be supershifted by an antibody to Ets1 but could be supershifted by the Sp1 MAb (Fig. 7C). The other case was a kidney transplant patient with nephropathy, who developed a generalized capillary leak pathology with fatal outcome (28). The BKPyV genome showed no evidence of NCCR rearrangements, but a point mutation in ETS1-2 (29). Our results now indicate that this mutation abrogates Ets1 binding similarly to the group 1 mutation generated experimentally, causing an activated EVGR expression. To evaluate the effect on EVGR and LVGR expression, the corresponding mutant NCCRs were placed into the pHRG and analyzed by flow cytometry (Fig. 7D). The results show that the clinically occurring point mutations result in increased EVGR expression compared to the archetype ww-NCCR, as expected for group 1 and 2 mutants.
DISCUSSION
The NCCR represents less than 10% of PyV genomes but effectively mediates key functions of polyomavirus biology inside the host cell. This includes silent persistence of the episomal viral genome and the appropriately timed (re)activation of the PyV life cycle, consisting of EVGR expression followed by viral genome replication and LVGR expression to generate infectious progeny. Besides a principle virological interest in the mechanisms mediating these functions within less than 400 base pairs (35), there is a strong clinical interest, since NCCR rearrangements have been associated with major human PyV pathologies, i.e., BKPyV-associated nephropathy and JCPyV progressive multifocal leukoencephalopathy (8, 13, 18, 25, 36, 37) and more recently an HPyV-7-associated proliferative keratinocytic skin disease (38). However, the multitude of TFBS and their considerable diversity in clinically identified NCCR rearrangements has hampered identification of their specific role in PyV biology and human pathology.
The discrete mutational analysis of the BKPyV archetype NCCR in this study now indicates that the basal bidirectional EVGR and LVGR expression is critically controlled by a hierarchy of TFBS, which can give rise to at least three phenotypic groups. Group 1 mutations include sp1-4 and ets1-2 inactivating a single Sp1 and a single Ets1 site close to the late TSS, respectively, and redirect expression to the EVGR at the expense of LVGR expression. Thereby, group 1 mutations appear to be functionally similar to previously characterized clinical rr-NCCR variants such as del(5.3) (8). The group 1 mutant NCCRs are functional in the viral genomic context, conveying increased LTag expression, higher viral loads, and infectious virus progeny.
Group 2 mutations show an intermediate phenotype regarding EVGR and LVGR reporter gene expression as well as viral replication rates. Group 2 mutations resulted from inactivating different TFBS located in the central P68-Q39-R63 sequence blocks and appear to modulate the EVGR-LVGR expression balance. Combining different group 2 mutations did not lead to a further increase in EVGR, suggesting that possibly similar or converging modulating functions might be affected by the respective TFBS mutations. In contrast, group 3 mutations interfered more prominently with the net expression of both EVGR and LVGR. Moreover, group 3 mutations were able to revert the activating effect of some group 1 and 2 mutations, suggesting the loss of an essential function required for gene expression in both directions from the NCCR. This contribution was most strikingly impaired when sp1-1 and sp1-2 were combined, which alone and in combination abrogated even the EVGR-activating group 1 mutation sp1-4. The group 3 recombinant viruses bearing the ww(sp1-1) and ww(sp1-2) NCCRs permitted no or little replication and infectivity. Thus, the archetype BKPyV ww-NCCR is mediated by multiple TFBS with differential abilities to direct, modulate, and facilitate EVGR and LVGR expression, with corresponding effects on viral replication.
Earlier studies examined the role of different TFBS on BKPyV EVGR or LVGR, but the most attention has been given to prototype strains bearing rearranged NCCR sequences. Moreover, the experimental approaches relied exclusively unidirectional reporter gene analyses. This precluded the unique detection of a loss-of-function phenotype for LVGR expression, resulting in a gain of function for EVGR expression, as shown here for group 1 mutations. By linker scanning deletions, Cassill et al. and Deyerle et al. identified a significant decline in LVGR reporter gene expression when a stretch presumably containing the SP1-2 site was deleted, which would be consistent with the essential role of group 3 sp1-2 reported here (39, 40). For the rr-NCCR present in the BKPyV Gardner strain, mutation of NCCR positions 147 to 156, also corresponding to SP1-2, was reported to reduce early transcription by 81%. Importantly, this SP1-2 deletion had no dramatic impact on mutant replication in COS-1 cells, which is able to provide EVGR functions in trans from the SV40 LTag (41, 42), similar to the COS-7 cells used for this purpose here. Markowitz and Dynan identified strong binding of Sp1 to the SP1-4 site when HeLa cell extracts were probed with the archetype ww-NCCR and reported binding of NF1 to multiple sites of the BKPyV NCCR, but they did not investigate functional aspects (43).
The hierarchy of TFBS revealed by our minimally disruptive mutation analysis of the archetype ww-NCCR extends these observations and provides a new perspective on naturally occurring NCCR mutants and their association with disease, where group 1 and group 2 sites are frequently altered (Fig. 1A) (8, 13, 26). The role of group 1 TFBS is underlined by independent clinical reports of Sp1-4 or Ets1-2 mutant NCCR variants in patients with significant disease, including hemorrhagic cystitis (27), nephropathy, and fatal capillary leak pathology (28). The latter case is particularly striking because of disseminated BKPyV infection taking a fatal course in the absence of previously known viral determinants (29). Our results provide the first experimental support for the notion that regulating ETS1-2 binding activates EVGR expression, thereby conferring a functional equivalent to rr-NCCRs in this specific case of BKPyV capillary leak pathology. Similarly, Priftakis et al. (27) reported an overrepresentation of mutations at the SP1-4 site which were associated with median urine viral loads that were approximately 1 log10 Geq/ml higher than those in patients with archetype virus (27). Here, we show experimentally that these previously reported sp1-4 mutations reduce or abrogate Sp1 binding and result in an activation of EVGR expression similarly to our designed sp1-4a and sp1-4b mutations. The weakest effect was seen with sp1-4(10CT), while the strongest appeared with the double mutation sp1-4(7CG/10CT) (27), suggesting that Sp1 affinity to the SP1-4 site might play a role. Interestingly, the related JCPyV NCCR also bears an SP1-4-like site in the D block close to the LVGR that is frequently lost in natural NCCR rearrangements in patients with progressive multifocal leukoencephalopathy (18, 44, 45). Other TFBS proposed to contribute to JCPyV EVGR activation include Spi-B, which is a member of the Ets family (46–48).
A review of Sp1 and Ets1 literature suggests that these factors act as versatile sensors of intrahost cell physiology and signaling (49–51), which can be exploited by BKPyV for steering its viral life cycle.
Ets1 was originally identified as the cellular proto-oncogene counterpart to the retroviral oncogene v-ets (E26 transforming sequence), which acts together with v-Myb protein in an avian erythroblastosis retrovirus, E26. The Ets family has more than 30 members, including Ets2, Spi-B, Spi1/Pu1, PEA3, and Elk1 and -2, and recognizes the 5′-GGAA/T-3′ core motif, thereby integrating responses in differentiation, proliferation, angiogenesis, and neoplasia, including in hematopoietic malignancies and prostate cancer.
Sp1, originally called specificity protein 1, is one of at least eight related Sp transcription factors that contain three C-terminal cysteine2-histidine2-type zinc finger DNA-binding domains (50, 51). Sp1 can either activate or repress the promoter activity of genes involved in environmental, nutritional, and hormonal responses (51, 52). This includes nuclear signaling downstream of insulin, corticosteroids, androgen, estrogen, parathyroid hormones and retinoic acid. Hormone receptor signals are captured by Sp1 binding to a single TFBS followed by superactivation involving Sp1 complexes and DNA looping through recruiting more distal Sp1 sites (51, 53). The sequential recruitment of TFIID and RNA polymerase II by Sp1 has also been linked to the efficient transcriptional initiation of TATA-less genes (50, 54, 55), which in many cases actually might be promoters with hidden TATA boxes in the form of TATA-like elements (56). In the BKPyV ww-NCCR, SP1-4 and ETS1-2 are located approximately 21 bp apart, forming a transcription factor module. Similar synergistic activation by Sp1 and Ets1 sites has been reported for the long terminal repeat (LTR) of the human T-cell leukemia virus type 1 (HTLV-1), where Sp1 and Ets1 form a ternary complex with the proviral DNA site and synergistically activate LTR-driven transcription (57). We and other researchers have reported that the BKPyV NCCR confers corticosteroid responses (8), which may explain the role of corticosteroids as risk factors for BKPyV replication and nephropathy beyond its immunosuppressive effects in kidney transplant recipients (58–61). In renal cells, ischemia and reperfusion have been linked to a lower Sp1 transcriptional activity (62). Also, Sp1 activity in response to insulin increases the expression of insulin receptor (51). It is therefore tempting to speculate that Sp1 and Ets1 sites in the BKPyV NCCR molecularly direct and integrate viral responses when hypoxia, ischemia/reperfusion injury, insulin resistance, and new-onset diabetes, as well as inflammation and stress signals, are sensed (58, 59, 63–65).
To summarize and integrate the experimental results, we propose the following working model (Fig. 8), in which Sp1 and Ets1 provide directional impact for LVGR and EVGR expression and group 2 factors like NF1 modulate this directionality. The model includes the TATA box for EVGR expression as well as a TATA-like element close to the LVGR TSS bearing mismatches (56), as suggested before (66, 67). Accordingly, host cell signals leading to viral (re)activation, such as stress, injury, or insulin signaling, can act by modulating Sp1 and Ets1 activity. Natural variants, including point mutations, deletions, and duplications, facilitate activated EVGR expression at the expense of LVGR. Thus, the EVGR-activating signals or group 1 and group 2 mutations could be a first step toward generation of more replication-competent variants in patients lacking sufficient immune responses (8, 18). Future studies need to examine in more detail the molecular mechanism(s) by which the NCCR rearrangements are generated. Presumably, rearrangements occur as errors during recircularization of the bidirectionally replicated polyomavirus DNA genome and select those variants that convey a replicative advantage in immunocompromised patients (8, 18, 68). In fact, the biological plasticity of a functional NCCR repair has been demonstrated in an experimental model of SV40 promoter rescue (69).
FIG 8.

Working model summarizing bidirectional regulation from the BK polyomavirus NCCR. The transcription factors Sp1, Ets1, and NF-1 determine the strength and direction of gene expression toward EVGR (early; red arrow) and LVGR (late; green arrow). (Top) The archetype ww(1.4) NCCR mediates basal activity with low EVGR expression (dashed arrow) and high LVGR expression (green arrow). Sp1 and Ets1 contribute a directional effect on neighboring Sp1 and Ets1, whereas NF1 has a stabilizing effect. (Center) Mutant NCCR phenotypes, according to EVGR and LVGR expression. The group 1 TFBS mediate a major contribution of Sp1 or Ets1 to LVGR expression, which is abrogated in their absence and results in a net increase of EVGR expression. Group 2 TFBS stabilize Sp1 and Ets1 locally and thereby contribute to LVGR and EVGR expression in a partly position-dependent way. Group 3 TFBS are essential for EVGR and LVGR expression, since in their absence, mutants show low viral expression. This suggests that binding to SP1-2 is of overall importance for EVGR and LVGR expression, with possible implications for latency. (Bottom) Patient isolates with rr del(5.3) and ins(7.3) NCCRs result in part from group 1 and 2 mutant effects. In addition, NF1 sites in the center of the NCCR may interfere in an insulator-like fashion with the presumed Sp1 action from SP1-2 on LVGR expression.
In conclusion, this study indicates that the basal BKPyV NCCR is critically controlled by a hierarchy of single TFBS in the archetype NCCR that direct, modulate, and execute bidirectional EVGR and LVGR expression. Our results provide new insights into how BKPyV NCCR functions as a viral sensor of host cell signals through TFBS, including Sp1, Ets1, and NF1. Future studies will demonstrate whether identical or similar hierarchy exists in other human PyVs.
Supplementary Material
ACKNOWLEDGMENTS
The NF1 antibody was a generous gift from Nicholas Mermod, Lausanne, Switzerland. The BKPyV agnoprotein-specific antiserum was kindly provided by Christine H. Rinaldo, Tromsø, Norway. We also thank Emmanuel Traunecker and Toni Krebs at the Department Biomedicine (Haus Hebelstrasse), University of Basel, for help and support regarding flow cytometry.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03625-14.
REFERENCES
- 1.Rinaldo CH, Hirsch HH. 2013. The human polyomaviruses: from orphans and mutants to patchwork family. APMIS 121:681–684. doi: 10.1111/apm.12125. [DOI] [PubMed] [Google Scholar]
- 2.Rinaldo CH, Tylden GD, Sharma BN. 2013. The human polyomavirus BK (BKPyV): virological background and clinical implications. APMIS 121:728–745. doi: 10.1111/apm.12134. [DOI] [PubMed] [Google Scholar]
- 3.Hirsch HH, Steiger J. 2003. Polyomavirus BK. Lancet Infect Dis 3:611–623. doi: 10.1016/S1473-3099(03)00770-9. [DOI] [PubMed] [Google Scholar]
- 4.Stolt A, Sasnauskas K, Koskela P, Lehtinen M, Dillner J. 2003. Seroepidemiology of the human polyomaviruses. J Gen Virol 84:1499–1504. doi: 10.1099/vir.0.18842-0. [DOI] [PubMed] [Google Scholar]
- 5.Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, Gosert R, Hirsch HH. 2009. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis 199:837–846. doi: 10.1086/597126. [DOI] [PubMed] [Google Scholar]
- 6.Kean JM, Rao S, Wang M, Garcea RL. 2009. Seroepidemiology of human polyomaviruses. PLoS Pathog 5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polo C, Perez JL, Mielnichuck A, Fedele CG, Niubo J, Tenorio A. 2004. Prevalence and patterns of polyomavirus urinary excretion in immunocompetent adults and children. Clin Microbiol Infect 10:640–644. doi: 10.1111/j.1469-0691.2004.00882.x. [DOI] [PubMed] [Google Scholar]
- 8.Gosert R, Rinaldo CH, Funk GA, Egli A, Ramos E, Drachenberg CB, Hirsch HH. 2008. Polyomavirus BK with rearranged noncoding control region emerge in vivo in renal transplant patients and increase viral replication and cytopathology. J Exp Med 205:841–852. doi: 10.1084/jem.20072097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dropulic LK, Jones RJ. 2008. Polyomavirus BK infection in blood and marrow transplant recipients. Bone Marrow Transplant 41:11–18. doi: 10.1038/sj.bmt.1705886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markowitz RB, Eaton BA, Kubik MF, Latorra D, McGregor JA, Dynan WS. 1991. BK virus and JC virus shed during pregnancy have predominantly archetypal regulatory regions. J Virol 65:4515–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Negrini M, Sabbioni S, Arthur RR, Castagnoli A, Barbanti-Brodano G. 1991. Prevalence of the archetypal regulatory region and sequence polymorphisms in nonpassaged BK virus variants. J Virol 65:5092–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li R, Sharma BN, Linder S, Gutteberg TJ, Hirsch HH, Rinaldo CH. 2013. Characteristics of polyomavirus BK (BKPyV) infection in primary human urothelial cells. Virology 440:41–50. doi: 10.1016/j.virol.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 13.Randhawa P, Zygmunt D, Shapiro R, Vats A, Weck K, Swalsky P, Finkelstein S. 2003. Viral regulatory region sequence variations in kidney tissue obtained from patients with BK virus nephropathy. Kidney Int 64:743–747. doi: 10.1046/j.1523-1755.2003.00132.x. [DOI] [PubMed] [Google Scholar]
- 14.Binggeli S, Egli A, Schaub S, Binet I, Mayr M, Steiger J, Hirsch HH. 2007. Polyomavirus BK-specific cellular immune response to VP1 and large T-antigen in kidney transplant recipients. Am J Transplant 7:1131–1139. doi: 10.1111/j.1600-6143.2007.01754.x. [DOI] [PubMed] [Google Scholar]
- 15.Ginevri F, Basso S, Hirsch HH, Fontana I, Cioni M, Botti G, Valente U, Perfumo F, Azzi A, Locatelli F, Comoli P. 2007. Reconstitution of BKV-specific immunity through immunosuppression reduction prevents BKV nephropathy in pediatric kidney recipients monitored prospectively. Transplant Int 20:80. [Google Scholar]
- 16.Schachtner T, Muller K, Stein M, Diezemann C, Sefrin A, Babel N, Reinke P. 2011. BK virus-specific immunity kinetics: a predictor of recovery from polyomavirus BK-associated nephropathy. Am J Transplant 11:2443–2452. doi: 10.1111/j.1600-6143.2011.03693.x. [DOI] [PubMed] [Google Scholar]
- 17.Hirsch HH, Babel N, Comoli P, Friman V, Ginevri F, Jardine A, Lautenschlager I, Legendre C, Midtvedt K, Munoz P, Randhawa P, Rinaldo CH, Wieszek A, ESCMID Study Group of Infection in Compromised Hosts . 2014. European perspective on human polyomavirus infection, replication and disease in solid organ transplantation. Clin Microbiol Infect 20(Suppl 7):S74–S88. doi: 10.1111/1469-0691.12538. [DOI] [PubMed] [Google Scholar]
- 18.Gosert R, Kardas P, Major EO, Hirsch HH. 2010. Rearranged JC virus noncoding control regions found in progressive multifocal leukoencephalopathy patient samples increase virus early gene expression and replication rate. J Virol 84:10448–10456. doi: 10.1128/JVI.00614-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO. 2012. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev 25:471–506. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan CS, Koralnik IJ. 2010. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol 9:425–437. doi: 10.1016/S1474-4422(10)70040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirsch HH, Kardas P, Kranz D, Leboeuf C. 2013. The human JC polyomavirus (JCPyV): virological background and clinical implications. APMIS 121:685–727. doi: 10.1111/apm.12128. [DOI] [PubMed] [Google Scholar]
- 22.Van Loy T, Thys K, Ryschkewitsch C, Lagatie O, Monaco MC, Major EO, Tritsmans L, Stuyver LJ. 2015. JC virus quasispecies analysis reveals complex viral population underlying PML and supports viral dissemination via the hematogenous route. J Virol 89:1340–1347. doi: 10.1128/JVI.02565-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imperiale MJ, Major EO. 2007. Polyomaviruses, p 2263–2298 InKnipe DM, Howley PM (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Wolters Kluwer, Philadelphia, PA, USA. [Google Scholar]
- 24.Olsen GH, Andresen PA, Hilmarsen HT, Bjorang O, Scott H, Midtvedt K, Rinaldo CH. 2006. Genetic variability in BK Virus regulatory regions in urine and kidney biopsies from renal-transplant patients. J Med Virol 78:384–393. doi: 10.1002/jmv.20551. [DOI] [PubMed] [Google Scholar]
- 25.Stoner GL, Alappan R, Jobes DV, Ryschkewitsch CF, Landry ML. 2002. BK virus regulatory region rearrangements in brain and cerebrospinal fluid from a leukemia patient with tubulointerstitial nephritis and meningoencephalitis. Am J Kidney Dis 39:1102–1112. doi: 10.1053/ajkd.2002.32795. [DOI] [PubMed] [Google Scholar]
- 26.Olsen GH, Hirsch HH, Rinaldo CH. 2009. Functional analysis of polyomavirus BK non-coding control region quasispecies from kidney transplant recipients. J Med Virol 81:1959–1967. doi: 10.1002/jmv.21605. [DOI] [PubMed] [Google Scholar]
- 27.Priftakis P, Bogdanovic G, Kalantari M, Dalianis T. 2001. Overrepresentation of point mutations in the Sp1 site of the non-coding control region of BK virus in bone marrow transplanted patients with haemorrhagic cystitis. J Clin Virol 21:1–7. doi: 10.1016/S1386-6532(00)00171-2. [DOI] [PubMed] [Google Scholar]
- 28.Petrogiannis-Haliotis T, Sakoulas G, Kirby J, Koralnik IJ, Dvorak AM, Monahan-Earley R, De Girolami PC, De Girolami U, Upton M, Major EO, Pfister LA, Joseph JT. 2001. BK-related polyomavirus vasculopathy in a renal-transplant recipient. N Engl J Med 345:1250–1255. doi: 10.1056/NEJMoa010319. [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Sharp PM, Fowkes M, Kocher O, Joseph JT, Koralnik IJ. 2004. Analysis of 15 novel full-length BK virus sequences from three individuals: evidence of a high intra-strain genetic diversity. J Gen Virol 85:2651–2663. doi: 10.1099/vir.0.79920-0. [DOI] [PubMed] [Google Scholar]
- 30.Dumoulin A, Hirsch HH. 2011. Reevaluating and optimizing polyomavirus BK and JC real-time PCR assays to detect rare sequence polymorphisms. J Clin Microbiol 49:1382–1388. doi: 10.1128/JCM.02008-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rinaldo CH, Traavik T, Hey A. 1998. The agnogene of the human polyomavirus BK is expressed. J Virol 72:6233–6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schreiber E, Matthias P, Muller MM, Schaffner W. 1988. Identification of a novel lymphoid specific octamer binding protein (OTF-2B) by proteolytic clipping bandshift assay (PCBA). EMBO J 7:4221–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schreiber E, Matthias P, Muller MM, Schaffner W. 1989. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res 17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seif I, Khoury G, Dhar R. 1979. The genome of human papovavirus BKV. Cell 18:963–977. doi: 10.1016/0092-8674(79)90209-5. [DOI] [PubMed] [Google Scholar]
- 35.Imperiale MJ, Jiang M. 3December2014. What DNA viral genomic rearrangements tell us about persistence. J Virol doi: 10.1128/JVI.01227-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amirhaeri S, Wohlrab F, Major EO, Wells RD. 1988. Unusual DNA structure in the regulatory region of the human papovavirus JC virus. J Virol 62:922–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loeber G, Dorries K. 1988. DNA rearrangements in organ-specific variants of polyomavirus JC strain GS. J Virol 62:1730–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho J, Jedrych JJ, Feng H, Natalie AA, Grandinetti L, Mirvish E, Crespo MM, Yadav D, Fasanella KE, Proksell S, Kuan SF, Pastrana DV, Buck CB, Shuda Y, Moore PS, Chang Y. 17September2014. Human polyomavirus 7-associated pruritic rash and viremia in transplant recipients. J Infect Dis doi: 10.1093/infdis/jiu524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cassill JA, Deyerle KL, Subramani S. 1989. Unidirectional deletion and linker scan analysis of the late promoter of the human papovavirus BK. Virology 169:172–181. doi: 10.1016/0042-6822(89)90053-6. [DOI] [PubMed] [Google Scholar]
- 40.Deyerle KL, Cassill JA, Subramani S. 1987. Analysis of the early regulatory region of the human papovavirus BK. Virology 158:181–193. doi: 10.1016/0042-6822(87)90252-2. [DOI] [PubMed] [Google Scholar]
- 41.Del Vecchio AM, Steinman RA, Ricciardi RP. 1989. An element of the BK virus enhancer required for DNA replication. J Virol 63:1514–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gluzman Y. 1981. SV40-transformed simian cells support the replication of early SV40 mutants. Cell 23:175–182. doi: 10.1016/0092-8674(81)90282-8. [DOI] [PubMed] [Google Scholar]
- 43.Markowitz RB, Dynan WS. 1988. Binding of cellular proteins to the regulatory region of BK virus DNA. J Virol 62:3388–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ault GS, Stoner GL. 1993. Human polyomavirus JC promoter/enhancer rearrangement patterns from progressive multifocal leukoencephalopathy brain are unique derivatives of a single archetypal structure. J Gen Virol 74:1499–1507. doi: 10.1099/0022-1317-74-8-1499. [DOI] [PubMed] [Google Scholar]
- 45.Daniel AM, Swenson JJ, Mayreddy RP, Khalili K, Frisque RJ. 1996. Sequences within the early and late promoters of archetype JC virus restrict viral DNA replication and infectivity. Virology 216:90–101. doi: 10.1006/viro.1996.0037. [DOI] [PubMed] [Google Scholar]
- 46.Marshall LJ, Moore LD, Mirsky MM, Major EO. 2012. JC virus promoter/enhancers contain TATA box-associated Spi-B-binding sites that support early viral gene expression in primary astrocytes. J Gen Virol 93:651–661. doi: 10.1099/vir.0.035832-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marshall LJ, Major EO. 2010. Molecular regulation of JC virus tropism: insights into potential therapeutic targets for progressive multifocal leukoencephalopathy. J Neuroimmune Pharmacol 5:404–417. doi: 10.1007/s11481-010-9203-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tada H, Rappaport J, Lashgari M, Amini S, Wong-Staal F, Khalili K. 1990. Trans-activation of the JC virus late promoter by the tat protein of type 1 human immunodeficiency virus in glial cells. Proc Natl Acad Sci U S A 87:3479–3483. doi: 10.1073/pnas.87.9.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greenall A, Willingham N, Cheung E, Boam DS, Sharrocks AD. 2001. DNA binding by the ETS-domain transcription factor PEA3 is regulated by intramolecular and intermolecular protein.protein interactions. J Biol Chem 276:16207–16215. doi: 10.1074/jbc.M011582200. [DOI] [PubMed] [Google Scholar]
- 50.Wierstra I. 2008. Sp1: emerging roles—beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun 372:1–13. doi: 10.1016/j.bbrc.2008.03.074. [DOI] [PubMed] [Google Scholar]
- 51.Solomon SS, Majumdar G, Martinez-Hernandez A, Raghow R. 2008. A critical role of Sp1 transcription factor in regulating gene expression in response to insulin and other hormones. Life Sci 83:305–312. doi: 10.1016/j.lfs.2008.06.024. [DOI] [PubMed] [Google Scholar]
- 52.Davie JR, He S, Li L, Sekhavat A, Espino P, Drobic B, Dunn KL, Sun JM, Chen HY, Yu J, Pritchard S, Wang X. 2008. Nuclear organization and chromatin dynamics—Sp1, Sp3 and histone deacetylases. Adv Enzyme Regul 48:189–208. doi: 10.1016/j.advenzreg.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 53.Chen PH, Tsao YP, Wang CC, Chen SL. 2008. Nuclear receptor interaction protein, a coactivator of androgen receptors (AR), is regulated by AR and Sp1 to feed forward and activate its own gene expression through AR protein stability. Nucleic Acids Res 36:51–66. doi: 10.1093/nar/gkm942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsushita K, Hagihara M, Sugiura Y. 1998. Participation of oligomerization through C-terminal D domain region of Sp1 in DNA binding. Biol Pharm Bull 21:1094–1097. doi: 10.1248/bpb.21.1094. [DOI] [PubMed] [Google Scholar]
- 55.Li L, He S, Sun JM, Davie JR. 2004. Gene regulation by Sp1 and Sp3. Biochem Cell Biol 82:460–471. doi: 10.1139/o04-045. [DOI] [PubMed] [Google Scholar]
- 56.Rhee HS, Pugh BF. 2012. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483:295–301. doi: 10.1038/nature10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gegonne A, Bosselut R, Bailly RA, Ghysdael J. 1993. Synergistic activation of the HTLV1 LTR Ets-responsive region by transcription factors Ets1 and Sp1. EMBO J 12:1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirsch HH, Knowles W, Dickenmann M, Passweg J, Klimkait T, Mihatsch MJ, Steiger J. 2002. Prospective study of polyomavirus type BK replication and nephropathy in renal-transplant recipients. N Engl J Med 347:488–496. doi: 10.1056/NEJMoa020439. [DOI] [PubMed] [Google Scholar]
- 59.Hirsch HH, Vincenti F, Friman S, Tuncer M, Citterio F, Wiecek A, Scheuermann EH, Klinger M, Russ G, Pescovitz MD, Prestele H. 2013. Polyomavirus BK replication in de novo kidney transplant patients receiving tacrolimus or cyclosporine: a prospective, randomized, multicenter study. Am J Transplant 13:136–145. doi: 10.1111/j.1600-6143.2012.04320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dadhania D, Snopkowski C, Ding R, Muthukumar T, Chang C, Aull M, Lee J, Sharma VK, Kapur S, Suthanthiran M. 2008. Epidemiology of BK virus in renal allograft recipients: independent risk factors for BK virus replication. Transplantation 86:521–528. doi: 10.1097/TP.0b013e31817c6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coleman DV, Mackenzie EF, Gardner SD, Poulding JM, Amer B, Russell WJ. 1978. Human polyomavirus (BK) infection and ureteric stenosis in renal allograft recipients. J Clin Pathol 31:338–347. doi: 10.1136/jcp.31.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu N, Siow YL, OK. 2010. Ischemia/reperfusion reduces transcription factor Sp1-mediated cystathionine beta-synthase expression in the kidney. J Biol Chem 285:18225–18233. doi: 10.1074/jbc.M110.132142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fishman JA. 2002. BK virus nephropathy—polyomavirus adding insult to injury. N Engl J Med 347:527–530. doi: 10.1056/NEJMe020076. [DOI] [PubMed] [Google Scholar]
- 64.Vincenti F, Friman S, Scheuermann E, Rostaing L, Jenssen T, Campistol JM, Uchida K, Pescovitz MD, Marchetti P, Tuncer M, Citterio F, Wiecek A, Chadban S, El-Shahawy M, Budde K, Goto N. 2007. Results of an international, randomized trial comparing glucose metabolism disorders and outcome with cyclosporine versus tacrolimus. Am J Transplant 7:1506–1514. doi: 10.1111/j.1600-6143.2007.01749.x. [DOI] [PubMed] [Google Scholar]
- 65.Kallio PJ, Okamoto K, O'Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L. 1998. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J 17:6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chakraborty T, Das GC. 1991. Proteins of the nuclear factor-1 family act as an activator of the late promoter in human polyomavirus BK in vitro. J Gen Virol 72:1935–1942. doi: 10.1099/0022-1317-72-8-1935. [DOI] [PubMed] [Google Scholar]
- 67.Kraus RJ, Shadley L, Mertz JE. 2001. Nuclear factor 1 family members mediate repression of the BK virus late promoter. Virology 287:89–104. doi: 10.1006/viro.2001.1024. [DOI] [PubMed] [Google Scholar]
- 68.Johnson EM, Wortman MJ, Dagdanova AV, Lundberg PS, Daniel DC. 2013. Polyomavirus JC in the context of immunosuppression: a series of adaptive, DNA replication-driven recombination events in the development of progressive multifocal leukoencephalopathy. Clin Dev Immunol 2013:197807. doi: 10.1155/2013/197807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Keiser S, Schmidt K, Bethge T, Steiger J, Hirsch HH, Schaffner W, Georgiev O. 10November2014. Emergence of infectious simian virus 40 whose AT tract in the replication origin/early promoter region is substituted by cellular or viral DNAs. J Gen Virol doi: 10.1099/vir.0.071274-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.