

Background: PTEN activity is enhanced by PI(4,5)P2 via either an allosteric or interfacial effect.
Results: 31P field cycling NMR with spin-labeled PTEN detects a PI(4,5)P2-binding site near but not in the active site.
Conclusion: A model for PTEN identifies Lys-13 and Arg-47 as part of the PI(4,5)P2 site.
Significance: Occupation of the PI(4,5)P2-binding site on PTEN stabilizes productive interfacial binding.
Keywords: Enzyme Kinetics, Nuclear Magnetic Resonance (NMR), Phosphatase and Tensin Homolog (PTEN), Phosphatidylinositol Phosphatase, Phosphoinositide, 31P Field Cycling, Micelles, Phospholipid-binding Site, Spin-labeled Protein
Abstract
The lipid phosphatase activity of the tumor suppressor phosphatase and tensin homolog (PTEN) is enhanced by the presence of its biological product, phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2). This enhancement is suggested to occur via the product binding to the N-terminal region of the protein. PTEN effects on short-chain phosphoinositide 31P linewidths and on the full field dependence of the spin-lattice relaxation rate (measured by high resolution field cycling 31P NMR using spin-labeled protein) are combined with enzyme kinetics with the same short-chain phospholipids to characterize where PI(4,5)P2 binds on the protein. The results are used to model a discrete site for a PI(4,5)P2 molecule close to, but distinct from, the active site of PTEN. This PI(4,5)P2 site uses Arg-47 and Lys-13 as phosphate ligands, explaining why PTEN R47G and K13E can no longer be activated by that phosphoinositide. Placing a PI(4,5)P2 near the substrate site allows for proper orientation of the enzyme on interfaces and should facilitate processive catalysis.
Introduction
The phosphatase and tensin homolog (PTEN)2 deleted on chromosome 10 is a tumor suppressor that regulates the PI3K/Akt signaling pathway. In cells where PTEN function is lost, excessive PI(3,4,5)P3 recruits and activates Akt and other downstream signaling molecules at higher levels, leading to abnormal cell growth and proliferation, which in turn causes many diseases (1). The deletion, insertion, and/or mutation of the PTEN gene are found in a wide variety of tumor cells, including endometrial cancer, glioblastoma, prostate cancer, breast cancer, and lung tumor as well as in many syndromes (1, 2). Overexpression of PTEN in tumor cells that have lost PTEN expression suppresses cell growth and tumorigenicity (3). Along with serving as a tumor suppressor, PTEN has roles in other cellular processes, including cell metabolism, cell polarity, stem cell self-renewing activity, and neuronal injury (1, 4). Most of the functions of PTEN are linked to its ability to dephosphorylate phosphoinositides.
Although PI(3,4,5)P3 is the best substrate in vitro, PTEN can also dephosphorylate PI(3,4)P2 and PI(3)P at the 3′-position of the inositol ring (5). The specific activity of PTEN toward PI(3,4,5)P3 is 50–1500-fold higher than for other substrates (depending on the assay system). This phosphatase can also dephosphorylate inositol 1,3,4,5-tetrakisphosphate, the water-soluble headgroup of PI(3,4,5)P3. However, the activity toward lipid substrates is significantly higher than that toward water-soluble inositol polyphosphates (6). PI(4,5)P2 has been shown to enhance PTEN membrane binding and activate the enzyme toward the PI(3)P and PI(3,4)P2 substrates (7, 8). An N-terminal loop containing a PI(4,5)P2 binding consensus sequence (N-terminal PI(4,5)P2 binding patch) (9) has been suggested as the putative PI(4,5)P2-binding site. However, the detailed mechanism for PI(4,5)P2 activation or the location of the bound product is not clear. It has been suggested that binding of PI(4,5)P2 induces a conformational change in PTEN, because mutation or deletion of this conserved patch eliminated PTEN membrane binding in a vesicle system and PI(4,5)P2 kinetic activation (10, 11).
The structure of a truncated form of PTEN (12) showed it is composed of the N-terminal catalytic phosphatase domain and a C-terminal lipid-binding C2 domain (Fig. 1A). The construct was missing 7 residues from the N terminus, 23 residues from a loop, and 52 residues from the C terminus. Relevant to phosphoinositide binding, residues 7–13 were not visible in the electron density map. Tartrate occupied the active site and was replaced with inositol 1,3,4,5-tetrakisphosphate in silico to have a sense of substrate binding. Molecular dynamics simulations of different dioctanoyl phosphatidylinositols bound to the catalytic domain of PTEN suggested that adjacent to the active site is another region that can play a role in monomer ligand binding (13). Those simulations suggested that there are specific interactions between the acyl chain of these lipids and the β2-α1 loop containing Arg-47. Mutants of Arg-47 showed much lower activity compared with the wild type protein regardless of the identity of the mutated amino acid, indicating the importance of this particular residue for substrate binding and/or catalysis (13). The interactions of the β2-α1 loop could facilitate the binding of several 3-deoxy-PI derivatives that inhibit enzyme activity (14). Detergent molecules, such as Triton X-100, were shown to reduce the kinetic inhibition caused by the deoxy-PI molecules and hence were proposed to bind in this hydrophobic pocket termed the “adjacent hydrophobic site” (AHS).
FIGURE 1.
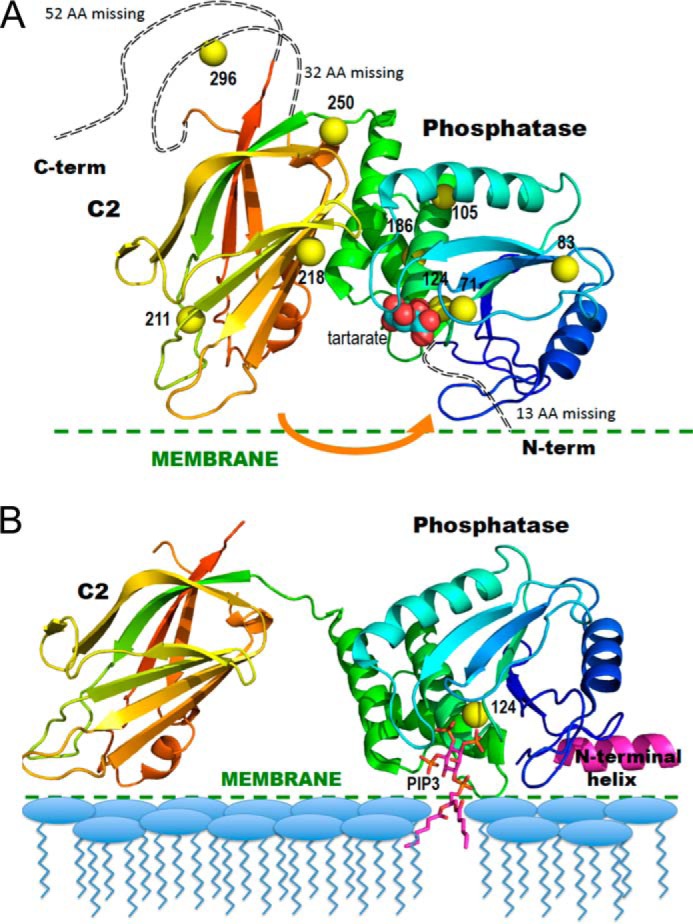
Ribbon diagrams of the crystal structure of PTEN in the absence and presence of substrate-containing membrane. A, PTEN crystal structure (Protein Data Bank code 1D5R) with colors from blue to red for N to C termini. Yellow spheres represent sulfur atoms of Cys residues that are spin-labeled in the NMR experiments. The tartrate molecule bound at the postulated active site is in van der Waals sphere representation. The arrow indicates a postulated movement of the two domains upon membrane binding. B, model for membrane-bound PTEN with the missing N-terminal peptide docked as an interfacial helix (pink). The sulfur of Cys-124 is depicted as a yellow sphere and a diC6PIP3 molecule (magenta) is docked in the active site. The postulated conformational change is based on that suggested by Kalli et al. (17).
Thus, there is a possibility that there are three functionally distinct regions in the catalytic domain of PTEN that can bind phospholipids, two quite specifically (the active site and PI(4,5)P2 site), whereas the AHS can be occupied by a number of amphiphiles. Discrete binding of the different phospholipids to PTEN could contribute to the regulation of this phosphatase. Several methods, including FRET (11), neutron reflectometry (15), and surface plasmon resonance (16), have been used to investigate PTEN or its isolated C2 domain binding to phospholipid vesicles. However, these methods are based on the overall partitioning of the protein onto the phospholipid surface. Location of an individual lipid-binding site on the protein is harder to define and usually relies on mutants that show altered kinetic or binding behavior. Coupled with these binding studies is the suggestion, based on recent MD simulations, that the protein undergoes a rotation of the phosphatase with respect to the C2 domain (17). The rotation in the membrane-bound conformation of PTEN allows it to access its substrate (Fig. 1B).
In this study, we provide evidence on how PTEN and its N-terminal point mutant (K13E) as well as an active site mutant (C124S) bind to phospholipid micelles. Both fixed high magnetic field and high resolution field cycling 31P NMR spectroscopy using spin-labeled protein (18–21), coupled with enzyme kinetics, are used to characterize binding of a series of phosphoinositides to PTEN. The results, which use short-chain phosphoinositides, identify a discrete site for a PI(4,5)P2 molecule close to, but distinct from, the active site on PTEN. A proposed structure for membrane-bound PTEN is generated by modeling the N-terminal segment as an interfacial helix (Fig. 1B) and using the constraints from the NMR results and kinetic behavior of mutant PTEN enzymes to identify this discrete binding site for a PI(4,5)P2 molecule. Its location near the active site would aid in keeping the protein at the surface as substrate diffuses in and product out of the active site. Thus, activator binding provides PTEN with an anchor for processive catalysis. We also demonstrate that K13E, which shows no kinetic activation, can still bind diC8PI in the active site (although in a slightly different orientation) with diC8PI(4,5)P2 able to bind to this protein as well. However, the NMR results are consistent with the activator molecule having a shorter residence time on the protein. These results suggest a mechanism for how kinetic activation of PI(3)P or PI(3,4)P2 by PI(4,5)P2 occurs.
EXPERIMENTAL PROCEDURES
Chemicals
The dioctanoyl phosphatidylinositol derivatives (diC8PI(3,4)P2, diC8PI(4,5)P2, and diC8PI(3)) were purchased from Echelon. The dihexanoyl phosphatidylinositol compounds (diC6PI and diC6PI(4,5)P2) were obtained from Cayman Chemicals. Long-chain phospholipids, including brain PI(4,5)P2, DOPI(3,4)P2, DOPI(4,5)P2, and POPC, as well as diC7PC were obtained from Avanti. The spin-labeling reagent, 2,2,5,5-tetramethyl-l-oxyl-3-methyl methane-thiosulfonate (MTSL), was from Toronto Research Chemicals. Other chemicals were from Sigma.
Overexpression of PTEN and Construction of PTEN Mutants
The pET28a-PTEN plasmid construction has been described previously (16). Generation of recombinant PTEN followed a previously published protocol with minor modifications (13, 14) that are summarized below. The plasmid was transformed into Escherichia coli BL21-CodonPlus® (DE3)-RIL cells from Stratagene, and the cells were grown at 37 °C until the A600 reached 0.8. Protein expression was induced with 0.1 mm isopropyl β-d-thiogalactoside, and the cultures were incubated at 16 °C for 20 h. Cells were harvested by centrifugation and stored at −20 °C until needed. His6-tagged PTEN was purified from the cell extracts using a Ni-NTA-agarose column (Qiagen). The purification process was conducted at 4 °C with 10 mm β-mercaptoethanol added to all buffers. Pure protein (>90% as judged by SDS-PAGE) was combined and dialyzed against 100 mm Tris-HCl, 1 mm DTT, pH 8.0 (a sample of the elution from the Ni-NTA affinity column is shown in Fig. 2, top); protein concentration was determined by UV absorbance as well as Lowry assay. The genes for PTEN mutants K13E, R47K, and C124S were constructed by site-specific mutagenesis using a mutagenesis kit from Stratagene. All mutations were confirmed by DNA sequencing (GeneWiz). Protein expression and purification of the mutant PTEN proteins followed the procedure for the wild type protein (13). The typical yield was 1–1.5 mg/liter culture. CD spectra of the mutant proteins showed that they all were well folded and had essentially the same wild type protein secondary structure (spectra for recombinant PTEN and R47K are shown in Fig. 2. bottom).
FIGURE 2.

Purification of recombinant PTEN and variants. Top, SDS-PAGE analysis of fractions from the Ni-NTA column; mass standards are indicated on the gel. Bottom, CD spectra for recombinant PTEN (–) and R47A (- - -) are indicated.
Enzyme Kinetics
The PTEN phosphatase assay for release of inorganic phosphate has been described previously (14). Generally, the assays were carried out at 37 °C in 50 mm Tris, pH 8.0, with 2 mm EDTA, 10 mm DTT, and substrates. In a “monomer” assay system, 0.5 mm diC8PI(3)P or 0.1 mm diC8PI(3,4)P2 was used (the critical micelle concentration (CMC) for each compound is ≥0.5 mm). The amount of protein added to each sample was chosen to obtain 5–20% substrate hydrolysis in 20 min. diC8PI(4,5)P2 and diC7PC concentrations in the assays were one-half, or equal to, that of the substrate. The Triton X-100 concentration was four times the substrate concentration. A mixed micelle system was also used with long-chain phosphoinositides DOPI(3,4)P2 and DOPI(4,5)P2 dissolved in Triton X-100 with a phospholipid to detergent ratio of 1:4. All these assays were done multiple times (and at least in duplicate) using different enzyme preparations. The specific activities presented are the mean ± S.D.
For assays using vesicles, large unilamellar vesicles (LUVs) containing 5 mm total lipids with the compositions DOPI(3,4)P2/POPC (5:95) or DOPI(3,4)P2/DOPI(4,5)P2/POPC (5:5:90) were made by mixing the designated amount of lipids in chloroform, removing the organic solvent under a stream of nitrogen, followed by lyophilization overnight, resuspending the dry film in 50 mm Tris, pH 8.0, and then extruding the suspension through a 0.1-μm polycarbonate membrane to form LUVs with an average diameter of 1000 Å. Enzyme assays used 1 mm (total phospholipid) LUVs as the substrate. These assays were conducted in duplicate, and the reported activity is the average of the two assays.
Fixed High Field 31P NMR
31P NMR spectra of short-chain phospholipids (diC8PI, various phosphorylated diC8PI molecules, and diC7PC) were obtained at 242.7 MHz on a Varian 600 MHz VNMRS system at 25 °C. The protein, in 100 mm Tris, 2 mm EDTA, pH 8.0, was added to the lipid mixture (0.25 to 1.0 mm phospholipid) to a final concentration of 0.5 mg/ml (10.1 μm). The peak width at half-height (Δν½) was measured, and the linewidth difference ΔΔν½ was calculated by subtraction of the linewidth of phospholipids alone from the linewidths of protein-bound phospholipid resonances. 31P spectra were acquired for every lipid concentration at least in duplicate and usually in triplicate using different samples and different protein preparations to obtain a mean value and standard deviation for the change in linewidth caused by addition of the protein.
Spin-labeling PTEN
Prior to labeling of PTEN cysteine groups, the protein (4 mg/ml protein solution in 100 mm Tris, pH 8.0) was incubated for 30 min with 10 mm DTT to reduce any disulfide bonds. The protein was then exchanged into 20 mm Tris, pH 7.5, and excess DTT was removed using a micro Bio-Spin 6 column (Bio-Rad). MTSL, from a 20 mg/ml stock in acetone, was added to the protein solution at a ratio of protein/MTSL = 1:4, based on the concentration of the protein prior to reduction and elution from the spin column. However, because about 50% of the protein is lost in that step, the ratio of spin-labeling reagent to protein was considerably higher (∼10:1). The incubation time, 1 h for PTEN and R47G and 1.5 h for K13E, was chosen so that greater than 80% of the PTEN activity was lost. Two spin columns removed excess MTSL and exchanged the protein back into 100 mm Tris, pH 8.0.
The remaining free cysteine residues were measured using Ellman's reagent (5,5′-dithiobis-(2-nitrobenzoic acid)), comparing the spin-labeled preparation to unlabeled PTEN. The labeling ratio was calculated by [Cys]free, PTEN-SL/[Cys]free, PTEN-WT·10 and showed that for the wild type recombinant protein 8 ± 1 cysteines were modified by the spin label.
Mass Spectrometry (MS) Detection of Modification Sites of Spin-labeled PTEN
LC-MS/MS was used to monitor the extent of MTSL modification of the 10 cysteines on PTEN. The protein, 10 μg of wild type or spin-labeled in 50 mm Tris, pH 8.5, was incubated in 8 m urea for 30 min. The sample was subsequently diluted eight times with 50 mm Tris, pH 8.5, and LysC endoproteinase (Wako) was added in a ratio of 1:50 (w/w) endoproteinase/PTEN. After overnight digestion at 37 °C, the peptide mixture was desalted on a C18 spin column (Pierce) and dried using a Speedvac (Thermo Scientific). The peptide sample was resuspended in 10 μl of buffer A (95% water, 5% acetonitrile, 0.1% formic acid). LC-MS/MS analysis was performed on an LTQ-Orbitrap Discovery mass spectrometer (Thermo Fisher) coupled to an Agilent 1200 series HPLC system. The LysC digest was pressure-loaded onto a 100-μm fused silica column (100 μm fused silica with a 5 μm tip) packed with 10 cm of Aqua C18 reverse phase resin (Phenomenex). The peptides were then eluted from the column using a 120-min gradient of 5–100% buffer B in buffer A (buffer A: 95% water, 5% acetonitrile, 0.1% formic acid; buffer B: 20% water, 80% acetonitrile, 0.1% formic acid) and injected into the mass spectrometer. The flow rate through the column was set to ∼0.25 μl/min, and the spray voltage was set to 2.75 kV. One full MS scan (Mr 400–1,800) was followed by seven data-dependent scans of the nth most intense ions with dynamic exclusion enabled. Tandem MS data were searched using the SEQUEST algorithm (22) using a concatenated target/decoy variant of the human International Protein Index database. A differential modification of +185.0874 on cysteine was specified to account for MTSL modification. SEQUEST output files were filtered with DTASelect 2.0 (23). Discriminant analyses were performed to achieve a peptide false-positive rate below 5%.
High Resolution Field Cycling 31P NMR
31P field-cycling spin-lattice relaxation rate (R1) measurements (18–21) were made at 20 °C on a Varian Unityplus 500 spectrometer using a standard 10-mm Varian probe in a custom-built device (24) that moves the sample, from the conventional sample probe location (at 11.7 T), to a higher position within, or just above, the magnet, where the magnetic field can be as low as 0.04 T; lower fields can be programmed to as low as 0.002 T with the use of a permanent magnet mounted at the top of the Dewar. Concentrations of the phosphorus-containing ligands were usually 3 mm for phosphatidylinositols and 5 mm for diC7PC; the protein concentration was usually 0.1 mg/ml (2.1 μm) for diC8PI samples and 0.25 mg/ml (5.3 μm) for other ligands. The sample buffer was 100 mm Tris, pH 8.0, with 2 mm EDTA. Controls used the same concentration of phospholipids but with 0.1 or 0.25 mg/ml unlabeled PTEN. The change in spin lattice relaxation rate due to the spin-labeled protein is ΔR1 = R1, spin label − R1, control. The data of R1 over a wide magnetic field range were fit to Equation 1,
where RP-e(0) is the low field limit of ΔR1; ωp is the angular frequency of 31P, and τP-e is the correlation time of the dipolar relaxation of 31P by nearby nitroxide unpaired electrons. The “c” term represents a limiting chemical shift anisotropy contribution due to the paramagnetic interaction and is not used in this analysis. The two parameters extracted are RP-e(0), related to proximity to the spin label, and τP-e, the correlation time for the protein/lipid interaction. As discussed previously (18, 19), if the protein contains a single spin label and the ligand binds to a discrete site on the protein, the average distance rP−e between a bound phospholipid phosphorus and the unpaired electron of the spin-labeled proteins can be extracted from both RP-e(0) and τP-e along with the concentration of the protein and phospholipid, [PTEN-SL] and [PL] as shown in Equation 2,
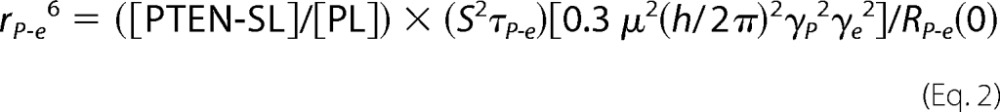 |
When more than one cysteine bears a spin label, as is the case for PTEN, the relaxation will be the sum of paramagnetic relaxation enhancement from the different label positions, and the distance calculated, rapp, reflects contributions from multiple spin labels. However, because RP-e(0) depends on rP-e−6, only very close spin labels will have a strong effect on the 31P relaxation of the ligand (20, 21). Because rapp6 is what is extracted, we will primarily use this parameter for comparisons with different samples.
For each combination of phospholipids reported, the field cycling experiments were carried out on two separately prepared samples (using a different batch of PTEN and often slightly different concentrations of protein or phospholipid). However, each plot of R1 versus magnetic field shown is for a single sample. Likewise, the entries in Table 3 are for a specific sample. In comparing two samples, the extracted r6 × 1054 values differed at most by 30%, leading to an error in rapp ± 4%.
TABLE 3.
Effect of spin-labeled PTEN on relaxation parameters of short-chain phospholipids as measured by high resolution 31P field cycling
| Ligand | [PTEN]/[PL] | τP-e | RP-e(0) | r6 × 1054 (m6) | rappa |
|---|---|---|---|---|---|
| ns | Å | ||||
| diC6PI | 0.0016 | 172 ± 21 | 0.88 ± 0.05 | 3.80 ± 0.46 | 12.5 ± 0.3 |
| diC6PI(4,5)P2 | 0.0017 | ||||
| P-1 | 37 ± 18 | 0.19 ± 0.04 | 4.0 ± 2.1 | 12.6 ± 1.1 | |
| P-4 | 35 ± 15 | 0.09 ± 0.01 | 8.0 ± 3.4 | 14.2 ± 1.1 | |
| P-5 | 32 ± 9 | 0.11 ± 0.01 | 6.0 ± 1.7 | 13.5 ± 0.6 | |
| diC8PIb | 0.0007 | 200a | 3.13 ± 0.36 | 0.54 ± 0.07 | 9.0 ± 0.2 |
| diC8PI/Triton X-100 (1:2)cc | 0.0007 | 68 ± 10 | 1.12 ± 0.07 | 0.52 ± 0.08 | 9.0 ± 0.2 |
| diC8PI(4,5)P2 | 0.0016 | ||||
| P-1 | 128 ± 26 | 1.53 ± 0.15 | 1.6 ± 0.3 | 10.8 ± 0.4 | |
| P-4 | 131 ± 18 | 0.52 ± 0.03 | 4.9 ± 0.7 | 13.0 ± 0.3 | |
| P-5 | 99 ± 22 | 0.58 ± 0.06 | 3.3 ± 0.7 | 12.2 ± 0.4 | |
| diC8PI/diC8PI(4,5)P2 | 0.0016 | ||||
| P-1 | 103 ± 20 | 1.47 ± 0.13 | 1.4 ± 0.4 | 10.5 ± 0.4 | |
| P-4 | 169 ± 21 | 0.45 ± 0.03 | 7.4 ± 0.4 | 14.0 ± 0.3 | |
| P-5 | 153 ± 19 | 0.53 ± 0.03 | 5.7 ± 0.4 | 13.4 ± 0.3 | |
| diC7PCd | 0.0011 | 120 ± 15 | 3.96 ± 0.24 | 0.40 ± 0.05 | 8.6 ± 0.2 |
| diC7PC/Triton X-100 (1:2) | 0.0011 | 118 ± 17 | 1.68 ± 0.15 | 0.94 ± 0.13 | 9.9 ± 0.2 |
| diC7PC/diC8PI(4,5)P2e | |||||
| P-1/PC | 110 ± 13 | 1.79 ± 0.11 | |||
| P-4 | 0.0018 | 93 ± 20 | 0.55 ± 0.03 | 3.7 ± 0.8 | 12.4 ± 0.4 |
| P-5 | 0.0018 | 101 ± 25 | 0.68 ± 0.08 | 3.2 ± 0.8 | 12.2 ± 0.5 |
a rapp is the 31P-e distance computed if the PR1E is due to a single closest spin label, although not a real distance because other cysteines are spin-labeled, it does serve for comparing how different ligands bind.
b The data for diC8PI required fitting with two dispersions: for the slower one, the value of τP-e is 200 ns, and the value from the low field dispersion measured for the system with unlabeled PI-PLC was used for determining RP-e(0). Error in τ was estimated as ±50 ns.
c PI concentration was 3 mm.
d The dispersion for diC7PC was fit with a single τP-e.
e The sample contained 5 mm diC7PC and 3 mm diC8PI(4,5)P2; overlap of both phosphodiester resonances and similar relaxation profiles makes ascribing rapp6 to a particular phospholipid impossible.
Modeling the N-terminal Region of PTEN and Locating the PI(4,5)P2-binding Site
The crystal structure is missing the 13-amino acid N-terminal portion of PTEN. Combined analysis of the sequence with the secondary structure prediction produced by Jpred (25) as well as SABLE programs suggests strongly that the initial 14-residue fragment, MTAIIKEIVSRNKR, is organized as an amphipathic helix. Lack of extensive contacts with the body of the phosphatase domain would create a highly mobile element suitable for interfacial membrane binding. The highly cationic character of this predicted helix would be expected to enhance membrane binding of the PTEN phosphatase domain, although it is possible that such a helix is not stable until the protein is proximal to the membrane surface. With this as a starting point, we used the program COOT (26) to create an idealized helix that had several hydrophobic residues on one side of the helix and several charged residues on the opposite site. The connectivity requirement (residue Arg-14 is observed in the crystal structure), shape, and charge compatibility of this helix with the rest of the phosphatase domain allows for a unique docking of the helix with the hydrophobic face directed toward the membrane (Fig. 1B). Such a model, although speculative, leads to the partial electrostatic neutralization of the acidic residues in the loop adjacent to the proposed N-terminal helix and also extends Lys-13 toward the active site of the enzyme. The new surface of the phosphatase domain created by docking the missing N-terminal peptide as an amphipathic helix contains two distinct indentations that are potential candidates for the binding sites of headgroups of phosphorylated inositol species. Structures with diC6PI(3,4,5)P3 and diC6PI(4,5)P2 molecules docked to these sites in the PTEN model were evaluated in PyMOL (27).
RESULTS
PI(4,5)P2 Effects on PTEN Enzymatic Activity
Monomer and micellar ligands are amenable to NMR analyses and can be used to provide information of how and where a PI(4,5)P2 molecule will bind on PTEN with respect to a substrate analog in the active site. However, for the biophysical results to be convincing, kinetic characterization of PTEN with these short-chain phosphoinositides is needed.
The effect of the PI(4,5)P2 on PTEN specific activity was assessed using both monomer and vesicle substrate systems. First, as expected, with 0.1 mm diC8PI(3,4)P2 as the substrate, 0.05 mm diC8PI(4,5)P2 increased recombinant PTEN activity 6-fold (Fig. 3A), comparable with what has been reported previously (8).
FIGURE 3.

Effect of PI(4,5)P2 and other amphiphiles on PTEN hydrolysis of different substrates. A, relative specific activities for PTEN toward 0.1 mm diC8PI(3,4)P2, DOPI(3,4)P2/POPC (0.05 mm/0.95 mm) LUVs, DOPI(3,4)P2/POPC (0.1 mm/0.9 mm) LUVs, and 0.5 mm diC8PI(3)P with PI(4,5)P2 are shown, with substrate/PI(4,5)P2 = 0.5 (■) or 1.0 (\\\\). B, effect of varying the chain length of substrate and activator. Substrates include 0.1 mm diC6PI(3,4)P2 or 0.1 mm diC8PI(3,4)P2 with dihexanoyl- or dioctanoyl-PI(4,5)P2 added, and substrate/PI(4,5)P2 = 0.5 (■) or 1.0 (\\\\). C, effect of PIP2 and Triton X-100 on PTEN hydrolysis of different substrates. Substrates include diC8PI(3,4)P2/Triton X-100 (0.1 to 0.4 mm), DOPI(3,4)P2/Triton X-100 (0.1 to 0.4 mm), and diC8PI(3)P/Triton X-100 (0.5 to 2 mm). The concentration of PI(4,5)P2 (either the dioctanoyl compound or DOPI(4,5)P2 depending on the substrate) was half (■) or the same (\\\\) as that of the substrate. The dashed lines in A–C would represent an activity with the PI(4,5)P2 equivalent to that in its absence. D, diC7PC inhibition of PTEN activity toward 0.5 mm diC8PI(3)P (■, □) or 0.1 mm diC8PI(3,4)P2 (●, ○) in the absence (filled symbols) or presence (open symbols) of Triton X-100 equivalent to four times the substrate concentration. Error bars here (and in other plots of enzyme activities) represent standard deviations from the repeats for each experimental condition.
With diC8PI(3)P as a substrate, the enzyme displays somewhat different behavior (Fig. 3A). The CMC of diC8PI(3)P was reported as 0.7 ± 0.2 mm (14), which means under assay conditions without protein, both substrate and activator would be mostly monomers and perhaps with some micelles. With 0.5 mm diC8PI(3)P as the substrate, 0.25 mm diC8PI(4,5)P2 reduced PTEN activity to 70%, and 0.5 mm diC8PI(4,5)P2 reduced activity to 40% of the rate in the absence of the “activating” lipid. Here, diC8PI(4,5)P2 is acting as an inhibitor. This could occur directly, by better binding of this PI(4,5)P2 molecule in the active site than substrate PI(3)P, or indirectly. For the latter, PI(4,5)P2 binding to an adjacent site may alter the local electrostatics so that a less anionic substrate like PI(3)P has reduced affinity for the active site.
The dioctanoyl lipids have CMCs around 0.5–1 mm, and PTEN could lower the CMC, differently for each lipid. To try and circumvent this potential complication, we also examined the effect of diC6PI(4,5)P2 on PTEN hydrolysis of diC6PI(3,4)P2 and diC8PI(3,4)P2. The specific activity was higher for the diC8PI(3,4)P2, (45 ± 4 nmol/mg/min) than for diC6PI(3,4)P2 (10 nmol/mg/min), possibly indicating that some protein-induced substrate aggregation occurs for systems poised around the CMC.
Acyl-chain length also modulates the magnitude of PI(4,5)P2 activation (Fig. 3B). The shorter molecule, diC6PI(4,5)P2, was less effective as an activator than diC8PI(4,5)P2 for both substrates. The effect of inositol 1,4,5-trisphophate, I(1,4,5)P3, the headgroup of PI(4,5)P2, was also examined for its effect on PTEN activity. No significant kinetic activation (or inhibition) was seen for either diC8PI(3,4)P2 or diC8PI(3)P as substrate in these assays.
Triton X-100 is a nonionic detergent that we have previously shown to be able to reduce the inhibition potency of short-chain deoxy-PI derivatives by binding to a hydrophobic site adjacent to the active site (AHS) and/or forming micelles that alter the on/off rates of the PI derivative in the active site (13). The presence of the nonionic detergent in the diC8PI(3)P/diC8PI(4,5)P2 assay system had little effect on the PIP2 activation of PTEN (Fig. 3C). However, the lack of activation of PTEN toward DOPI(3,4)P2 by DOPI(4,5)P2 in Triton X-100 mixed micelles (as well as the enhanced inhibition in diC8PI(3)P/diC8PI(4,5)P2 assays) could suggest that occupation of the AHS can alter both substrate and activator binding.
PC is often used as the matrix lipid in preparing vesicles. However, in monomer/micelle assay systems, short-chain PC is an inhibitor of PTEN (Fig. 3D). Comparable concentrations of Triton X-100 have little effect on PTEN enzymatic activity (13). For both diC8PI(3)P or diC8PI(3,4)P2 as the substrate, the addition of diC7PC below its CMC of 1.5 mm significantly reduced the enzyme activity, although somewhat less so for the better substrate. Adding the nonionic detergent Triton X-100 into the mixture reduced the extent of the diC7PC inhibition of PTEN. These results indicate that a short-chain PC molecule may interact with the protein AHS and in so doing alter access to the nearby active site (13). Triton X-100, in contrast, appears to bind almost exclusively to the AHS with little effect on substrate access to the active site, although it can preclude strong activation by PI(4,5)P2.
Lys-13 has been suggested as a key residue for PI(4,5)P2 activation in PTEN (10, 11). Conversion of this cationic side chain to an anionic one should alter electrostatic interactions of protein and ligand. The K13E enzyme was less active than wild type PTEN toward diC8PI(3)P or diC8PI(3,4)P monomers (less than 10% of wild type PTEN activity) and toward DOPI(3,4)P2 in PC vesicles (25% of the activity of wild type PTEN). K13E could not be activated either by diC8PI(4,5)P2 or long-chain DOPI(4,5)P2 (Fig. 4). The ability of PI(4,5)P2 to activate PTEN has been lost.
FIGURE 4.

Effect of PI(4,5)P2 on the activity of recombinant PTEN and variants K13E, R47K, and R47G in monomer and vesicle assay systems. The ratio of the specific activity with PI(4,5)P2 added to the activity in its absence is shown for WT and the three mutant proteins. In the monomer substrate mixture diC8PI(3,4)P2 is 0.1 mm with either 0.05 mm (■) or 0.1 mm (///) diC8PI(4,5)P2. Vesicles (▩) were composed of DOPI(3,4)P2/POPC (0.05 to 0.95 mm) with 0.05 mm DOPI(4,5)P2. The specific activities of wild type PTEN toward each substrate in the absence of PIP2 are 45 ± 4 nmol/mg min for diC8PI(3,4)P2 and 0.59 ± 0.02 nmol/mg min for DOPI(3,4)P2/POPC LUVs.
Arg-47 was previously shown to be a key residue of the AHS (13). Substituting Arg with Gly, Trp, Leu, or Lys reduced PTEN activity in all assay systems examined (Table 1), although the effect was most pronounced for monomer and micelle systems. The relative activity compared with recombinant PTEN was only decreased 3–4-fold for LUV assay systems, likely because vesicle binding is weak even with wild type enzyme (11). The loss of activity is consistent with this key residue of the AHS affecting the interaction of substrate with the active site. The much higher relative activity for R47K with diC8PI(3,4)P2 as substrate compared with diC8PI(3)P might suggest that it is substrate binding that is affected with this mutation. Keeping this residue cationic is also preferable to placing a small and flexible residue at that position (R47G).
TABLE 1.
Relative activities of Arg-47 mutants compared to wild type PTEN acting on different PI(3)P substrate systems
The specific activities in nanomoles/mg min of wild type PTEN toward each substrate, determined at room temperature, are 14.2 ± 0.4 (diC8PI(3)P), 45 ± 4 (diC8PI(3,4)P2), 11.5 ± 0.7 (diC16PI(3)P/Triton X-100), 0.27 ± 0.03 (diC16PI(3)P/POPC), and 0.59 ± 0.02 (DOPI(3,4)P2/POPC). Mutants activities were measured in duplicate with errors <20% of the tabulated value.
| diC8PI(3)P 0.5 mm | diC8PI(3,4)P2 0.1 mm | diC16PI(3)P/ Triton X-100 (1:4)a | diC16PI(3)P/ POPC (1:9)b | DOPI(3,4)P2/POPC (1:19)c | |
|---|---|---|---|---|---|
| R47G | 0.040 | 0.019 | 0.049 | 0.063 | 0.23 |
| R47W | 0.010 | –d | 0.015 | 0.052 | – |
| R47L | 0.014 | – | 0.041 | 0.24 | – |
| R47K | 0.006 | 0.12 | 0.018 | 0.28 | 0.29 |
a The concentration of diC16PI(3)P was 0.1 mm.
b The total phospholipid concentration was 1 mm.
c The DOPI(3,4)P2 concentration was 0.1 mm.
d – indicates not determined.
Mutation of Arg-47 also alters activation by PI(4,5)P2. For R47G, there was virtually no activation by PI(4,5)P2 (Fig. 4). When the Arg was replaced by a Lys, the relative increase in activity provided by PI(4,5)P2 was comparable with wild type protein in the monomer assay systems but was significantly lower with vesicles. This kinetic result could suggest that the PI(4,5)P2 activator site is near the AHS, and the positive charge on Arg-47 is important for PI(4,5)P2 binding and subsequent activation of PTEN.
All these kinetics are consistent with three distinct but perhaps partially overlapping lipid-binding sites as follows: the catalytic site, the PIP2-activating site (likely involving Lys-13 and Arg-47), and the adjacent hydrophobic site (which also appears to involve Arg-47 (13)). However, are these “sites” truly discrete?
Fixed Field 31P NMR Study of Phospholipids Binding to Recombinant PTEN and Variants
Because the different phospholipids used here have multiple phosphorus atoms and also different chemical shifts, 31P NMR would seem an obvious choice to explore binding of the different short-chain phospholipids to PTEN. Initially, changes in the 31P linewidths (ΔΔν½) of ligands induced by the presence of the protein were used for qualitative characterization of lipid binding to PTEN at 14.02 T. Concentrations of the phosphoinositol compounds were below or close to their CMC in the absence of protein, although the protein was 10 μm. The dihexanoylphospholipids exhibited minimal changes in linewidth (<2.5 Hz). In contrast, the dioctanoylphosphoinositides showed significantly larger linewidths in the presence of PTEN. The d-diC8PI and l-3,5-dideoxy-diC8PI, which bind to the active site (13, 14), exhibited a 25–30 Hz increase in linewidth under the experimental conditions used. diC7PC also exhibited a large increase in linewidth (28 ± 4 Hz for 1 mm of the phospholipid), consistent with occupation of the AHS and part of the active site as suggested by the kinetics. In contrast to the active site ligands, diC8PI(4,5)P2 showed much smaller line broadening in the presence of PTEN (∼5 Hz increase with 0.25 mm lipid).
Interpreting these linewidth changes is difficult because 31P at this field strength has contributions from chemical shift anisotropy as well as dipolar interactions that are also coupled with exchange of molecules into a protein-binding site, and perhaps formation of small micelle aggregates with protein. When Triton X-100 was added to the sample with 0.5 mm diC8PI, the change in linewidth due to PTEN was significantly reduced from 25 ± 1 to 12 ± 3 Hz. Because that nonionic detergent is not an inhibitor (14), this suggests that either the diC8PI-PTEN complex may be large or perhaps that the exchange of ligand on and off the protein is in an intermediate regime and that Triton X-100, by virtue of providing a micelle reservoir, may accelerate such exchange. Adding Triton X-100 to diC7PC samples with PTEN reduced line broadening from 30 to 3 Hz. The larger decrease in linewidth (compared with what is observed for Triton X-100 added to diC8PI with PTEN) could be explained by competitive binding between diC7PC and Triton X-100 in the AHS.
PTEN was also studied with both diC8PI(4,5)P2 and l-3,5-dideoxy-diC8PI in the same sample. The phosphodiester peaks of these two lipids have slightly different chemical shifts, and therefore linewidth changes that can be measured separately. A large increase in linewidth was detected for the 0.25 mm deoxy-PI bound in the active site (31 ± 1 Hz) with 5.7 ± 0.2 Hz for 0.25 mm diC8PI(4,5)P2. The values of ΔΔν½ are comparable with what is observed for either separately.
The ΔΔν½ of 31P resonances in the presence of K13E and Arg-47 mutants was also measured. diC8PI in the presence of these three mutant enzymes still showed a large increase in linewidth (30–40 Hz) with a much smaller linewidth increase (6 Hz) for diC8PI(4,5)P2. diC7PC was also broadened in a similar fashion to diC8PI for the R47G or R47K mutants (increases of 42 ± 7, 37 ± 4, and 33 ± 5 Hz, for K13E, R47K and R47G, respectively), and when Triton X-100 (2 mm) was added, the increase in diC8PI linewidth caused by PTEN was reduced (to 6.3 ± 0.5, 11.2 ± 3.1, and 8.8 ± 0.9 Hz for K13E, R47K, and R47G), as observed for wild type PTEN (reduction to 11.5 ± 2.8 Hz). diC8PI(4,5)P2 (0.25 mm) linewidths in the presence of K13E, R47K, or R47G were also comparable with the effect of wild type PTEN with linewidth increases of 6.1 ± 0.4, 6.9 ± 0.7, and 6.6 ± 0.9 Hz for K13E, R47K, and R47G, again suggesting this lipid still binds to the protein. At least for the short-chain phospholipid assay system, the loss of activity and PI(4,5)P2 activation for these mutant enzymes does not appear to be caused by a lack of PI(4,5)P2 interacting with PTEN. The difficulty is to disentangle somewhat nonspecific interactions of the activating phospholipid with occupation of a discrete site on PTEN that is different from the active site.
Although these fixed field 31P linewidths provide a fingerprint for phospholipid molecules in the active site (large line-width change with PTEN added), adjacent hydrophobic site (large change in linewidth that is reduced to less than a few Hz by the addition of Triton X-100), and activator site (small linewidth change unaffected by adding molecules that occupy the active site), there is no information on where such sites might be and whether they are indeed spatially distinct.
Spin-labeled PTEN and Measuring the PR1E by Field Cycling
Modifying a protein to introduce a specific spin label, followed by NMR experiments, is useful to estimate proximity of ligands to discrete sites. The large electron dipole can dominate dipolar relaxation of nearby nuclei (in this case 31P of different amphiphiles) leading to an increase in the relaxation rate (paramagnetic relaxation enhancement (PRE)). A large number of reports of line-broadening effects of this kind have been reviewed recently (28). We use paramagnetic effects on NMR kinetic rates between populations in different spin levels (denoted as R1). Effects of spin labels on 31P nuclear spins at high field are almost negligible. This hybrid method, named “PR1E,” is particularly useful in characterizing the location of specific phospholipid-binding sites on proteins (19, 20).
Both the more conventional PRE and the PR1E are proportional to the inverse sixth power of the distance, but the 31P R1 effects that are seen at very low fields (by high resolution field cycling) are completely clear of interference by relaxation from chemical shift anisotropy. Therefore, much lower concentrations of spin-labeled PTEN are required. The method requires that the 31P species be in moderately fast exchange between free- and enzyme-bound species. The inverse sixth power behavior provides a reasonable degree of specificity as well as a molecular yardstick. Fig. 5 shows how RP-e(0) will vary with distance of the 31P from the spin label, rP-e, at two different ratios of protein to phospholipid. Also shown is how the overall micelle-protein complex correlation time describing that interaction, τP-e, affects RP-e(0). The longer the correlation time, the larger the parameter RP-e(0). The r−6 dependence of the dipolar interaction means that spin labels more than 15 Å away from discrete phospholipid-binding sites will not contribute significantly to R1 at the ratios of protein to amphiphile shown in Fig. 4 (these are the same ratios used for PTEN and different ligands). Furthermore, the response can be tuned by adjusting the amount of protein added to a fixed amount of amphiphile. Although two binding sites may be close, for example one 10 Å and the other 15 Å away from the spin label, the PR1E will be much larger for the 31P of the amphiphile in the closer site.
FIGURE 5.

Dependence of RP-e(0) on the distance of a 31P nucleus from an unpaired electron and the τP-e correlation time predicted for two different ratios of protein to the 31P-containing amphiphile. Different τP-e values are 25 (– – –), 50 (—), 100 (- - -), 150 (···), and 200 (— — —) ns.
PTEN might not seem an easy target for the PR1E method because it has 10 cysteines, 5 on the phosphatase domain, and 5 on the C2 domain. Under the spin-labeling conditions (which used loss of enzyme activity to ensure modification of Cys-124), 9 of the 10 cysteines were modified by the spin-label reagent as monitored by MS, although not all completely (Table 2). The distribution of modified peptides also agreed well with the overall cysteine labeling determined with Ellman's reagent. Previous work (29) has shown that membrane binding is uncoupled from enzyme activity, so the use of an inactive enzyme should still provide information on ligand binding.
TABLE 2.
MS identification of PTEN cysteine modified by the 2,2,5,5-tetramethyl-l-oxyl-3-methyl methane-thiosulfonate spin label
The peptides in the tryptic digest containing spin-labeled Cys (C*) and unmodified Cys (C) are indicated. The number of the peptides detected in the sample is also shown.
| Peptide | Cys no. | Digested peptides | |
|---|---|---|---|
| IYNLCAERHYDTAK | Cys-71 | 1 | |
| IYNLC*AERHYDTAK | 1 | ||
| FNCRVAQYPFEDHNPPQLELIK | Cys-83 | 1 | |
| FNC*RVAQYPFEDHNPPQLELIK | 4 | ||
| PFC*EDLDQWLSEDDNHVAAIHC* | Cys-105 | Cys-124 | 1 |
| PFC*EDLDQWLSEDDNHVAAIHC*K | 7 | ||
| MMFETIPMFSGGTCNPQFVVCQLK | Cys-211 | Cys-218 | 2 |
| MMFETIPMFSGGTC*NPQFVVC*QLK | 6 | ||
| FMYFEFPQPLPVCGDIK | Cys-250 | 1 | |
| FMYFEFPQPLPVC*GDIK | 10 | ||
| VENGSLCDQEIDSICSIERADNDK | Cys-296 | Cys-304 | 3 |
| VENGSLC*DQEIDSIC*SIERADNDK | 3 | ||
The N-terminal peptide of PTEN has been suggested to contribute to PI(4,5)P2 binding, which localizes it near the phosphatase domain. In the PTEN crystal structure, the distances of the C2 domain cysteine sulfur atoms to tartrate carbons are >20 Å. Therefore, spin labels at those sites should not contribute much to the relaxation of 31P nuclei of the bound PI phospholipids. Spin labels attached to Cys-83 and Cys-105, located in the phosphatase domain, are far from the suggested membrane binding interface (12) and protein active site and should be only minor contributors to 31P dipolar relaxation. The distances of the Cys-83 and Cys-105 sulfur atoms to C(1)/C(4) of tartrate are 23/20 and 18/19 Å, respectively. Thus, the major contributors to relaxation of the phospholipids that dock into the active site should be spin labels on Cys-71 and Cys-124. The distances of the Cys-71 and Cys-124 sulfur atoms to C(1)/C(4) of tartrate are 10/6.7 and 6.0/3.6 Å, respectively. If the site for PI(4,5)P2 is near the active site, it is likely to be relaxed by these spin labels; if it is further away (but still around the face that is thought to interact with the membrane interface) then relaxation by spin-labeled PTEN will be smaller than for the PI bound in the active site.
The NMR-detectable effect of spin-labeled PTEN on different short-chain phosphoinositides was first explored at 242.7 MHz to ensure that ligands still bound in the same manner. Although the contribution of dipolar relaxation to relaxation of 31P at high fields is small (30), the large dipole of the unpaired electron on the spin label should enhance transverse relaxation of the 31P nuclei in a distance-dependent manner. At a ratio of diC8PI to spin-labeled PTEN of 25:1, there was an additional 7–10 Hz increase in the linewidth over the increase produced by unlabeled PTEN; for diC8PI(4,5)P2, the linewidth change due to the spin labels on the protein was not significant under these conditions. The lack of an additional increase in observed linewidth upon addition of the spin-labeled PTEN suggests the following: (i) it is not in the active site, and (ii) it is further away from spin labels attached to the protein. Therefore, although the protein is heavily spin-labeled, it has not lost the capacity to bind either active site ligands or diC8PI(4,5)P2.
PTEN Effects on Monomeric Phospholipids and I(1,4,5)P3
diC6PI has a CMC around 10–14 mm (31); adding phosphates to the inositol ring will increase the CMC so that at the concentrations used in the field cycling experiment (3 mm), the lipids are monomeric in the absence of protein. In the field cycling NMR experiments, the ratio of phospholipid to PTEN was always greater than 500:1, providing an excess of phospholipid so that observed high field phospholipid linewidths were not dramatically affected. As expected, unlabeled PTEN had no effect on the 31P relaxation behavior of either compound at low fields at these ratios of protein to phospholipid. The profile for diC6PI interacting with PTEN is shown in Fig. 6A with the field dependence of the 31P PR1E (ΔR1) shown in the inset. Clearly, there is a significant enhancement of the diC6PI 31P R1 by the presence of spin-labeled PTEN at fields below 1 T.
FIGURE 6.

Field dependence of the PR1E for diC6PI and diC6PI(4,5)P2. A, R1 for the 31P of diC6PI (3 mm) caused by 4.9 μm spin-labeled PTEN; the inset shows the ΔR1 of this phosphodiester from 0.004 to 4 T after subtraction of the profile obtained using nonlabeled PTEN. B, R1 for the 31P resonances of diC6PI(4,5)P2 (3 mm) in the presence of 5.1 μm spin-labeled PTEN: P-1 (●), P-4 (▵), and P-5 (▴). The inset in B shows the R1 profile for 3 mm I(1,4,5)3 in the presence of 4.9 μm spin-labeled PTEN: P-4 (●), P-1 phosphate (○), and P-5 phosphate (□). Note that none of these 31P resonances exhibit the low field rise in R1 indicative of a small molecule-protein complex. Error bars for R1 values (here and in subsequent plots) were obtained from a least squares fit of the ln(intensity) versus time determining R1 at that particular field.
There are several interesting features of the protein-diC6PI complex PR1E data (Table 3). First, τP-e is 172 ± 21 ns, a time significantly larger than expected for rotational diffusion of a single PTEN molecule complexed to a single phospholipid. This indicates that the presence of the diC6PI ligand in the active site causes protein/phospholipid aggregates to form, even though by itself the diC6PI is monomeric at this concentration. Second, the parameter rapp6 is about 4.0 × 10−54 m6, suggesting moderate proximity of the lipid to spin labels on the protein. A second sample of spin-labeled PTEN and slightly different PTEN and lipid concentrations led to a value of 4.1 × 10−54 m6 for rapp6, indicating the reproducibility of the experiments. If a single spin label were responsible for this PR1E, rP-e would be 12.5 Å. However, Cys-71 and Cys-124, both fairly well labeled, should be close enough to generate a larger PR1E for diC6PI. The modest effect on relaxation strongly suggests that the active site of the PTEN is not saturated by 3 mm diC6PI.
For diC6PI(4,5)P2, the magnitude of the relaxation enhancement from the spin-labeled PTEN, RP-e(0), was considerably smaller than for diC6PI (Fig. 6B and Table 3). However, τP-e was also noticeably shorter, 35 versus 170 ns for diC6PI. It appears from our data that binding of diC6PI causes large protein/lipid aggregates to form, whereas the phosphorylated activator molecule has a much smaller effect.
I(1,4,5)P3 is a soluble molecule that mimics the PI(4,5)P2 headgroup and might be expected to bind into the active site or perhaps a discrete PI(4,5)P2 site on the protein. However, there was no low field rise in R1 indicative of that small molecule (3 mm) binding to the spin-labeled protein (4.9 μm) and similar to what was seen with diC6PI(4,5)P2 (Fig. 6B, inset). This indicates that inositol 1,4,5-trisphosphate (I(1,4,5)P3) binds poorly to the spin-labeled protein, consistent with the observation that IP3 had virtually no inhibitory or activating effect on enzymatic activity. It appears that acyl or alkyl chains are needed for the ligands to bind well to PTEN (at least at low millimolar concentrations of the inositol compound used here).
PTEN-induced PR1Es, Micellar Phosphoinositides
The effect of spin-labeled PTEN on micellar diC8PI and diC8PI(4,5)P2 was much larger than for the dihexanoyl chain compounds. The field cycling profile for diC8PI mixed with unlabeled PTEN (Fig. 7A, open circles) showed a low field component significantly greater than for diC8PI micelles alone (14). This indicated that PTEN interacts with the longer chain PI and forms larger micelles than the diC8PI by itself. Analysis of this curve yielded two distinct dispersions with different correlations times as follows: 5 ns (postulated to represent fast motions, e.g. phospholipid rotation or wobbling (30, 32)) and 190 ns (representing diffusive rotation of the entire protein/phospholipid aggregate (33)). The field dependence of the PR1E, obtained by subtracting R1 for diC8PI mixed with unlabeled protein from the rates obtained with spin-labeled protein, is shown in Fig. 7B. These data were also not well fit with a single correlation time. To extract parameters for the low field dispersion, a 200 ± 50 ns correlation time, comparable with the value seen for the same phospholipid interacting with the unlabeled PTEN sample, was used and a value of 5 ns for the faster dispersion (comparable with the value seen for unlabeled protein mixed with the micelles). As shown in Table 3, the value of rapp6 was 0.52 × 10−54 m6, a value significantly smaller than for diC6PI. Analyzing the diC8PI dependence of ΔR1 on field with a single dispersion yielded rapp6 = 0.49 × 10−54 m6, indicating that the rapp6 value is fairly robust with large ΔR1 at low fields. Clearly, the 31P atom of diC8PI bound to PTEN is close to the spin labels.
FIGURE 7.

Field dependence of the PR1E for diC8PI and diC8PI(4,5)P2. A, R1 for the 31P of diC8PI (3.0 mm) in the presence of 2.0 μm unlabeled (○) or spin-labeled (●) PTEN is shown as a function of magnetic field. B, ΔR1 representing the difference due to the spin-labeled PTEN is shown; the line represents a fit with two dispersions with τP-e values of 5 and 200 ns. C, ΔR1 for diC8PI(4,5)P2 (3.0 mm) in the presence of 4.9 μm spin-labeled PTEN (solid symbols): P-1 (●), P-4 (▴), and P-5 (■).
The dispersion with a 5-ns correlation time can also yield information about amphiphile binding. The τP-e and RP-e(0) extracted generated a value of rapp6 as 0.06 × 10−54 m6. On this short time scale, there are many interactions of the spin labels on the protein with PI molecules in the micelle; the bulk of these reflect nonspecific interactions (i.e. not occupation of a discrete binding site) because they do not persist for the length of the correlation time of the entire complex (∼200 ns). By comparison, the large PR1E for diC8PI at the lower fields means a phospholipid must occupy a discrete binding site on the protein for at least 200 ns.
The effect of spin-labeled PTEN on 3 mm of the activator molecule diC8PI(4,5)P2 was also examined (Fig. 7C). For these micelles, more spin-labeled protein was needed to get effective relaxation of the 31P nuclei. Unlike what was observed for diC8PI, there was no measurable effect of unlabeled PTEN on the diC8PI(4,5)P2 relaxation rates at fields below 1 T. This is expected because small micelles (14) tumble more rapidly making τP shorter so that the whole relaxation dispersion is pulled out to higher fields. The excess relaxation caused by the spin-labeled protein was fit with a single exponential. The average correlation time from the three 31P resonances for the phospholipid-protein complex, τP-e, was 119 ± 17 ns. The values of rapp6 for the three phosphorus resonances follow the same pattern seen for diC6PI(4,5)P2 (Table 3); rapp6 is smallest for P-1 with P-4 and P-5 significantly larger. A key point is that P-1 has the largest PR1E, meaning it is closest to the spin labels on the protein.
The effect of the spin-labeled PTEN is much easier to detect and quantify for diC8PI(4,5)P2 because the micelles interacting with the protein form a larger complex with a longer τP-e. The higher value of rapp6 for the phosphodiester of diC8PI(4,5)P2 compared with diC8PI suggests it is occupying a different site on the protein and is further away from spin-label sites that efficiently relax diC8PI in the active site.
The similarity between the observed rates (values of RP-e(0) in Table 2), particularly for the two phosphomonoester 31P spins, suggested to us the possibility of diffusion of spin magnetization (usually called “spin diffusion” in the NMR literature) that could complicate the analysis. The rate for this transfer between two 31P spins separated, for example, by 2.5 Å, with tumbling correlation times of 100 ns, is about 40 s−1 (34). This is fast compared with the observed RP-e(0) rates of 1.5 s−1, at most, for the phosphodiester 31P of diC8PI(4,5)P2 samples.
In this case, the distances from the 31P nuclei and the spin labels for the monoesters are equal within 10%, and the two 31P nuclei would be expected to relax at nearly the same rate with or without spin diffusion, so therefore it can be ignored. In cases where the distance from the spin label to the nearest 31P is less than 80% as large as the distance from the spin label to the most distant 31P, spin diffusion could distort the rates and confuse the model building process. However, this is not the case here for the phosphodiester. Therefore, the distances we extract should not be severely distorted by spin diffusion.
Enzyme kinetics with short-chain phospholipid monomers below the CMC strongly showed that diC8PI(4,5)P2 at moderate concentrations can act as an inhibitor. If the inhibition arises from that PTEN product binding to the active site, then some of the relaxation of PI(4,5)P2 31P resonances could be caused by its binding into the active site. To assess this, we examined a sample of diC8PI, diC8PI(4,5)P2, and spin-labeled PTEN. If the PI(4,5)P2 relaxation is primarily caused by binding to the active site, then the presence of the diC8PI should reduce R1 substantially. We know from measuring linewidths at high field that in mixtures of the two phosphoinositides the linewidth increases were comparable with what was observed for either alone. In the field cycling experiment, the diC8PI resonance relaxed too quickly at low fields to obtain τP-e and RP-e(0). However, the field dependence profiles of the two phosphomonoesters with this enzyme preparation were basically the same in the absence and presence of diC8PI, indicating that PI(4,5)P2 is binding to a site spatially distinct from the active site but near the labeled cysteine residues of PTEN.
Localizing the diC8PI(4,5)P2 Site with C124S
Spin labels on Cys-71 and Cys-124 are the major contributors to relaxing diC8PI. However, they or other spin labels may contribute to the PR1E for PI(4,5)P2. One approach to obtaining a value of rP-e from a particular spin label is to remove one of the active site spin labels. To this end, we spin-labeled the variant C124S. When modified, this variant will still have a nitroxide on Cys-71 near the active site.
For diC8PI binding to spin-labeled C124S, the profile shows a 50% decrease in the PR1E (Fig. 8). The diC8PI profiles have two components, and the low field component was used for comparing effects of spin-labeled wild type and C124S proteins. That difference in RP-e(0), 1.7 s−1 (adjusting for the slightly higher concentration of C124S), represents the contribution of spin-labeled Cys-124 to the relaxation of the diC8PI. At the concentrations of PTEN (∼0.71 μm) and diC8PI (3 mm) used, the value for the active site-bound diC8PI phosphodiester 31P distance to the spin label attached to Cys-124 is 10.0 Å.
FIGURE 8.

Field dependence of the 31P PR1E for diC8PI and diC8PI(4,5)P2 in the presence of spin-labeled PTEN C124S. A, effect of 4.3 μm spin-labeled C124S on 3 mm diC8PI(4,5)P2: P-1 (●); P-4 (□); (▴) P-5. B, comparison of PRE for diC8PI (3 mm) produced by spin-labeled 2.0 μm native PTEN (○) or 2.2 μm spin-labeled variant C124S (●); the field dependence was fit with τP-e = 200 ns, and the arrow approximates the amount of R1 contributed by a spin label on Cys-124.
The RP-e(0) values for the three phosphorus nuclei in diC8PI(4,5)P2 binding to spin-labeled C124S are also reduced compared with spin-labeled wild type protein. The ΔRP-e(0) contributions from a spin label on Cys-124 (subtracting the value obtained for C124S from the wild type PTEN value of RP-e(0) adjusted to the same protein concentration) were 0.39, 0.24, and 0.35 s−1 for P-1, P-4, and P-5. From the fitted correlation time of 160 ns and these RP-e(0) values, the distances between each 31P and the unpaired electron of the nitroxide on Cys-124 are estimated as 13.8, 15.0, and 14.1 Å for P-1, P-4, and P-5 of diC8PI(4,5)P2. For the phosphomonoesters, RP-e(0) is fairly small, and differences in τP-e (which is less well defined) could also add some uncertainty in the ΔRP-e(0) value needed to estimate rP-e for PI(4,5)P2 with respect to the spin label on Cys-124. However, the ΔRP-e(0) values are unlikely to be off by as much as a factor of 2, which would translate to ≤10% error in rP-e. Thus, the rP-e values extracted for diC8PI(4,5)P2 indicate that the activator site is close to the active site.
Occupation of the AHS
Our kinetics with the synthetic short-chain PIs showed that diC7PC is inhibitory, whereas Triton X-100 has little effect on the observed hydrolysis rate by itself but is able to counteract the inhibition by diC7PC. With this in mind, we explored the effect of spin-labeled PTEN on zwitterionic diC7PC micelles in the absence and presence of Triton X-100 and diC8PI(4,5)P2. For 5 mm diC7PC micelles interacting with 5.3 μm spin-labeled PTEN, there was a large effect of the protein on the 31P resonance; rapp6 was 0.40 × 10−54 m6 for this sample (Table 3), a value comparable with the value for diC8PI and consistent with the diC7PC phosphocholine moiety binding in the PTEN active site, as suggested by the kinetics.
diC7PC also interacts with the AHS because inclusion of Triton X-100 in the micelles dramatically altered its relaxation by the spin-labeled protein. When 5 mm diC7PC was mixed with 10 mm Triton X-100, the effectiveness of the spin-labeled PTEN in relaxing the diC7PC was reduced about 2-fold (Table 3). The kinetics, carried out with substrate concentration below its CMC, suggest that a 4-fold excess of Triton X-100 rescues about half the activity lost by diC7PC binding in/near the active site, so not all of the PC is likely to be removed from near the active site. The PR1E reduction by Triton X-100 is consistent with competition between the nonionic detergent alkyl chain and one or both chains of diC7PC binding to the AHS.
For comparison, the effect of Triton X-100 on diC8PI binding to spin-labeled PTEN was also examined. Protein binding to mixed micelles of diC8PI/Triton X-100 (3:6 mm) exhibited a smaller average size as indicated by a shorter correlation time for diC8PI in the presence of unlabeled protein. Once the control was subtracted, the dependence of the diC8PI PR1E on field was best fit with a correlation time of 68 ± 10 ns; rapp6 was 0.52 × 10−54 m6 (Table 3), comparable with what is observed with diC8PI alone. Thus, the polar headgroup interactions of the PI are more than adequate to compensate for disruption by Triton X-100 of any hydrophobic interactions of acyl chain moieties with the AHS.
If diC7PC partially occupies the active site and the AHS, does it have an effect on the PI(4,5)P2 interaction with PTEN? To answer this, we obtained a field cycling profile for 3 mm diC8PI(4,5)P2, 5 mm diC7PC, and 5.3 μm spin-labeled protein. Although the PC phosphodiester overlapped with the PI(4,5)P2 phosphodiester resonance, the behavior of the phosphomonoesters in the absence or presence of the micellar PC can act as sensors of the added PC. The two PI(4,5)P2 phosphomonoester peaks exhibited little change in τP-e or RP-e(0)with the excess of diC7PC added, again consistent with a discrete PI(4,5)P2 site on PTEN different from the active site (Table 3).
Modeling PIP2 Binding to PTEN
The field cycling NMR studies provide distances for placement of the activating PI(4,5)P2 molecule with respect to spin-labeled Cys-124. These distances, the mutagenesis data implicating Lys-13 and Arg-47 in the PI(4,5)P2 activation process, and the model for PTEN bound to a substrate-containing membrane (Fig. 1B) were used to identify a discrete site on the protein that is close to but does not compete with substrate occupying the active. As shown in Fig. 9A, diC6PI(4,5)P2 can be docked so that its 31P nuclei are 10.2, 12.8, and 10.4 Å, respectively, from the spin-label on Cys-124. Lys-13 and Arg-47 are critical for the orientation with P-4. These distances are all shorter than the values estimated by the NMR experiment, although the model tracks which 31P is closest to the spin label (P-1, with P-5 only slightly different) and which is decidedly further away (P-4). Movement of the spin label and faster dynamics of the short-chain molecules could easily reduce the effectiveness of the PR1E, but trends would be preserved.
FIGURE 9.
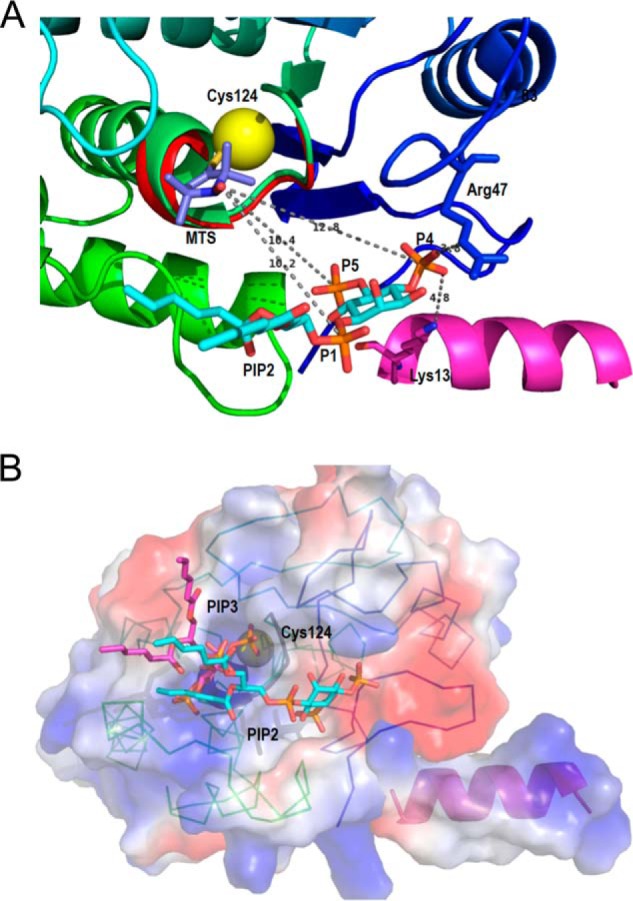
Ribbon diagrams of the model of diC6PI(4,5)P2 binding to PTEN at a discrete site near the active site are compatible with the 31P electron distances to the Cys-124 spin label. A, view of the phosphatase domain as seen from the membrane. The sulfur of Cys-124 is the yellow sphere to which the 2,2,5,5-tetramethyl-1-oxyl-3-methyl methanethiosulfonate is attached. The PI(4,5)P2 molecule, placed according to the surface features and to the relative distances determined by NMR, forms hydrogen-bonded contacts with Arg-47 and Lys-13, both critical residues for the PIP2 activation to be observed. B, surface representation of the phosphatase domain, in a similar view to that in A, colored by the electrostatic potential (red, negative, and blue, positive) with both substrate and activator docked to their sites. The PI(3,4,5)P3 molecule (magenta) is docked at the active site marked by the dark loop and the yellow sphere depicting Cys-124. The PI(4,5)P2 molecule (in cyan) occupies the same site as in A. The N-terminal helix is docked into the phosphatase domain, and its positive electrostatic potential complements the charges of the domain to create a positive membrane binding profile.
The model also provides an explanation for the difference in kinetic versus NMR effects of what happens with higher concentrations of PI(4,5)P2 and PI(3)P. Enzyme kinetics show that increasing PI(4,5)P2 can inhibit PTEN acting on PI(3)P as the substrate. However, in the NMR both PI(4,5)P2 and PI (the surrogate substrate) clearly bind to PTEN at discrete sites at the same time. The spin label in the active site would make it difficult for PI(3)P to bind well in the active site cleft. However, PI, lacking that phosphorus, can still fit in the cleft.
Both substrate and activator phospholipids are shown docked to the phosphatase domain (without the spin label) in Fig. 9B. Here, the surface electrostatic potential of PTEN is shown. Given the choice of PI(3,4,5)P3 and PI(4,5)P2, the higher electropositive character of the active site will preferentially attract PI(3,4,5)P3. Modeling the missing N-terminal peptide as a helix provides a ligand for P-4 of PI(4,5)P2 but also creates a surface with a higher cationic character to enhance membrane binding. The two phospholipids are in close van der Waals contact with each other as well as the active site loop. Thus, occupation of the PI(4,5)P2 site will enhance anchoring of PTEN to membranes allowing substrate and product to enter and exit the active site cleft.
Perturbation of the Activator Site in K13E
The model presented for PI(4,5)P2 binding to PTEN shows that Lys-13 as well as Arg-47 interact with the phosphates of the activating ligand. Our kinetic studies show that the variant K13E is not activated by PI(4,5)P2, indicating that the activator site has been altered, i.e. either it has been disrupted or its communication with the active site has been affected. The fixed field studies showed that the increased linewidth for each phospholipid was the same whether wild type or K13E spin-labeled protein was used. However, does PI(4,5)P2 still bind in the same way? The field cycling experiments provide insights for the mechanism behind this kinetic effect (Fig. 10).
FIGURE 10.

Effect of spin-labeled K13E PTEN (4.1 μm) on the field dependence of R1 for phospholipid mixed micelles of 3 mm diC8PI (○) and 3 mm diC8PI(4,5)P2 (P-1 (●), P-4, (Δ), P-5 (■)).
diC8PI binding to spin-labeled K13E exhibited a similar PR1E to that for binding to native PTEN as reflected in RP-e(0). There is a statistically real decrease in rapp6 for diC8PI in the mutant that could represent a very small shift in the position of the phosphodiester with respect to the spin labels. Changes in extracted parameters were also observed for diC8PI(4,5)P2. In particular, the rapp6 for P-5 is now larger than that for P-4, meaning its orientation with respect to the active site spin labels has changed (Table 4). This suggests that the diC8PI(4,5)P2 inositol ring is adopting a different orientation when bound to spin-labeled PTEN K13E. In the model presented for diC6PI(4,5)P2 binding to PTEN (Fig. 9), P-5 is closer to the spin label in the active site than P-4, but it is P-4 that interacts with Lys-13. If Lys-13 is mutated, a major constraint on orienting the activating PI(4,5)P2 inositol ring has been removed. In the micelle system, the phosphoinositide headgroup can still bind in this site, but it must be reoriented.
TABLE 4.
Comparison of low field relaxation parameters for phosphoinositides interacting with spin-labeled PTEN K13E
The ratio of protein/phospholipid was 0.00071 for diC8PI alone; for the sample containing 3 mm of both phospholipids, the protein/phospholipid ratio was 0.00141.
| Ligand | τP-e | RP-eP-e(0) | r6 × 1054 (m6) |
|---|---|---|---|
| ns | s−1 | ||
| diC8PI | 144 ± 26 | 3.87 ± 0.40 | 0.32 ± 0.06 |
| diC8PI/diC8PI(4,5)P2 | |||
| P-1 | 46 ± 10 | 0.72 ± 0.07 | 1.1 ± 0.2 |
| P-4 | 32 ± 14 | 0.21 ± 0.04 | 2.6 ± 1.1 |
| P-5 | 46 ± 16 | 0.13 ± 0.03 | 6.0 ± 2.1 |
| PI | 87 ± 24 | 3.81 ± 0.79 | 0.39 ± 0.10 |
Another intriguing difference between the effects of spin-labeled native PTEN and K13E is that the correlation times for the diC8PI(4,5)P2 bound to K13E are shorter than for normal PTEN, particularly with diC8PI present (Table 4). With both phospholipids present, the average τP-e for the diC8PI(4,5)P2 resonances was 43 ± 8 ns (Table 4) compared with 142 ± 34 ns when the same two phospholipids are mixed with spin-labeled wild type PTEN (Table 2). The bound diC8PI resonance in the same sample with K13E had a τP-e twice that of the activator molecule. If one assumes the longer rotational correlation time (for the diC8PI) is for the overall protein-micelle complex, then the diC8PI(4,5)P2 molecule has a shorter residence time on the enzyme. These changes in relaxation parameters for both diC8PI and diC8PI(4,5)P2 are consistent with a change in the protein conformation of K13E that weakens diC8PI(4,5)P2 binding (and likely alters the orientation of the inositol ring) and slightly alters the placement of diC8PI in the active site. This indicates that the orientation of PI(4,5)P2 when bound to PTEN must be very specific for kinetic activation to occur. Because the two lipids are within van der Waals contact in the model, the loss of activation with K13E is consistent with a requirement of close contact of the two phospholipids for optimal membrane binding and catalysis.
DISCUSSION
What Does PIP2 Binding Do?
A number of model membrane binding studies (11, 16) as well as cell studies (29, 35, 36) have shown that PI(4,5)P2 (but phosphatidylserine as well) enhances membrane anchoring of PTEN. Deletion or mutation of cationic residues in the first 15 residues reduces membrane binding of the protein and can relocalize the protein to the cytosol. These results clearly demonstrate that PI(4,5)P2 is critical for tight PTEN binding to membranes. However, there is no definitive evidence that an individual PI(4,5)P2 molecule binds to the N-terminal binding motif of PTEN, evidence for a specific conformational change, or a defined position for the N-terminal peptide with respect to the active site. The NMR studies in this work clearly show that there is a very specific site for a PI(4,5)P2 molecule binding to the protein near the cleft where substrate binds. Molecular modeling with the NMR and mutagenesis constraints allowed us to identify such a site (Fig. 9A).
One of the intriguing results of a recent MD simulation study was that productive membrane binding of PTEN required a rotation of the two domains. Substrate PI(3,4,5)P3 promoted correct orientation at the membrane surface, and Kalli et al. (17) suggested that perhaps PI(4,5)P2, at significantly higher concentrations in cells, could have the same effect. However, the simulation used the crystal structure of PTEN with both C- and N-terminal peptides missing.
In our model, the N-terminal peptide is docked as an interfacial helix and is critical in creating the PI(4,5)P2 site (Lys-13 as a ligand for P-4). This portion of the protein could be inherently flexible and require a phosphoinositide to adopt a stable helix oriented for membrane interactions. As a helix, the electrostatics of this segment would contribute to overall tighter membrane binding. Thus, PI(4,5)P2 binding at the site would lead to better binding of the protein on membranes. A PI(4,5)P2 molecule adjacent to the cleft occupied by substrate could also enhance processive catalysis through a van der Waals contact that helps substrate or product navigate in and out of the cleft.
PTEN Aggregates with Monomeric Phosphoinositides
The phosphoinositide monomer and micelle systems used in this study provide an easier way to characterize the PI(4,5)P2-binding site than working with vesicles. The micelles are dynamic with small linewidths (for micelles, less than 8 Hz at 14.08 T in the absence of protein) compared with phospholipids in vesicles, allowing experiments with low millimolar concentrations of lipids. Fixed high field studies of concentrations of the short-chain phosphoinositides below their CMC provide a marker for molecules binding in the active site, i.e. large linewidth increases occur for molecules known to bind to the active site and inhibit PTEN activity, while significantly smaller increases in linewidth are observed for diC8PI(4,5)P2. The large linewidth changes for molecules that bind to the active site could reflect aggregation of the PTEN, but they could also be caused by exchange broadening. The use of high resolution field cycling, under conditions where there is a very large excess of phospholipids compared with protein (typically phospholipid/PTEN is 1500:1 for a PI and 500–600:1 for PIP2), provides unambiguous information on the size of PTEN·short-chain phosphoinositide complexes, as well as an averaged distance between the 31P of interest and unpaired electrons on nitroxides introduced in the protein.
Although diC6PI is monomeric in the absence of protein (31), it is in a large complex characterized by a correlation time of ∼170 ns when it is bound to PTEN. That large rotational correlation time strongly suggests that the complex consists of more than one or two PTEN molecules, along with many diC6PI. In contrast, a much smaller τP-e (35 ns) was obtained for the PTEN-diC6PI(4,5)P2 complex. The experimental rotational diffusion constant, τP-e, is related to an isotropic rotational diffusion constant by τP-e = 1/(6 × D) where D = kT/(8 πη r3), and r is the hydrodynamic radius. From this, we estimate the average radius of the PTEN·diC6PI(4,5)P2 complex as 32 Å. For comparison, a monomer of PTEN, assuming it can be approximated by a sphere, would have a radius r = {3M/(4πρNo)}1/3, where M is the molecular weight; ρ is the average protein density, and No is Avogadro's number. The extracted value is 24 Å for a monomer PTEN binding a single phospholipid at the activator site. This would be increased 1–2 Å with a hydration layer. Again, considering the protein as globular, a PTEN dimer would have a rotational correlation time of 30–32 Å. Recently, it has been shown that PTEN can form dimers and that this is the active species of the enzyme (37). The experimental τP-e for the diC6PI(4,5)P2·PTEN complex is consistent with diC6PI(4,5)P2 binding to a PTEN dimer in solution.
If PTEN/diC6PI(4,5)P2 is dimeric in solution, why is the complex with diC6PI so much larger? The dimeric PTEN structure is suggested to have a more “open” or relaxed conformation of PTEN when it is bound on the membrane (37). This could resemble the model shown in Fig. 1B. A more extensive membrane-binding surface in the dimer would likely attract a significant number of the short-chain phosphoinositides with it and lead to the observed large correlation time for the complex. That a larger complex is observed for the PTEN with diC6PI suggests that a molecule binding to the active site is critical for this more extended conformation.
The rapp6 extracted for diC6PI compared with that for diC8PI provides another useful piece of information. rapp6 is 7-fold larger for diC6PI, indicating that the active site is not saturated with the ligand. This is consistent with productive substrate binding requiring a very specific conformational change of PTEN that is enhanced by the presence of an interface.
In contrast, the rapp6 parameters for diC6PI(4,5)P2 were only 1.4–2.2-fold larger than the values for diC8PI(4,5)P2 and very different from those for diC8PI. The interaction of this monomeric phosphoinositide with PTEN also leads to a complex larger than for a single molecule binding to PTEN. However, the similar values for rapp6 for the two different chain length PI(4,5)P2 molecules are consistent with a discrete and moderately tight site on the protein for the PI(4,5)P2. These results also suggest an explanation for the acyl chain dependence of PI(4,5)P2 activation kinetics (Fig. 3B). diC6PI(4,5)P2 is poor at activating PTEN toward diC6PI(3)P because, by analogy to what we see with diC6PI versus diC8PI binding, less substrate is bound at the active site. diC8PI(4,5)P2 is also not very effective at enhancing hydrolysis of diC6PI(3)P for the same reason.
In summary, we have shown that there are three functionally distinct but spatially close sites for amphiphiles to bind to PTEN, at least for ∼100 ns: (i) the active site, (ii) the PI(4,5)P2“allosteric” site, and (iii) the AHS. A discrete binding site for PI(4,5)P2 involving the N-terminal peptide of PTEN (specifically Lys-13) and Arg-47 has been proposed and is consistent with the NMR data. Its placement adjacent to the active site provides a pathway to stabilize the conformation of PTEN needed for optimal processive catalysis. The interplay/synergism of the three sites detected by field cycling NMR also provides ways to regulate PTEN activity. Like many other signaling proteins that cycle on/off the membrane surface, PTEN possesses a C2 domain that should transiently anchor it to the membranes via PS interactions. However, the presence of a hydrophobic site and PIP2 activator site in the vicinity of the active site on the phosphatase domain provides novel ways to direct the PTEN conformational change needed for catalysis.
Acknowledgment
We thank Dr. Wonhwa Cho for the original plasmid containing the gene for PTEN.
This work was supported by National Science Foundation Grant MCB-0950331.
- PTEN
- phosphatase and tensin homolog
- AHS
- adjacent hydrophobic site
- CMC
- critical micelle concentration
- DOPI(3,4)P2
- dioleoylphosphatidylinositol 3,4-bisphosphate
- DOPI(4,5)P2
- dioleoylphosphatidylinositol 4,5-bisphosphate
- LUV
- large unilamellar vesicle
- MTSL
- 2,2,5,5-tetramethyl-l-oxyl-3-methyl methanethiosulfonate
- PI
- phosphatidylinositol
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PI(3,4)P2
- phosphatidylinositol 3,4-bisphosphate
- PI(3,4,5)P3
- phosphatidylinositol 3,4,5-trisphosphate
- PI(3)P
- phosphatidylinositol-3-phosphate
- I(1,4,5)P3
- inositol 1,4,5-trisphosphate
- diC6-
- dihexanoyl-
- diC8-
- dioctanoyl-
- diC7PC
- 1,2-diheptanoyl-sn-glycero-3-phosphocholine
- POPC
- 1,2-diheptanoyl-sn-glycero-3-phosphocholine
- PR1E
- paramagnetic spin lattice relaxation enhancement
- R1
- spin-lattice relaxation rate
- Ni-NTA
- nickel-nitrilotriacetic acid
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- T
- tesla
- PRE
- paramagnetic relaxation enhancement
- IP3
- inositol 1,4,5-trisphosphate.
REFERENCES
- 1. Song M. S., Salmena L., Pandolfi P. P. (2012) The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 [DOI] [PubMed] [Google Scholar]
- 2. Salmena L., Carracedo A., Pandolfi P. P. (2008) Tenets of PTEN tumor suppression. Cell 133, 403–414 [DOI] [PubMed] [Google Scholar]
- 3. Leslie N. R., Downes C. P. (2004) PTEN function: how normal cells control it and tumour cells lose it. Biochem. J. 382, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chang N., El-Hayek Y. H., Gomez E., Wan Q. (2007) Phosphatase PTEN in neuronal injury and brain disorders. Trends Neurosci. 30, 581–586 [DOI] [PubMed] [Google Scholar]
- 5. Maehama T., Dixon J. E. (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378 [DOI] [PubMed] [Google Scholar]
- 6. Myers M. P., Pass I., Batty I. H., Van der Kaay J., Stolarov J. P., Hemmings B. A., Wigler M. H., Downes C. P., Tonks N. K. (1998) The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proc. Natl. Acad. Sci. U.S.A. 95, 13513–13518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McConnachie G., Pass I., Walker S. M., Downes C. P. (2003) Interfacial kinetic analysis of the tumour suppressor phosphatase, PTEN: evidence for activation by anionic phospholipids. Biochem. J. 371, 947–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Campbell R. B., Liu F., Ross A. H. (2003) Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 278, 33617–33620 [DOI] [PubMed] [Google Scholar]
- 9. Maehama T., Taylor G. S., Dixon J. E. (2001) PTEN and myotubularin: novel phosphoinositide phosphatases. Annu. Rev. Biochem. 70, 247–279 [DOI] [PubMed] [Google Scholar]
- 10. Walker S. M., Leslie N. R., Perera N. M., Batty I. H., Downes C. P. (2004) The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem. J. 379, 301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Redfern R. E., Redfern D., Furgason M. L., Munson M., Ross A. H., Gericke A. (2008) PTEN phosphatase selectively binds phosphoinositides and undergoes structural changes. Biochemistry 47, 2162–2171 [DOI] [PubMed] [Google Scholar]
- 12. Lee J. O., Yang H., Georgescu M. M., Di Cristofano A., Maehama T., Shi Y., Dixon J. E., Pandolfi P., Pavletich N. P. (1999) Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–334 [DOI] [PubMed] [Google Scholar]
- 13. Wang Q., Wei Y., Mottamal M., Roberts M. F., Krilov G. (2010) Understanding the stereospecific interactions of 3-deoxyphosphatidylinositol derivatives with the PTEN phosphatase domain. J. Mol. Graph. Model. 29, 102–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y. K., Chen W., Blair D., Pu M., Xu Y., Miller S. J., Redfield A. G., Chiles T. C., Roberts M. F. (2008) Insights into the structural specificity of the cytotoxicity of 3-deoxyphosphatidylinositols. J. Am. Chem. Soc. 130, 7746–7755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shenoy S. S., Nanda H., Lösche M. (2012) Membrane association of the PTEN tumor suppressor: electrostatic interaction with phosphatidylserine-containing bilayers and regulatory role of the C-terminal tail. J. Struct. Biol. 180, 394–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Das S., Dixon J. E., Cho W. (2003) Membrane-binding and activation mechanism of PTEN. Proc. Natl. Acad. Sci. U.S.A. 100, 7491–7496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kalli A. C., Devaney I., Sansom M. S. (2014) Interactions of phosphatase and tension homologue (Pten) proteins with phosphatidylinositol phosphates: insights from molecular dynamics simulations of PTEN and voltage-sensitive phosphatase. Biochemistry 53, 1724–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pu M., Feng J., Redfield A. G., Roberts M. F. (2009) Enzymology with a spin-labeled phospholipase C: soluble substrate binding by P-31 NMR from 0.005 to 11.7 T. Biochemistry 48, 8282–8284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pu M., Orr A., Redfield A. G., Roberts M. F. (2010) Defining specific lipid binding sites for a peripheral membrane protein in situ using subtesla field-cycling NMR. J. Biol. Chem. 285, 26916–26922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gradziel C. S., Wang Y., Stec B., Redfield A. G., Roberts M. F. (2014) Cytotoxic amphiphiles and phosphoinositides bind to two discrete sites on the Akt1 PH domain. Biochemistry 53, 462–472 [DOI] [PubMed] [Google Scholar]
- 21. Cai J., Guo S., Lomasney J. W., Roberts M. F. (2013) Ca2+-independent binding of anionic phospholipids by phospholipase Cδ1 EF-hand domain. J. Biol. Chem. 288, 37277–37288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yates J. R., 3rd, Eng J. K., McCormack A. L., Schieltz D. (1995) Method to correlate tandem mass-spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 23. Tabb D. L., McDonald W. H., Yates J. R. (2002) DTASelect and contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Redfield A. G. (2012) High resolution NMR field-cycling device for full-range relaxation and structural studies of biopolymers on a shared commercial instrument. J. Biomol. NMR 52, 159–177 [DOI] [PubMed] [Google Scholar]
- 25. Cole C., Barber J. D., Barton G. J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC, New York [Google Scholar]
- 28. Clore G. M., Iwahara J. (2009) Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 109, 4108–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rahdar M., Inoue T., Meyer T., Zhang J., Vazquez F., Devreotes P. N. (2009) A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc. Natl. Acad. Sci. U.S.A. 106, 480–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roberts M. F., Redfield A. G. (2004) High-resolution P-31 field cycling NMR as a probe of phospholipid dynamics. J. Am. Chem. Soc. 126, 13765–13777 [DOI] [PubMed] [Google Scholar]
- 31. Lewis K. A., Garigapati V. R., Zhou C., Roberts M. F. (1993) Substrate requirements of bacterial phosphatidylinositol-specific phospholipase C. Biochemistry 32, 8836–8841 [DOI] [PubMed] [Google Scholar]
- 32. Klauda J. B., Roberts M. F., Redfield A. G., Brooks B. R., Pastor R. W. (2008) Rotation of lipids in membranes: MD simulation, 31P spin-lattice relaxation, and rigid-body dynamics. Biophys. J. 94, 3074–3083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roberts M. F., Redfield A. G. (2004) Phospholipid bilayer surface configuration probed quantitatively by 31P field-cycling NMR. Proc. Natl. Acad. Sci. U.S.A. 101, 17066–17071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kowalewski J., Maler L. (2006) Nuclear Spin Relaxation in Liquids: Theory, Experiments, and Applications. Taylor & Francis Group, CRC Press [Google Scholar]
- 35. Vazquez F., Matsuoka S., Sellers W. R., Yanagida T., Ueda M., Devreotes P. N. (2006) Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 103, 3633–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Iijima M., Huang Y. E., Luo H. R., Vazquez F., Devreotes P. N. (2004) Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J. Biol. Chem. 279, 16606–16613 [DOI] [PubMed] [Google Scholar]
- 37. Papa A., Wan L., Bonora M., Salmena L., Song M. S., Hobbs R. M., Lunardi A., Webster K., Ng C., Newton R. H., Knoblauch N., Guarnerio J., Ito K., Turka L. A., Beck A. H., Pinton P., Bronson R. T., Wei W., Pandolfi P. P. (2014) Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 157, 595–610 [DOI] [PMC free article] [PubMed] [Google Scholar]