

ABSTRACT
Viral infection results in the generation of massive numbers of activated effector CD8+ T cells that recognize viral components. Most of these are short-lived effector T cells (SLECs) that die after clearance of the virus. However, a small proportion of this population survives and forms antigen-specific memory precursor effector cells (MPECs), which ultimately develop into memory cells. These can participate in a recall response upon reexposure to antigen even at protracted times postinfection. Here, antiapoptotic myeloid cell leukemia 1 (MCL1) was found to prolong survival upon T cell stimulation, and mice expressing human MCL1 as a transgene exhibited a skewing in the proportion of CD8+ T cells, away from SLECs toward MPECs, during the acute phase of vaccinia virus infection. A higher frequency and total number of antigen-specific CD8+ T cells were observed in MCL1 transgenic mice. These findings show that MCL1 can shape the makeup of the CD8+ T cell response, promoting the formation of long-term memory.
IMPORTANCE During an immune response to a virus, CD8+ T cells kill cells infected by the virus, and most die when the infection resolves. However, a small proportion of cells survives and differentiates into long-lived memory cells that confer protection from reinfection by the same virus. This report shows that transgenic expression of an MCL1 protein enhances survival of memory CD8+ T cells following infection with vaccinia virus. This is important because it shows that MCL1 expression may be an important determinant of the formation of long-term CD8+ T cell memory.
INTRODUCTION
Upon exposure to infectious agents, T cells undergo changes in gene expression that promote the generation and survival of effector cells, followed by the emergence of long-lived memory cells. The acute phase of virus infection results in the following sequence of events in CD8+ T cells. A primary phase of clonal expansion generates cytolytic effector cells to facilitate elimination of the pathogen. This is followed by a contraction phase, during which a large number of potentially damaging cytotoxic effector cells undergo apoptosis. However, a number of cells survive this contraction and form the precursors of memory cells. Finally, during the memory phase, a small subset of antigen-specific CD8+ T cells is maintained for an extended period, providing memory for later recall responses (1).
While short-lived effector cells (SLECs) are important for the resolution of infection, memory precursor effector cells (MPECs) differentiate into the long-lived memory population (2). MPECs exhibit differences from SLECs in terms of phenotype and function (3). While both populations elaborate effector functions, MPECs have more subdued effector activity than SLECs (1, 4, 5). MPECs exhibit lower cell surface expression of the killer cell lectin-like receptor subfamily G member 1 (KLRG1) but higher expression of CD127 (IL-7Rα) (3). In contrast, SLECs exhibit higher KLRG1 but lower CD127 expression. In addition, interleukin-2 (IL-2) production is largely restricted to the MPEC CD8+ population and is necessary for memory cells to mount efficient recall responses (6). The formation of memory versus effector CD8+ T cells depends on multiple factors, including the strength of T cell receptor (TCR) signaling, engagement of costimulatory molecules, and responsiveness to cytokines such as IL-2, IL-10, IL-12, and IL-21 (7, 8).
BCL2 family members control the viability of T cells during development and in response to foreign antigens (9, 10). MCL1 is a viability-promoting member of this family that contains the signature BCL2 homology (BH) domains in its carboxyl portion (11). MCL1 also exhibits characteristics different from those of BCL2 and is unique in containing a long N-terminal regulatory region. Accordingly, a salient characteristic of MCL1 is its ability to undergo rapid upregulation/stabilization in response to environmental stimuli, such as cytokines/growth factors and antigen signaling (11, 12). MCL1 also binds proapoptotic family members, such as Noxa, that do not interact extensively with BCL2. While MCL1 was identified in myeloid leukemia cells stimulated to differentiate, it has effects in lymphoid cells at various stages of development. These effects were first seen in transgenic mice expressing a human MCL1 minigene in hematolymphoid tissues, where transgene expression is in the range of that normally seen in response to stimulation (13). Lymphoid cells from the spleens of transgenic mice exhibit enhanced survival in tissue culture. However, the mice do not exhibit an increase in circulating lymphocyte numbers, presumably because of homeostatic regulatory influences. Knockout experiments have shown that MCL1 has an important role in T cell development, as this lineage is reduced upon conditional knockout in thymic cells at early or later stages (14). Congruently, the MCL1 transgene can promote survival in early thymic progenitors (15, 16). MCL1 also has a role in the response of T cells to foreign antigens. TCR ligation leads to MCL1 stabilization and promotes the survival of high-affinity clones, by neutralizing proapoptotic family members (prominently Noxa) (17). In recent studies, knockout of MCL1 during infection with lymphocytic choriomeningitis virus was found to result in a severe decrease in the production of virus-specific T cells (18, 19).
It is not yet clear how the survival of memory T cells is regulated. BCL2 is expressed during memory formation, but knockout of BCL2 or BCLX does not abolish T-cell memory (20–24). Recent experiments are consistent with the possibility that MCL1 could have a role, as an association was observed between MCL1 stabilization and enhanced memory precursor frequency, induced through NKG2D signaling (25).
The present studies used transgenic mice expressing a human MCL1 minigene to assess the effect of the transgene on memory CD8+ T cell formation. The findings obtained show that MCL1 prolongs the survival of antigen-stimulated cells and that MCL1 transgenic mice exposed to vaccinia virus exhibit an increase in antigen-specific CD8+ T cells with the MPEC phenotype. In addition, the frequency and total number of antigen-specific CD8+ T cells increase during the memory stage. By demonstrating that MCL1 transgene expression favors the MPEC phenotype and memory cell formation, our data show that MCL1 can contribute to the development and maintenance of this important cell population.
MATERIALS AND METHODS
Animals.
C57BL/6 mice bearing a human MCL1 transgene have been described (16). These mice were bred and maintained in the animal facility at the Geisel School of Medicine at Dartmouth. Genotyping was carried out with primers that are specific to the human MCL1 transgene but do not recognize the endogenous mouse homologue (26), by using the RedExtract-N-AMP tissue PCR kit (Sigma-Aldrich). Mice found to lack the MCL1 transgene, from the same or contemporaneous litters, served as nontransgenic controls. Animal studies were performed in accordance with a protocol approved by the institutional animal care and use committee of the Geisel School of Medicine at Dartmouth and with the National Institutes of Health recommendations set forth in the Guide for the Care and Use of Laboratory Animals.
Vaccinia virus primary infection and Listeria recall infection.
Primary infection with the vaccinia virus Western Reserve strain (VV-WR) (1,000 PFU) was administered via the intranasal (i.n.) route under anesthesia with isoflurane. For secondary infections, replication-defective Listeria monocytogenes expressing the VV-WR dominant epitope, B8R20–27 (TSYKFESV)/Kb (here referred to as B8R), was administered (2 × 106 CFU) via the intravenous route under anesthesia with ketamine-xylazine.
Antibodies and flow cytometry.
To obtain single-cell suspensions, spleens were passed through a 70-μm cell strainer with a syringe plunger. Red blood cells were lysed with Gey's solution. Cell counts were monitored with a Coulter counter as in previous studies (16). This method is optimized to exclude debris, which may include debris representing dying cells. Cell surface staining was performed by incubation with antibodies in cell staining buffer (phosphate-buffered saline [PBS] containing 2% fetal bovine serum [FBS]) on ice for 20 min. The fluorochrome-conjugated antibodies used included fluorescein isothiocyanate (FITC)-CD127, FITC-CD44, phycoerythrin (PE)-KLRG1, PE–IL-2, PE-tumor necrosis factor alpha (TNF-α), and allophycocyanin (APC)-gamma interferon (IFN-γ) from BioLegend, PE-granzyme B from Invitrogen, and peridinin chlorophyll protein-eFluor 710 CD8α from eBioscience. To measure apoptosis, cells were washed once with annexin V binding buffer and stained with PE-annexin V (eBioscience). Samples were analyzed with an Accuri C6 flow cytometer or a FACSCalibur (BD Biosciences) in the Dartlab core facility at the Geisel School of Medicine at Dartmouth. Data were analyzed with FlowJo (Tree Star) or Accuri CFlow plus software.
Tetramer and intracellular cytokine/molecule staining.
Peptide major histocompatibility complex class I tetramers consisting of B8R20-27/Kb conjugated to APC were obtained from the NIH tetramer core facility. Splenocytes were either directly stained with tetramer together with Fc block (2.4G2) or, in some cases, stimulated with 1 μg/ml B8R peptide for 5 h at 37°C in complete medium containing 10 U/ml recombinant IL-2 and 10 μg/ml brefeldin A in 96-well U-bottom plates. Cells were stained with antibodies detecting surface markers (20 min at 4°C) and then fixed (2% formaldehyde at room temperature for 20 min). They were then incubated in permeabilization buffer (PBS containing 2% FBS and 0.5% saponin) for 10 min and maintained in this buffer for staining with antibodies directed against intracellular cytokines/molecules (IFN-γ, TNF-α, granzyme B, IL-2) by incubation for 30 min at 4°C.
Plaque assay.
To assess the lung virus load after vaccinia virus infection, lung tissue homogenate was diluted, added to 143B cells in 12-well plates, and incubated for 1 h at 37°C to allow absorption before the addition of 2 ml of prewarmed medium containing carboxymethyl cellulose. Cultures were incubated at 37°C for 2 days, fixed with methanol, and then stained with Giemsa stain. Plaques were counted, and the viral titer was calculated by multiplying by the relevant dilution.
CFSE cell proliferation assay.
Assays measuring carboxyfluorescein succinimidyl ester (CFSE) dye dilution (as a measure of proliferation) and 7-aminoactinomycin D (7-AAD) dye exclusion (as a measure of cell viability) were carried out as follows. Splenocytes were cultured with 10 μM CFSE (CellTrace CFSE cell proliferation kit; Life Technologies) at 37°C for 10 min. After being washed twice with cold RPMI 1640 medium, 2.5 × 106 splenocytes were dispensed into each well of 24-well plates. Cells were then stimulated with 2 μg/ml CD3ε and CD28 soluble antibodies. On day 3, cells were harvested, washed once with cell staining buffer, and stained with 1 μg/ml 7-AAD. CFSE dye dilution was detected as a measure of proliferation and 7-AAD exclusion to measure cell viability by flow cytometry on a FACSCalibur. CFSE data were processed by using the proliferation function in FlowJo software.
Western blot assay.
Splenocytes were cultured in RPMI 1640 medium supplemented with 10% FBS (24-well plates, 37°C) in the absence or presence of 2 μg/ml anti-CD3ε and -CD28 antibodies. Protein was extracted by sonication in radioimmunoprecipitation assay buffer, and the protein concentration was measured by bicinchoninic acid assay. Equal amounts of protein were loaded onto 12.5% polyacrylamide gels and probed with a rabbit polyclonal anti-MCL1 antibody (S-19; Santa Cruz). The blots were then reprobed with anti-glyceraldehyde 3-phosphate dehydrogenase antibody.
Statistical analysis.
Statistical analysis was carried out, after testing for normality, with the Student t test or by analysis of variance (SigmaStat Software), and a P value of <0.05 was considered significant.
RESULTS
Anti-CD3 and anti-CD28 antibodies increase MCL1 transgene expression and prolong survival in cultured splenocytes.
In transgenic mice expressing human MCL1 under the control of its endogenous promoter, splenocytes of both B- and T-cell origins exhibit enhanced survival in tissue culture, with the transgene being expressed at levels comparable to those seen upon the stimulation of endogenously expressing cells (13). Enhanced survival might therefore also occur upon exposure to anti-CD3ε and anti-CD28 antibodies to simulate T cell activation, especially since normal T cells, when activated, exhibit MCL1 induction and prolonged survival (14). Transgene expression was indeed increased when splenocytes were cultured in the presence of anti-CD3ε and anti-CD28 antibodies (two transgenic mice are shown in Fig. 1A, lanes 8 versus 6 and 12 versus 10). Such an effect was not seen with IL-2, and IL-2 did not further enhance the effect seen with anti-CD3 and anti-CD28 antibodies. As predicted, survival was also enhanced in the presence of the transgene plus anti-CD3 and anti-CD28 antibodies (Fig. 1B). Survival from day 1 to day 3 was greater in stimulated than in unstimulated cultures (white versus black bars), and the survival of transgenic splenocytes was enhanced to a greater extent than that of their nontransgenic counterparts. Three days after stimulation, the viable cells remaining in transgenic and nontransgenic cultures exhibited equivalent dilutions of CFSE, used to measure cell proliferation (Fig. 1C). Transgenic and nontransgenic cells also exhibited equivalent levels of expression of CD25 (IL-2Rα), a marker of activation (Fig. 1D). In sum, more cells remained present on day 3 upon the stimulation of transgenic cultures, but this did not appear to be due to greater proliferation or activation. Instead, the MCL1 transgene appeared to be capable of enhancing survival in the presence of T cell activation.
FIG 1.

Anti-CD3 and anti-CD28 antibodies increase MCL1 transgene expression in splenocytes, prolonging their survival in culture. Splenocytes from MCL1 transgenic (TR) and nontransgenic (Non TR) mice were explanted into tissue culture and exposed to anti-CD3 and anti-CD28 antibodies. (A) Transgene expression was monitored (Western blotting) on days 1 and 3 in the absence or presence of recombinant murine IL-2. The lack of signal in cells from nontransgenic animals (lanes 1 to 4) was consistent with the fact that human (but not mouse) MCL1 is detected by the antibodies under the conditions used. (B) Viable cell numbers were monitored by 7-AAD staining. The percentage of viable cells remaining on day 3, relative to day 1, is shown, and represents the mean value of eight independent pairs of transgenic and nontransgenic animals. *, P < 0.05 (for the difference between transgenic and nontransgenic cultures; ANOVA). (C) Cell proliferation (CFSE) was examined 3 days after the application of anti-CD3 and anti-CD28 antibodies. The filled gray histogram represents nontransgenic splenocytes, and the open histogram represents MCL1 transgenic splenocytes, in the 7AAD− (viable-cell) gate. (D) Expression of the T cell activation marker CD25 was examined on day 1.
The MCL1 transgene enhances the viability of antigen-specific CD8+ T cells during the acute phase of virus infection.
Given the above findings, we tested if the MCL1 transgene would prolong T cell survival during the acute phase of virus infection. We chose to study the CD8+ T cell response to VV-WR, administered by the i.n. route, as CD8+ T cells are crucial for protection by this route of infection (27). We infected mice i.n. with VV-WR and measured the response to the immunodominant endogenous, H-2Kb-restricted TSYKFESV epitope (B8R). Antigen-specific CD8+ T cells were monitored by staining with B8R tetramers at day 10 after virus infection, which is the peak of the antigen-specific CD8+ T cell response. Naive MCL1 transgenic mice had spleen cell numbers similar to those of nontransgenic B6 control mice. After infection, MCL1 transgenic and nontransgenic mice exhibited comparable numbers of total splenocytes and CD8+ T cells (data not shown), as well as B8R-specific CD8+ T cells in the spleen (Fig. 2A) at day 10 postinfection. Similar data were obtained from the mediastinal lymph node, which drains the lung (data not shown). This agreed with the observation that the lung virus loads were similar (Fig. 2B), as measured by plaque assay, as were the titers of serum antibodies recognizing VV-WR (measured by virus neutralization assay; data not shown). Next, we tested the frequency of B8R-specific CD8+ T cells undergoing apoptosis by measuring annexin V expression. Lower frequencies of both total and B8R-specific CD8+ T cells expressed annexin V in MCL1 transgenic mice than in nontransgenic mice (Fig. 2C) at day 10 postinfection. In sum, the MCL1 transgene not only enhanced the survival of CD3/CD28-stimulated cells in in vitro cultures (Fig. 1) but also reduced apoptosis in virus-specific CD8+ T cells upon infection in vivo.
FIG 2.
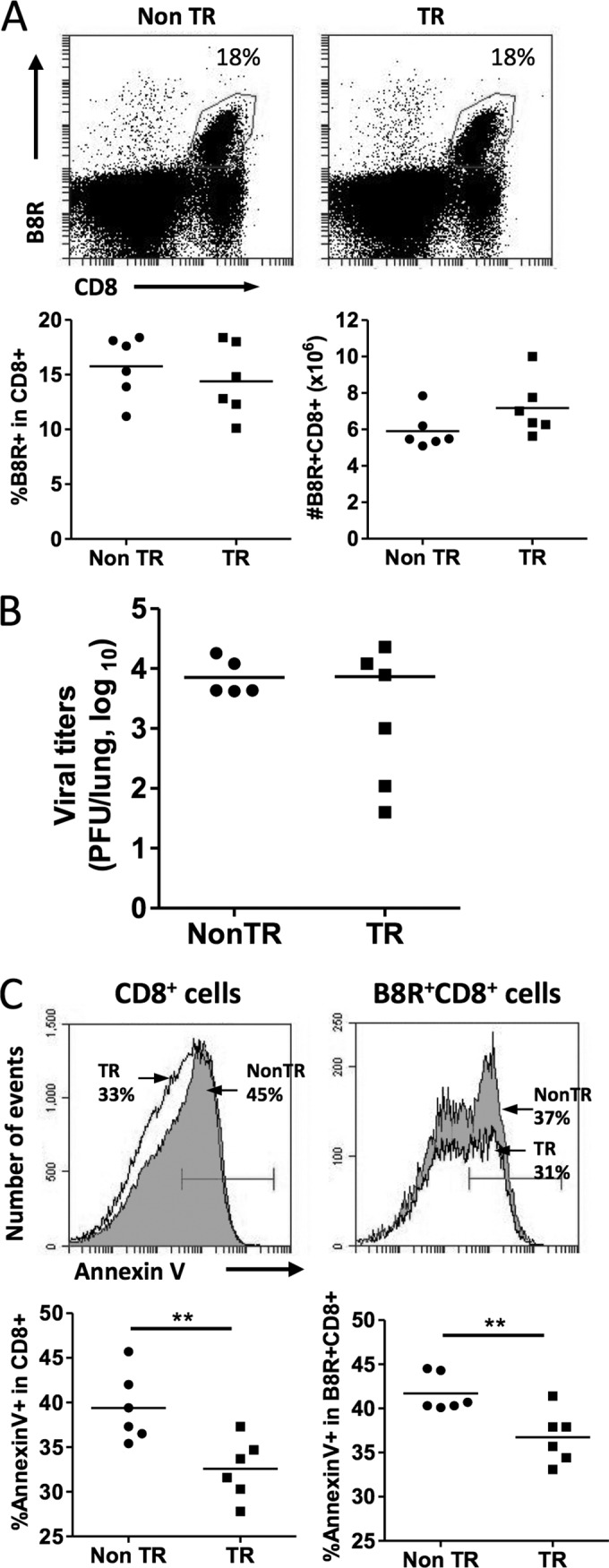
The MCL1 transgene decreases apoptosis of antigen-specific CD8+ T cells during the acute phase of virus infection. Mice were infected i.n. with VV-WR, and splenocytes were harvested at 10 days postinfection. (A) Flow cytometry was used to monitor the expression of CD8 and B8R, an H-2Kb-restricted VV-WR epitope. The percentages of B8R-specific cells shown are those within the CD8+ gate. TR, transgenic; Non TR, nontransgenic. (B) Lung virus titers were measured at 10 days postinfection. (C) Apoptosis of CD8+ splenocytes and B8R-specific CD8+ T cells was assessed by annexin V staining. The percentages of annexin V+ cells (filled histogram, nontransgenic; open histogram, transgenic) are the values obtained after gating on CD8+ (left panels) or B8R+ CD8+ (right panels) cells. The data shown are representative of two independent experiments, where each point in the graphs represents one mouse and the mean value of the six mice is represented by the horizontal line. **, P < 0.001 (for the difference between transgenic and nontransgenic animals; Student's t test).
The MCL1 transgene promotes memory precursor effector cell (MPEC) formation.
Upon initial virus infection, CD8+ T cells become activated, expand, and differentiate into effector T cells. While some of these effector cells terminally differentiate and ultimately die, a subset of activated CD8+ T cells survives and continues to differentiate to become precursors of long-lived memory CD8+ T cells. Knowing that the MCL1 transgene enhanced antigen-specific CD8+ T cell viability, we proposed that this may result in a larger population of MPECs. Characteristics of MPECs are increased expression of the cytokine receptor CD127 (IL-7Rα) and decreased expression of KLRG1 (28). Indeed, at 10 days postinfection, the spleen B8R-specific CD8+ T cells in MCL1 transgenic mice displayed a lower percentage of KLRG1+ cells, as well as higher CD127 expression (Fig. 3A), indicative of a skewing toward the MPEC phenotype. Examination of the mediastinal lymph nodes showed that the frequency of KLRG1 was significantly lower in B8R+ CD8+ T cells of MCL1 transgenic mice, although CD127 expression did not exhibit a statistically significant difference (Fig. 3B).
FIG 3.

The MCL1 transgene promotes antigen-specific CD8+ T cell skewing to an MPEC phenotype during primary viral infection. Mice were infected with VV-WR, and spleens and mediastinal lymph nodes were harvested on day 10 postinfection. (A) By flow cytometry, B8R+ CD8+ T cells from the spleen were assayed for expression of KLRG1 and CD127 (filled histogram, nontransgenic [Non TR]; open histogram, transgenic [TR]). The values shown are percentages of KLRG1+ cells or mean fluorescence intensity for CD127 in the B8R+ CD8+ gate. (B) B8R+ CD8+ T cells from mediastinal lymph node cells were assayed for expression of CD127 and KLRG1. The data shown are representative of two independent experiments with four mice per experiment. *, P < 0.05; **, P < 0.001 (for the difference between transgenic and nontransgenic animals; Student's t test).
CD8+ T cell effector functions remain intact in the presence of the MCL1 transgene.
We wished to determine if expression of the MCL1 transgene would affect effector CD8+ T cell functions during the acute phase of virus infection. We therefore stimulated splenocytes from infected animals with the B8R peptide and used intracellular staining to measure IFN-γ, TNF-α, granzyme B, and IL-2 production in CD8+ T cells. Transgenic splenocytes did not exhibit any significant difference in the percentage of IFN-γ+ cells in CD44+ CD8+ T cells (Fig. 4A). No difference was seen in terms of the expression of TNF-α (Fig. 4B) or granzyme B in the IFN-γ+ CD8+ T cell population (Fig. 4C). Interestingly, the percentage of IL-2-producing cells within the IFN-γ+ CD8+ T cell population was higher in MCL1 transgenic splenocytes than in nontransgenic splenocytes (Fig. 4D). The ability to produce IL-2 increases progressively during memory CD8+ T cell differentiation and is one of the characteristics of MPECs. The greater capacity to produce IL-2 in MCL1 transgenic peptide-responding CD8+ T cell subsets likely reflects a propensity for transition to memory cells. These data, together with surface marker expression, demonstrate that increased MPEC formation in MCL1 transgenic mice during the acute phase of virus infection occurred without a loss of effector capacity.
FIG 4.

Effector function during primary viral infection remains intact in the presence of the MCL1 transgene. Mice were infected with VV-WR, and their spleens were harvested at day 10 postinfection. Splenocytes were restimulated with or without B8R peptide for 5 h in the presence of brefeldin A. Intracellular staining was performed to determine IFN-γ production by CD44+ CD8+ T cells (A). No peptide stimulation is shown as a negative control. The percentages in the representative fluorescence-activated cell sorter plots and graphs are those within the CD44+ CD8+ gate. (B) Percentages of IFN-γ+ CD8+ cells that were TNF-α+. (C) Granzyme B (GzmB) mean fluorescence intensity in IFN-γ+ CD8+ cells (filled, nontransgenic [Non TR]; open, transgenic [TR]). Antibody isotype (ISO) for GzmB (light gray line) was used as a negative control for staining. (D) Percentages of IFN-γ+ CD8+ cells that were IL-2+. The data shown are representative of two independent experiments with four mice per experiment. ***, P < 0.0001 (for the difference between transgenic and nontransgenic animals; Student's t test).
MCL1 transgenic mice exhibit increased numbers of antigen-specific CD8+ T cells upon a secondary challenge.
Given the MPEC skewing seen in MCL1 transgenic mice 10 days after infection, we wished to test for longer-term effects. The memory recall response was examined 2 months after primary VV-WR infection by challenging mice with the intracellular pathogen L. monocytogenes engineered to express the B8R epitope. A marked secondary expansion of B8R-specific CD8+ T cells was seen in transgenic and nontransgenic animals, with B8R-specific cells representing a much larger proportion of the CD8 population than had been seen during the acute phase of infection (nontransgenic, 16% primary versus 48% secondary; MCL1 transgenic, 15% primary versus 54% secondary; averages from Fig. 2A and 5A). Interestingly, the total number of B8R-specific CD8+ T cells was significantly higher in MCL1 transgenic mice than in nontransgenic mice (Fig. 5A). This appeared to relate, at least in part, to the presence of a larger spleen during the recall response in transgenic mice than in nontransgenic mice, whereas the proportion of B8R-specific cells among CD8+ T cells did not exhibit a statistically significant difference. B8R peptide stimulation revealed similar proportions of CD44+ CD8+ T cells that were IFN-γ+ and TNF-α-, granzyme B-, and IL-2-producing CD8+ T cells that were IFN-γ+ in splenocytes from these MCL1 transgenic and nontransgenic mice (Fig. 5B).
FIG 5.

MCL1 transgenic mice exhibit an increased number of B8R-specific CD8+ T cells during recall in response to a secondary challenge. Mice were infected with VV-WR, and at 55 days after receiving primary infection, they were administered 2 × 106 CFU of L. monocytogenes expressing the B8R epitope. At 7 days after a secondary challenge, their spleens were harvested. (A) Splenocytes were stained with B8R tetramer and anti-CD8 antibody and analyzed by flow cytometry. The percentages of B8R-specific cells shown are those within the CD8+ gate. (B) Splenocytes were restimulated with B8R peptide for 5 h in the presence of brefeldin A. Intracellular staining was performed to measure the percentage of CD44+ CD8+ T cells that were IFN-γ+, the percentage of IFN-γ+ CD8+ cells that were TNF-α+, the GzmB mean fluorescence intensity in IFN-γ+ CD8+ cells, and the percentage of IFN-γ+ CD8+ cells that were IL-2+. The data shown are representative of two independent experiments with four mice per experiment. **, P < 0.001 (for the difference between transgenic [TR] and nontransgenic [Non TR] animals; Student's t test).
MCL1 transgenic mice have a higher frequency of virus-specific memory CD8+ T cells.
The above data showing a larger recall response in MCL1 transgenic mice raised the question of whether there was a larger “resting” memory population of virus-specific cells in these mice. Mice were therefore examined at 69 days after primary infection, without a secondary challenge. MCL1 transgenic mice exhibited an increase in the total number of splenocytes and CD8+ T cells (Fig. 6A). Importantly, the frequency of B8R+ cells in the CD8+ T cell pool was 2-fold higher in MCL1 transgenic mice than in nontransgenic mice (3% versus 1.5%) (Fig. 6B, left panel). With an increase in both the total splenocyte number and the proportion of B8R+ CD8+ T cells, the number of antigen-specific T cells remaining 2 months after primary infection was increased 3-fold in the presence of the MCL1 transgene (Fig. 6B, right panel). Overall, these findings showed that an MCL1 transgene can contribute to the long-term survival of virus-specific T cells following infection.
FIG 6.
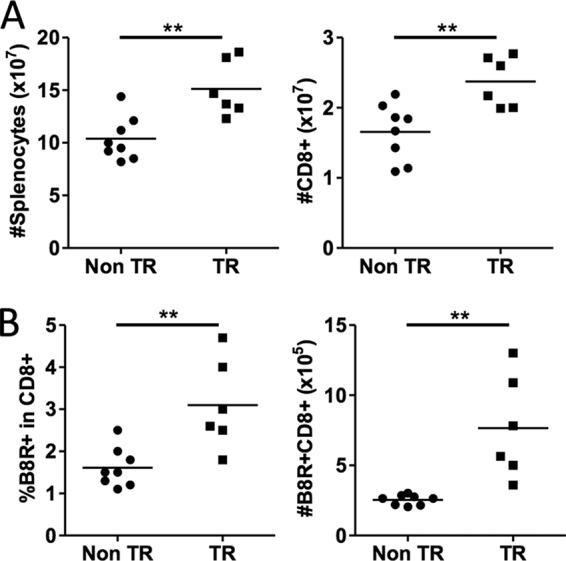
The MCL1 transgene increases the frequency and total number of memory B8R-specific CD8+ T cells at late times following infection. Mice were infected with VV-WR, and their spleens were harvested at day 69 postinfection. (A) Total splenocytes and CD8+ T cell numbers were determined. (B) Splenocytes were stained with B8R tetramer and anti-CD8 antibody and analyzed by flow cytometry. The data shown are representative of two independent experiments with six to eight mice per experiment. **, P < 0.001 (for the difference between transgenic and nontransgenic animals; Student's t test).
DISCUSSION
MCL1 promotes viability in a host of hematopoietic and lymphoid cells at various stages of differentiation and in response to different stimuli (14, 29–31). In T cells, MCL1 controls death during development (14, 16) and is upregulated in response to cytokines that promote T cell survival (32). In the present work, using a transgenic mouse model expressing a human MCL1 minigene, we found that expression of exogenous human in addition to endogenous mouse MCL1 extended the survival of mouse splenocytes in vitro after T cell activation by TCR ligation (Fig. 1). This observation led us to hypothesize that the transgene might promote the survival of CD8+ T cells after activation in vivo during infection and might have an effect in memory T cells. In support of this concept, MCL1 has been shown to be essential for B cell memory (12).
Our studies of primary infection with vaccinia virus showed that, in the acute phase of infection, antigen-specific CD8+ T cells were shifted toward the MPEC phenotype in the presence of the MCL1 transgene. Cells exhibited higher expression of CD127, lower expression of KLRG1 (Fig. 3A), and higher IL-2 secretion after restimulation with antigen (Fig. 4D). The increased proportion of MPECs did not impair CD8 effector functions, as IFN-γ, TNF-α, and granzyme B production was normal and the virus loads in the lungs of transgenic and nontransgenic mice during the acute phase were comparable (Fig. 4A to C). MCL1 transgenic mice exhibited a reduced proportion of antigen-specific CD8+ T cells entering apoptosis (Fig. 2C). Thus, while the number and percentage of antigen-specific CD8+ T cells were equivalent in nontransgenic and transgenic animals at 10 days, in transgenic animals, a lower proportion of these cells expressed annexin V, a marker that appears with the onset of apoptosis. In other words, while transgenic animals exhibit unaltered total numbers of antigen-specific cells, a greater proportion of these cells were viable. This is consistent with antigen-specific cells being generated in equal numbers at 10 days (and proliferating to a similar extent), with a smaller proportion of these cells being destined to die in the case of the transgenic animals. This reduction in the number of cells destined to die likely relates to the observation that a higher frequency of memory cells was present at 2 months after infection.
A previous body of work examining the effects of MCL1 in a variety of tissues has led to a model in which MCL1 promotes cell viability but does not abolish cell death, often resulting in an increase in the percentage of viable cells for a limited period of time; nonetheless, by having this effect in specific cells at key points in their life span, MCL1 can significantly influence cell pool sizes and even organ sizes over the life span of the organism (11, 16). This model may be applicable to the present studies of T cell memory. Expression of transgenic MCL1 in SLECs may not be sufficient to promote their survival significantly in vivo. However, MCL1 transgene expression in MPECs, which are more resistant to apoptosis because of higher-level expression of BCL2 and BCL-XL, may further increase the survival of these cells during infection. Thus, the lower level of annexin V staining seen in cells from transgenic animals than in those from nontransgenic animals at 10 days may spare a proportion of the cells that otherwise would have died. The maintenance of these cells may result in the observed increase in the population of long-term memory cells seen at 2 months postinfection.
Previous studies have shown that CD8+ T cells from patients with chronic hepatitis B virus infection were highly susceptible to apoptotic cell death (33). However, the surviving cells expressed higher levels of CD127 and MCL1. The survival of these cells in vitro was enhanced by inhibition of Bim-mediated apoptosis, consistent with MCL1 expression favoring the survival of memory cells in vivo. Proapoptotic Noxa may also negatively regulate the memory T cell population size, as seen in influenza virus and cytomegalovirus infection experiments (34). In addition, a recent report on NKG2D gene knockout mice showed that NKG2D potentiates IL-15-mediated signaling through phosphoinositol 3-kinase, leading to increased MCL1 protein levels and enhanced survival of memory precursors (25). These findings, along with those presented here, suggest that, in addition to its functions promoting the survival of immature T cells and primary T cells, MCL1 may contribute to memory cell formation and/or survival.
Following resolution of the primary virus infection, we detected a significantly larger memory CD8+ T cell population in MCL1 transgenic mice. This is likely due to the preferential survival of a larger population of memory precursor cells within the effector population in MCL1 mice. The MCL1 transgene appears to promote antigen-specific CD8 T cell survival after pathogen recognition. It is also possible that MCL1 affects cellular processes distinct from those involved in cell survival. Some studies suggest that forms of MCL1 may play a role in normal mitochondrial physiology (35) and autophagy (36), both of which affect T cell differentiation (32, 37). Further studies are necessary to elucidate the molecular mechanisms underlying these effects regarding skewing toward the MPEC phenotype.
It has been shown that the apoptosis threshold set by MCL1 and its antagonist Noxa determined the selection of high-affinity T cell clones. Effector T cells activated in Noxa knockout mice displayed decreased antigen affinity and functionality (17). Similarly, MCL1 transgenic mice might support the survival of suboptimal clones competing for low-affinity antigen, which results in an effector pool with a high proportion of cells with weak avidity for cognate antigen. While this was not directly addressed in our studies, neither control of vaccinia virus upon a primary challenge nor control of L. monocytogenes upon a secondary challenge was significantly compromised. This indicates that the CD8+ T cell response was of sufficient avidity to recognize and eliminate infected cells. However, we cannot rule out changes in CD8+ T cell avidity that did not prevent efficient recognition of target cells and therefore control of the infection.
In conclusion, this report complements other studies involving knockout of MCL1 (14, 18, 32) and shows that the presence of an MCL1 transgene promotes the survival and differentiation of memory CD8+ T cells. This finding adds a further dimension to our understanding of the ability of the MCL1 protein to act at multiple stages during T cell development and the response to viral antigens.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI069943 and CA103642 to E.J.U., CA057359 to R.W.C., and CA062275 to E.D.
REFERENCES
- 1.Wherry EJ, Ahmed R. 2004. Memory CD8 T-cell differentiation during viral infection. J Virol 78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, Kaech SM. 2009. Transcriptional repressor Blimp-1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. 2007. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaech SM, Hemby S, Kersh E, Ahmed R. 2002. Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111:837–851. doi: 10.1016/S0092-8674(02)01139-X. [DOI] [PubMed] [Google Scholar]
- 5.Williams MA, Bevan MJ. 2007. Effector and memory CTL differentiation. Annu Rev Immunol 25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- 6.Feau S, Arens R, Togher S, Schoenberger SP. 2011. Autocrine IL-2 is required for secondary population expansion of CD8+ memory T cells. Nat Immunol 12:908–913. doi: 10.1038/ni.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaech SM, Cui W. 2012. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allie SR, Zhang W, Fuse S, Usherwood EJ. 2011. Programmed death 1 regulates development of central memory CD8 T cells after acute viral infection. J Immunol 186:6280–6286. doi: 10.4049/jimmunol.1003870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunkle A, He YW. 2011. Apoptosis and autophagy in the regulation of T lymphocyte function. Immunol Res 49:70–86. doi: 10.1007/s12026-010-8195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang N, Hartig H, Dzhagalov I, Draper D, He YW. 2005. The role of apoptosis in the development and function of T lymphocytes. Cell Res 15:749–769. doi: 10.1038/sj.cr.7290345. [DOI] [PubMed] [Google Scholar]
- 11.Craig RW. 2002. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia 16:444–454. doi: 10.1038/sj.leu.2402416. [DOI] [PubMed] [Google Scholar]
- 12.Vikstrom I, Carotta S, Luthje K, Peperzak V, Jost PJ, Glaser S, Busslinger M, Bouillet P, Strasser A, Nutt SL, Tarlinton DM. 2010. Mcl-1 is essential for germinal center formation and B cell memory. Science 330:1095–1099. doi: 10.1126/science.1191793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou P, Qian L, Bieszczad CK, Noelle R, Binder M, Levy NB, Craig RW. 1998. Mcl-1 in transgenic mice promotes survival in a spectrum of hematopoietic cell types and immortalization in the myeloid lineage. Blood 92:3226–3239. [PubMed] [Google Scholar]
- 14.Dzhagalov I, Dunkle A, He YW. 2008. The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J Immunol 181:521–528. doi: 10.4049/jimmunol.181.1.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gui J, Mustachio LM, Su DM, Craig RW. 2012. Thymus size and age-related thymic involution: early programming, sexual dimorphism, progenitors and stroma. Aging Dis 3:280–290. [PMC free article] [PubMed] [Google Scholar]
- 16.Gui J, Morales AJ, Maxey SE, Bessette KA, Ratcliffe NR, Kelly JA, Craig RW. 2011. MCL1 increases primitive thymocyte viability in female mice and promotes thymic expansion into adulthood. Int Immunol 23:647–659. doi: 10.1093/intimm/dxr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wensveen FM, van Gisbergen KP, Derks IA, Gerlach C, Schumacher TN, van Lier RA, Eldering E. 2010. Apoptosis threshold set by Noxa and Mcl-1 after T cell activation regulates competitive selection of high-affinity clones. Immunity 32:754–765. doi: 10.1016/j.immuni.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Tripathi P, Koss B, Opferman JT, Hildeman DA. 2013. Mcl-1 antagonizes Bax/Bak to promote effector CD4+ and CD8+ T-cell responses. Cell Death Differ 20:998–1007. doi: 10.1038/cdd.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ottina E, Pellegrini M, Villunger A. 2013. Guarding effector T-cell survival: all for one, Mcl-1 for all? Cell Death Differ 20:969–971. doi: 10.1038/cdd.2013.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang N, He YW. 2005. The antiapoptotic protein Bcl-xL is dispensable for the development of effector and memory T lymphocytes. J Immunol 174:6967–6973. doi: 10.4049/jimmunol.174.11.6967. [DOI] [PubMed] [Google Scholar]
- 21.Dunkle A, Dzhagalov I, Gordy C, He YW. 2013. Transfer of CD8+ T cell memory using Bcl-2 as a marker. J Immunol 190:940–947. doi: 10.4049/jimmunol.1103481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurtulus S, Tripathi P, Moreno-Fernandez ME, Sholl A, Katz JD, Grimes HL, Hildeman DA. 2011. Bcl-2 allows effector and memory CD8+ T cells to tolerate higher expression of Bim. J Immunol 186:5729–5737. doi: 10.4049/jimmunol.1100102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wojciechowski S, Tripathi P, Bourdeau T, Acero L, Grimes HL, Katz JD, Finkelman FD, Hildeman DA. 2007. Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J Exp Med 204:1665–1675. doi: 10.1084/jem.20070618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grayson JM, Zajac AJ, Altman JD, Ahmed R. 2000. Cutting edge: increased expression of Bcl-2 in antigen-specific memory CD8+ T cells. J Immunol 164:3950–3954. doi: 10.4049/jimmunol.164.8.3950. [DOI] [PubMed] [Google Scholar]
- 25.Wensveen FM, Lenartic M, Jelencic V, Lemmermann NA, ten Brinke A, Jonjic S, Polic B. 2013. NKG2D induces Mcl-1 expression and mediates survival of CD8 memory T cell precursors via phosphatidylinositol 3-kinase. J Immunol 191:1307–1315. doi: 10.4049/jimmunol.1300670. [DOI] [PubMed] [Google Scholar]
- 26.Zhou P, Levy NB, Xie H, Qian L, Lee CY, Gascoyne RD, Craig RW. 2001. MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood 97:3902–3909. doi: 10.1182/blood.V97.12.3902. [DOI] [PubMed] [Google Scholar]
- 27.Goulding J, Bogue R, Tahiliani V, Croft M, Salek-Ardakani S. 2012. CD8 T cells are essential for recovery from a respiratory vaccinia virus infection. J Immunol 189:2432–2440. doi: 10.4049/jimmunol.1200799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 29.Pierson W, Cauwe B, Policheni A, Schlenner SM, Franckaert D, Berges J, Humblet-Baron S, Schonefeldt S, Herold MJ, Hildeman D, Strasser A, Bouillet P, Lu LF, Matthys P, Freitas AA, Luther RJ, Weaver CT, Dooley J, Gray DH, Liston A. 2013. Antiapoptotic Mcl-1 is critical for the survival and niche-filling capacity of Foxp3+ regulatory T cells. Nat Immunol 14:959–965. doi: 10.1038/ni.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marriott HM, Bingle CD, Read RC, Braley KE, Kroemer G, Hellewell PG, Craig RW, Whyte MK, Dockrell DH. 2005. Dynamic changes in Mcl-1 expression regulate macrophage viability or commitment to apoptosis during bacterial clearance. J Clin Invest 115:359–368. doi: 10.1172/JCI200521766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dzhagalov I, St John A, He YW. 2007. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 109:1620–1626. doi: 10.1182/blood-2006-03-013771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunkle A, Dzhagalov I, He YW. 2011. Cytokine-dependent and cytokine-independent roles for Mcl-1: genetic evidence for multiple mechanisms by which Mcl-1 promotes survival in primary T lymphocytes. Cell Death Dis 2:e214. doi: 10.1038/cddis.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lopes AR, Kellam P, Das A, Dunn C, Kwan A, Turner J, Peppa D, Gilson RJ, Gehring A, Bertoletti A, Maini MK. 2008. Bim-mediated deletion of antigen-specific CD8 T cells in patients unable to control HBV infection. J Clin Invest 118:1835–1845. doi: 10.1172/JCI33402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wensveen FM, Klarenbeek PL, van Gisbergen KP, Pascutti MF, Derks IA, van Schaik BD, Ten Brinke A, de Vries N, Cekinovic D, Jonjic S, van Lier RA, Eldering E. 2013. Pro-apoptotic protein Noxa regulates memory T cell population size and protects against lethal immunopathology. J Immunol 190:1180–1191. doi: 10.4049/jimmunol.1202304. [DOI] [PubMed] [Google Scholar]
- 35.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, Youle RJ, Green DR, Opferman JT. 2012. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, Opferman JT, Slack RS. 2011. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J 30:395–407. doi: 10.1038/emboj.2010.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. 2012. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]