

Abstract
Autism spectrum disorder (ASD) affects as many as 1 in 68 children and is said to be the fastest-growing serious developmental disability in the United States. There is currently no medical cure or diagnostic test for ASD. Furthermore, the U.S. Food and Drug Administration has yet to approve a single drug for the treatment of autism’s core symptoms. Despite numerous genome studies and the identification of hundreds of genes that may cause or predispose children to ASD, the pathways underlying the pathogenesis of idiopathic ASD still remain elusive. Post-mortem brain samples, apart from being difficult to obtain, offer little insight into a disorder that arises through the course of development. Furthermore, ASD is a disorder of highly complex, human-specific behaviors, making it difficult to model in animals. Stem cell models of ASD can be generated by performing skin biopsies of ASD patients and then dedifferentiating these fibroblasts into human-induced pluripotent stem cells (hiPSCs). iPSCs closely resemble embryonic stem cells and retain the unique genetic signature of the ASD patient from whom they were originally derived. Differentiation of these iPSCs into neurons essentially recapitulates the ASD patient’s neuronal development in a dish, allowing for a patient-specific model of ASD. Here we review our current understanding of the underlying neurobiology of ASD and how the use of stem cells can advance this understanding, possibly leading to new therapeutic avenues.
Keywords: autism, autism spectrum disorder, stem cells, induced pluripotent stem cells
Introduction
Autism spectrum disorder (ASD) affects as many as 1 in 68 children and is said to be the fastest-growing serious developmental disability in the United States [1]. The disorder is characterized by social and communication deficits as well as restricted interest and repetitive behaviors [2]. Variations in clinical presentation and disease progression across ASD patients is tremendously high. In addition, patients often present with a mixture of accompanying conditions such as epilepsy, intellectual disability, gastrointestinal problems, anxiety, and depression [2]. Such comorbidities are especially common in syndromic forms of ASD — cases in which the ASD appears as part of a rare syndrome with a known genetic cause. In correspondence with the diversity of phenotypes seen in affected individuals, the genetic component underlying the pathogenesis of autism is also highly heterogeneous across patients. The primary pathological abnormality in all of these forms of ASD is thought to be altered development of the neuronal circuitry through early changes in synaptic plasticity and function and/or disruptions in the excitation-inhibition (E-I) balance, conditions which, until now, were very hard to model in vivo and in vitro systems [3].
Induced pluripotent stem cells (iPSC) are cells that have undergone an in vitro deprogramming process that renders them capable of giving rise to all cells of an organism. These cells are immortal and can be reprogrammed to differentiated cell types, including brain cells. While post-mortem studies and advances in neuroimaging have allowed us to examine the ASD brain phenotype in some detail, it is hard to discriminate cause from consequences and experimental artifacts. iPSCs would allow us in principle to examine how and why aberrations in brain structure and composition develop initially.
The advent of advanced stem cell differentiation technologies allows us to artificially grow in vitro miniature organs resembling the brain, known as cerebral organoids. Organoids are created by growing human pluripotent stem cells in a 3D culture system. Cerebral organoids resemble certain regions of the brain in their layer/tissue cytoarchitecture and cell types. Because they retain the unique genetic signature of the individual from whom they were originally derived, these cerebral organoids can be used as a means of comparing the early brain structure and composition of an individual with ASD and his/her unaffected family member.
The Neurobiological Substrates of ASD
ASD Genetic Subtypes
Family and twin studies have confirmed the high heritability of ASD. The concordance rate in monozygotic twins is between 60 and 91 percent, making ASD one of the most heritable psychiatric conditions as defined by this measure [4]. Furthermore, the recurrence risk in families with one child with ASD may be as high as 20 percent [5]. However, despite these indications of a strong genetic component to the pathogenesis of ASD, only ~15 percent of total ASD cases have a known genetic cause [6]. In fact, Gaugler et al. [7] suggest that the most genetic risk for ASD comes from common genetic variation. Of the 15 percent of cases with identified genetic causes, more than 50 percent are monogenic forms of ASD known as syndromic autisms — ASD cases in which the ASD appears as part of a rare syndrome with a known genetic cause [7]. The most common syndromic autisms appear as part of Fragile X syndrome, tuberous sclerosis, Rett syndrome, Type I neurofibromatosis, and Cowen syndrome [7], although association has not been always confirmed [8]. Apart from syndromic autisms, rare mutations that can lead to ASD have been identified in synaptic genes, including members of the neuroligin [9,10], neurexin [11], and SHANK [12-15] families of proteins. Once again, though, by definition, each of these mutations accounts for <1 percent of total ASD cases. Similarly, genomic variants (e.g., cytogenetic abnormalities such as the maternal duplication) at the 15q11-13 locus and the deletions or duplications at the 16p11 locus account for approximately 1 to 3 percent of total ASD cases [16]. The remaining ~85 percent of total ASD cases with no known genetic cause are referred to as idiopathic ASD.
Insights from Rare Mutation-Associated and Syndromic Autisms
In an effort to further illuminate the causative molecular and cellular mechanisms of ASD more broadly, groups have studied the neurobiological underpinnings of syndromic and rare mutation-associated autisms through various animal models. Additionally, with the advent of more advanced technologies that make sequencing both more affordable and reliable, numerous genome-wide association (GWAS), candidate gene re-sequencing, and exome-sequencing studies have been performed. These studies collectively indicate the existence of hundreds of genetic variants that contribute to ASD risk, indicating the need to shift focus to elucidate common and converging pathways among these genes. It is the hope that the identification of such key molecular and functional pathways will connect both common and rare forms of ASD. Perhaps stem cell models will aid in this effort by allowing researchers to zoom into the pathways disturbed in early brain development. Those pathways are the most likely to be influential in the pathogenesis of a neurodevelopmental disorder such as ASD.
Several groups have already conducted studies along the lines of pathway analyses on genetic or transcriptome data. O’Roak et al. demonstrated that ~ 40 percent of all the most disruptive de novo mutations converged onto one highly connected beta-catenin/chromatin remodeling protein-protein interaction (PPI) network controlling neuronal differentiation and synaptic formation [17]. Iossifov et al. suggested the involvement of genes encoding proteins associated with Fragile X syndrome protein (FMRP), based on enrichment of de novo variants specifically in these genes, suggesting the involvement of synaptic plasticity [18]. In accordance with both studies, Neale et al. showed that de novo variants were functionally related to each other and showed functional enrichment for synaptic genes [19]. However, other genetic studies suggested the involvement of a wider set of neurodevelopmental functions in ASD. Through bioinformatic pathway analysis of genetic association data, Talkowski et al. showed that rare de novo copy number variations (CNVs) occurring in ASD cases are primarily found in genes related to synapse development, neuron motility, and axon targeting [20]. Similarly, Pinto et al. [21] and Glessner et al. [22] found significant CNV enrichment in affected individuals compared to controls in numerous synaptic plasticity and neural cell adhesion genes, as well as genes involving cellular proliferation, projection, and motility. Sanders et al. [23] suggested an excitation-inhibition (E-I) unbalance in individuals with ASD by implicating SCN1A, a voltage-gated sodium ion channel, to be a high risk gene for ASD. The largest studies so far have implicated transcription and chromatin regulation and embryonic cortical neuron differentiation in ASD, suggesting the involvement of early embryonic etiological factors involved in the regulation of cell fate, in addition to synaptic dysfunction [24-26]. Finally, in an elegant study of autistic brain pathology, Voineagu et al. [27] demonstrated that the typical regional differences found in the gene expression profiles of the frontal and temporal lobes were attenuated in the brains of ASD patients. In a data-driven network analysis, the authors related one of the differentially expressed modules of genes to interneurons and, more generally, to genes involved in synaptic function. This module of genes was down-regulated in ASD individuals compared to controls. On the other hand, a module with a functional enrichment of immunity and microglial activation was up-regulated in ASD brains compared to controls. Collectively, these studies indicate that the underlying neurobiology of autism is likely rooted in alterations in synaptic function and plasticity and neural structural connectivity and cell fate. Engineering of the above genetic network aberrations into cerebral organoids will allow us to further validate the bioinformatically driven predictions of cellular and molecular phenotypes and might even allow us to establish a causal link.
Animal model studies of rare mutation-associated and monogenic forms of ASD have yielded very similar results. Mutations in TSC1 and TSC2 (tuberous sclerosis), NF1 (neurofibromatosis), and PTEN (Cowen syndrome) all affect signal transduction through the PI3K-mTOR signaling pathway, which, in turn, regulate essential functions such as cell proliferation, cell survival, protein synthesis, and cell/synaptic pruning. Furthermore, FMRP is specifically involved in mRNA translation at the synapse [3,28,29]. The neuroligin, neurexin, and SHANK families of proteins, rare mutations that lead to ASD, are involved in the formation of excitatory and inhibitory synapses [30], once again suggesting an E-I imbalance. However, the direction and ratios of these imbalances in protein synthesis and excitation when modeled in animals remain unclear. While rodent models of Rett syndrome and tuberous sclerosis show decreased density of dendrites, models of Fragile X syndrome show increased dendritic density [31,32]. Both increased and decreased glutamate currents, as well as increased and decreased inhibitory currents, have been observed in various SHANK [33] and neuroligin family member mutant models [34]. Additionally, Toro et al. [35] point out that several of these genes associated with ASD are regulated by neuronal activity itself, suggesting that an individual’s specific circuitry, connectivity, and E-I ratio may be species-, experience-, and/or environment-dependent.
Other animal models include inbred mouse strains such as BTBR mice and models that utilize intrauterine exposure to toxic chemicals or immune challenges to induce cognitive and behavioral deficits. These models of autism often have great construct and face validity, but their predictive validity is unmeasurable due to lack of any molecules actually approved for the treatment of core symptoms in ASD. Furthermore, while researchers can fairly easily monitor behaviors such as self-grooming and basic social interactions, other more complex, uniquely human behaviors involving theory of mind and complex language formation are extremely difficult to model in animals. The neurobiological roots of these complex, uniquely human behaviors are also likely to lie in the inherent complexity and sophisticated architecture of human brains that rodent brains do not possess.
Theories and Inferences from Neuroimaging Studies
Neuroimaging studies, due to technological limitations, have focused on large-scale structural abnormalities such as brain volume or white matter connectivity, with a particular focus on neural systems that are most likely relevant to the behavioral and neuropsychological deficits characteristic of autism in the mature brain [36]. Interestingly, differences in these regions are correlated with symptom severity in that particular domain. For example, differences in frontotemporal regions and amygdalae have been associated with deficits in processing relevant social cues [37], while differences in volumes in the frontostriatal system have been associated with repetitive and stereotypical behavior [38,39]. Total brain volume of ASD patients is increased between ages 2 and 5 [40-43], but whether this difference surfaces earlier than age 2 (see below) and whether it maintains between age-matched controls into childhood and subsequent adolescence and adulthood remains unclear [39]. Regardless, these studies indicate abnormalities in early development and brain maturation that persist into at least early childhood.
The current standing theory for the neuropathology of autism is that the brain undergoes a period of accelerated growth in early childhood followed by a deceleration in age-related growth. Evidence for this precocious growth during early postnatal life comes from studies of head circumference, a proxy for brain size [44-47]. Thus, confirmation of this theory via an actual longitudinal study, rather than just a cross-sectional study, is still required. Furthermore, stem cell studies will provide novel insight into the prenatal brain growth patterns of ASD patients and may even demonstrate volumetric abnormalities that existed prior to and at birth in these individuals.
It also has not yet been determined whether these increases reflect differences in cortical thickness or surface area (since cortical volume is a product of both) and whether they are predominately driven by changes in grey matter or white matter volume. Hazlett et al. suggest that the accelerated brain growth is the result of a precocious non-uniform expansion of cortical surface area, rather than an increase in cortical thickness [48]. This precocious expansion of cortical surface area is likely to disproportionately involve white matter [49]. Though interestingly, while grey matter might play a proportionately smaller role in early growth, differences in grey matter seem likely to persist through adolescence and adulthood (unlike white matter differences, whose persistence into adulthood remains unclear) [41,50-52]. These enlargements in grey/white matter volumes in the brains of ASD patients mostly have been reported in the frontal lobes [50,52-54].
Altered structural connectivity has been observed, through diffusion tensor imaging (DTI), in limbic and language pathways, frontostriatal circuitry, and the corpus callosum [38,55,56]. Currently, the general notion, which also has been confirmed by functional connectivity MRI [57,58], is a global hyper-connectivity in the brains of individuals with ASD, consistent with many of the above-stated genetic observations of increased synaptic and dendritic density, the more generally increased mRNA translation, and the observed early brain overgrowth and increased gray and white matter volumes. While this notion is largely based on cortical white matter connectivity, Ecker et al. [59] have shown that the brain’s intrinsic connectivity (that is, cortical grey matter connectivity/corticocortical connections) also significantly differs in individuals with ASD from age-matched controls. Intrinsic wiring costs, the minimum length of horizontal connections required to form corticocortical connections, were significantly reduced in individuals with ASD, indicating an increased propensity for forming corticocortical connections. This phenomenon was observed predominately in the frontotemporal regions. Interestingly, this decreased wiring cost correlated with symptom severity, particularly in the category of repetitive behaviors [59]. Imaging of patient-derived cerebral organoids hopefully will provide further evidence of this altered structural connectivity theory at earlier stages in prenatal brain development, as well as elucidate the cause of this abnormality.
Magnetic resonance spectroscopy and PET have proven invaluable in validating interest in E-I imbalance hypothesis for ASD pathogenesis. Similar to the genetic results, though, the actual direction of the imbalance varies tremendously across studies and patients. Some neuroimaging studies in humans indicate that ASD individuals have a higher neurotransmitter density and consequent up-regulation of the glutamatergic system [60], as well as reduced expression of GABA receptors and consequent down-regulation of the GABAergic system [61,62], while others report the exact opposite profile in different regions of the brains of the same individual or the same region in different individuals [63].
Findings from Neuropathological Studies
Neuropathological studies have been limited by the lack of post-mortem ASD brains and closely matched controls. Poor quality of the tissue and the adoption of qualitative, biased, non-systematic methods of analysis have plagued many of these studies. Furthermore, it should be noted that the brain regions represented by these studies are in no way indicative of the selective regions in the brain affected by ASD. Several regions of the brain have never been studied. Early studies found that cell packing density was increased and neuronal size decreased in the limbic system. Similarly, the number of Purkinje cells is decreased in the cerebellum [64,65]. However, these studies relied on histological stains that are sensitive to post-mortem artifacts. Indeed, more recent studies from the same group, using immunocytochemistry and stereological analyses, found no difference in overall density of cerebellar Purkinje cells in ASD [66].
Several groups have examined alterations in several regions of the cerebral cortex. Cytoarchitectonical abnormalities have been found in several regions, such as irregular definition of layers and displacement of neurons in the white matter, which have been called “focal cortical dysplasias” [67-70]. While intriguing, it should be noted that such abnormalities were not noted in all brains, and their origin (whether due to some artifact of brain processing or reflecting actual pathology) remains controversial. More generally accepted are an increased number of neurons, a decrease in neuronal size, and a smaller size and increased density of neuronal minicolums in the neocortex [71-73]. Cortical volume was not assessed in these neuropathological studies; however, the white matter volume was noted to be increased, a finding consistent with increased neuron number. Increased white matter volume for the shorter subcortical tracts and overall increased brain volume and cortical surface area have been noted in structural neuroimaging studies (see above).
Very few studies have examined alterations in the composition of cellular subtypes in ASD. Using stereological analyses, Bauman, Blatt and colleagues recently found an increased density of GABAergic neuronal subtypes in the hippocampus, but not in the posterior cingulate cortex, of patients with ASD [68,74]. Other studies found an increased number of microglial cells, suggestive of inflammation [75,76]. Finally, a very recent study showed an increased density of synapses in the neocortex of ASD individuals [77], which was attributed to a decrease in the physiological process of pruning thought to occur mostly in the postnatal period.
These studies suggest that an increased neuronal/synapse number may represent an important feature of ASD; however, the scarcity of the tissue and the lack of wide replication of these findings leave the exact proportion of ASD patients with features of neuron/synapse overgrowth uncertain. In balance, it seems prudent to conclude that an excess production or a lack of regressive/pruning phenomena characterizes a portion of, but perhaps not all, cases with ASD. Of course, these hypotheses are on the basis of post-mortem studies, and it is important that we begin to develop a similar understanding of ASD pathology in the developing brain through stem cells. However, it is likely that stem cell models will suffer from the same problem of variability.
The Use of iPSCs to Study Neurodevelopmental Disorders
While exome sequencing studies identify loci that could contribute to ASD risk, they do not actually assess the functional consequences of associated variants in those areas. Transcriptome analyses (with a comparison between disease and control groups) on the other hand, while they do quantify mRNA output and thus assess gene function by extension, are subject to environmental influence and other confounding variables. Furthermore, brain transcriptome studies cannot distinguish primary causes from secondary consequences of disease. Ideally, studies should simultaneously assess DNA sequence variations and gene expression levels in a disease-relevant tissue. However, achieving the necessary statistical power in these experiments is understandably difficult due to the limited sample availability of post-mortem brain tissue from ASD patients. Furthermore, post-mortem brain tissue is subjected to a number of artifacts and is obtained many years from the beginning of the pathological process. Of course, observing the brain in early human growth and development has been more difficult, until recently.
New approaches have emerged so that researchers now can model human neurodevelopment in vitro and actually re-enact the altered trajectory of brain development as seen in disorders such as autism. Fibroblasts obtained from a skin biopsy of any individual can be “de-programmed” into human-induced pluripotent stem cells (hiPSCs) (Figure 1). Since the discovery in 2007 that iPSC can be reproducibly generated by overexpressing a set of trascription factors in fibroblasts in vitro [78], this field has been undergoing exponential expansion. These cells closely resemble the multipotent stem cells isolated from the inner cell mass of a blastocyst, otherwise known as embryonic stem cells (see Table 1). It has been discovered that iPSCs can be derived from a variety of somatic cells, including keratynocytes [79], lymphoblasts, and bladder epithelial cells [80], that can be converted in vitro into a variety of cell types — most importantly, cerebral cortical neurons of the central nervous system (CNS). Using hiPSCs allows us to harness the power of human embryonic stem cells in studying human development while avoiding the ethical controversies associated with killing an embryo to acquire embryonic stem cells. These hiPSCs can be differentiated into neuronal progenitors and more mature neurons in vitro, thus allowing the generation of patient-specific models of neurological disorders. Our lab has shown that patient-derived hiPSCs retain the unique genetic signature of patients from whom they were originally derived [81]. This allows these cells to serve as a window into understanding the cellular and physiological level consequences of an individual’s genetic idiosyncrasies.
Figure 1.
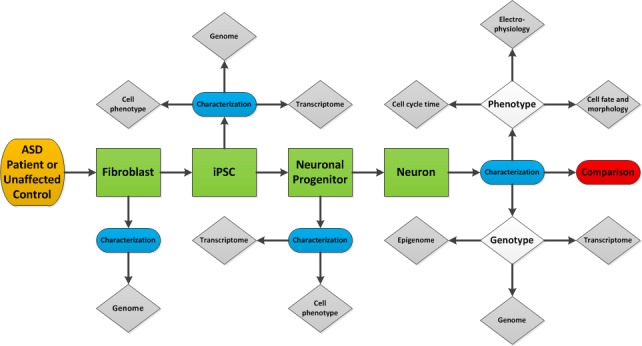
A potential workflow through which iPSCs can be used to further elucidate the mechanisms underlying the pathogenesis of ASD.
Table 1. Stem Cell Term Glossary. Adapted from [108].
| Term | Definition |
| Stem cell | Stem cells are uncommitted cells capable of a) self-renewal, i.e., cell division results in another stem cell; and b) specialization, i.e., can become tissue- or organ-specific cells. They are integral to human and animal development/growth, and function in many tissues as an internal repair system. Stem cells also can be induced experimentally. |
| Pluripotent stem cell | Pluripotent stem cells are capable of specializing into any of the three germ layers, i.e., all of the cell types that make up the embryo proper. However, unlike totipotent cells (i.e., zygotes), they cannot give rise to extra-embryonic or placental tissue. |
| Differentiation | Differentiation is the process by which a stem cell or less specialized cell becomes a more specialized cell type. Differentiation is a physiological process as well as one that can be induced experimentally. |
| Dedifferentiation | Dedifferentiation is the process of reverting a specialized cell, i.e., a tissue- or organ-specific cell to a stem cell or less specialized cell type state. Dedifferentiation is a physiological process that also can be induced experimentally (also referred to as deprogramming). |
| Reprogramming | Reprogramming refers to the process of erasing and remodeling of a cell’s epigenetic signatures. In the context of stem cells, it often refers to artificially/experimentally inducing dedifferentiation and/or differentiation. |
| Embryonic stem cell (ESC) | Embryonic stem cells are pluripotent stem cells isolated from the inner cell mass of early developing blastocysts. They can give rise to all embryonic lineages and adult cell types. |
| Induced pluripotent stem cell (iPSC) | Induced pluripotent stem cells are pluripotent stem cells resembling embryonic stem cells that can be generated through reprogramming of an adult somatic cell. |
| Neural stem cell (NSC) | Neural stem cells are stem cells capable of generating cells of the nervous system. |
| Neural progenitor cell (NPC) | A neural progenitor cell, or neuronal progenitor, is the intermediate state between a stem cell and a neuron. These cells are more differentiated than stem cells and are primed to differentiate further into a specific neural cell type. They have limited replication capacity compared to stem cells. |
| Cerebral organoid | Organoids are created by growing human pluripotent stem cells in a 3D culture system. Cerebral organoids are miniature organs resembling certain regions of the brain in their layer/tissue cytoarchitecture and cell types. |
| Mesenchymal stem cell (MSC) | Mesenchymal stem cells are stem cells found largely in the bone marrow that are capable of giving rise to bone, fat, and cartilage cells. |
In addition to their patient-specific capacity, hiPSCs are also crucial to understanding in general the pathogenesis of neurobiological disorders. In principle, they can elucidate the relationship between specific gene mutations/variants/epigenetic modifications and expression levels of that specific gene, as well as expression levels of all genes (interaction networks) [82-84]. Although a comprehensive investigation of the changes in epigenetic signature of iPSC and how that parallels the normal epigenetic changes occurring in brain development has not yet been done, these studies are doable and indeed planned in several laboratories. They can help evaluate the effects of altering the expression of specific genes (known or unknown) on the typical development of human neural cells. Furthermore, observations can be made at different time points and in different neuronal precursors, as well as across structural variants (such as copy number variations) and sequence variants (such as single nucleotide variations) of each gene. Until now, our knowledge and understanding of neurodevelopmental disorders has been largely limited to the findings of genome-wide association studies and rodent models [82-84]. While rodent models do provide insight into the fundamental (and evolutionarily conserved) processes underlying neuronal development, they fail to provide similar insights into human disease pathogenesis and therapeutic avenues, perhaps due to the increased complexity, size, and diversity of cell types found in the human brain. Currently, hiPSC models present us with a unique and powerful means of delving deeper into the molecular and cellular underpinnings of human brain development, as well as the abnormalities in this development, that give rise to disorders like ASD. The hope is that this power can be even further exploited to result in a model for drug discovery.
However, there are some potential disadvantages to iPSCs. There have been concerns about genetic and phenotypic stability of iPSCs over time in culture, i.e., the accumulation of mutations (CNVs and structural variations [SVs]) as well as the formation of mosaic lines. To further address this concern, our lab performed whole-genome sequencing of clonally amplified iPSC lines. Our data discovered line-manifested CNVs in iPSC lines apparently not found in parental fibroblasts that were confirmed by quantitative polymerase chain reaction (qPCR). Interestingly, however, there was no correlation between lineage manifested copy number variation (LM-CNV) number and iPSC passage number. Furthermore, at least 50 percent of LM-CNVs could be traced back to the original fibroblast population; that is, they were present in a small minority of fibroblast cells, a phenomenon called somatic mosaicism. We estimated that 30 percent of skin fibroblast cells carried large somatic CNVs absent in the germline. Fibroblast mosaicism varied widely (between 0.3 and 15 percent). These results collectively indicate that reprogramming of fibroblasts to hiPSCs does not necessarily introduce de novo CNVs, but rather, the mutations seen in iPSCs reveal an extraordinary extent of genomic mosaicism in normal skin fibroblasts [81].
The variability between iPSCs lines and robustness of the iPSC system itself are also a large concern. While theoretically, iPSCs are in a completely undifferentiated state, iPSCs occasionally may differ in their differentiation potential. While the cause for such intrinsic variability is not known, efforts can be made to minimize known sources of variability, such as genomic variation (due to mosaicism or integration sites of viral vectors, discussed above) and epigenetic influences due to environmental stresses and improper handling of the lines. This can be also combatted through comprehensive characterization and confirmation of the pluripotent state of iPSCs used in studies, thus excluding partially reprogrammed/not completely undifferentiated lines. Furthermore, neuronal differentiation protocols do not necessarily mimic natural processes of development and/or allow for cells’ own regulatory control, thus allowing for further variability.
Finally, thus far, it has been difficult to achieve the regional specificity and recapitulation of gene expression patterns within the various regions of the CNS using iPSCs, owing to the fact that differentiation protocols are still very primitive in this emerging field. The authenticity of iPSC-derived neurons has been, in some cases, verified by transcriptome comparisons with the post-mortem human brain [85], as well as their ability to synatically integrate [86] and project to correct targets after transplantation into the mouse brain [87]. However, converging evidence suggests that human neurons differentiate according to their own pace, and, hence, iPSC-derived neurons correspond at best to first-trimester embryonic human neurons [85,86]. Groups have tried three-dimensional culture methods, i.e., cerebral organoids, which seem to be a relatively accurate representation of the human telencephalic regions at early embryonic stages [85,88]. However, once again, the result is still neurons at immature fetal stages. Currently, studies are under way to utilize synthetic hydrogel systems and microfluidic chambers [89] to further advance the functionality, maturity, and connectivity of these neurons.
Direct conversion of somatic cells into “neurons” by transcription factor overexpression has been explored with the aim of overcoming the complexities of recapitulating neuronal development. While in theory this approach is attractive, in practice it is very limited, since the efficiency is very low and the nature of neurons obtained with this approach is unclear. Finally, overexpressing transcription factors may bypass and thus mask the very same pathogenetic processes we wish to study in ASD. It is our belief that this model would be most useful when recapitulating the process of development, together with its homeostatic and regulatory mechanisms, if we wish to clarify abnormalities that give rise to disorders like ASD. Such studies will make the development of an iPSC-based drug discovery platform far more feasible, since viable molecular drug targets likely cannot be identified in immature fetal stages of brain development.
Potential Uses of Stem Cells in ASD
Use of Stem Cells for In Vivo Cell Replacement/Repair
Transplantation studies in animal models of CNS disorders have suggested potential roles of neural stem cells and their derivatives in repair of damaged neuronal circuitry. While stem cells often are unable to regenerate lost neurons and glia, they appear to secrete trophic factors that decrease inflammation and promote endogenous compensatory growth. Whether ASD may benefit from this type of mechanisms, however, is unknown.
Currently, among classes of stem cells, mesenchymal stem cells (MSCs) seem to hold the most promise for cell therapies for ASD, likely owing to their immunomodulatory capacities. Furthermore, MSCs have a rapid self-renewal proliferation rate, do not elicit a host versus graft rejection upon transplantation, and are non-tumorigenic. It is hypothesized that they would function in ASD treatment through the secretion of anti-inflammatory cytokines and survival-promoting growth factors and integrating into neural networks and restoring plasticity [90,91]. Interestingly, several groups have implicated immune dysregulation in ASD patients [76,92-96]. There are, however, no pre-clinical studies of the use of MSCs in ASD models. Early clinical trials of human cord blood mononuclear cell (CBMNCs) and/or human umbilical cord-derived mesenchymal stem cells (UCMSCs) transplantation have shown some efficacy in treating ASD [97].
Neural stem cells (NSCs) or neural progenitor cells (NPCs) have proved useful in other CNS disease models due to their potential to integrate and replace damaged neural tissue and reconstruct neuronal circuitry. However, these potential mechanisms of benefit do not necessarily concur with our hypotheses of the cause of ASD, i.e., global hyperconnectivity, increased synaptic translation, and E-I balance. Embryonic stem cell/induced pluripotent stem cell (ESC/iPSC) transplantation seems unlikely to have clinical benefit in ASD patients for the same the reason: Damaged neural tissue/neuronal loss does not seem to be a prevailing mechanism in ASD pathogenesis. Therefore, in summary, the normal regenerative and restorative beneficial capacities of stem cells are largely irrelevant in ASD treatment. Whether the immunomodulatory properties of other mesenchymal-related stem cells are useful remains to be seen.
In Vitro Stem Cell Models of ASD
Despite the caveats described above, iPSCs have been used to model human neurological and psychiatric disorders. With particular relevance for the ASD field, studies have used iPSC to model monogenic disorders such as Rett syndrome, Fragile X syndrome, and Timothy syndrome. In these studes, one or more aberrant developmental effects thought to arise from the mutation were observed in iPSC-derived neuronal cells, allowing to screen for agents able to revert, or compensate for, the aberrant phenotype. Although there is no guarantee that agents active in vitro are going to be beneficial for the disorder in vivo, iPSCs offer the distinct advantage (with respect to animals) of modeling a disorder within the human genetic background. As such, iPSCs as a neurological disorder model system are already proving quite useful in further elucidating the pathogenesis of ASD and in the future may be valuable in ASD drug discovery efforts. Several studies have examined iPSC models of Rett syndrome [98-100]. These studies collectively show reduced spine density, synapse number, soma size, nuclear size, neuronal soma size, and expression of mature neuronal cellular markers. They also show alterations in calcium signaling and electrophysiological properties. These findings continue to echo those from genetic, neuropathological, and neuroimaging studies — evidence of an E-I imbalance and alterations in synaptic plasticity and function. While they are consistent among themselves regarding the direction of these abnormalities, they cannot be used as any indication of the direction in the context of idiopathic ASD, given the several genotypic and phenotypic differences between the disorders.
Sheridan et al. and Liu et al. generated Fragile X syndrome iPSC models and showed fewer and shorter neurites, decreased synaptic proteins and density, alterations in calcium signaling, and response to glutamate uptake [101,102]. Interestingly, Pasca et al., in a Timothy syndrome iPSC model, also show impairments in calcium signaling and electrophysiology, as well as abnormalities in activity-dependent gene expression, differentiation, and production of norepinephrine and dopamine (increased) [103]. It is important to remember that in Rett syndrome, Fragile X syndrome, and Timothy syndrome, the autism appears as part of a larger disorder that arises as a consequence of a mutation in a single gene or a set of penetrant gene mutations. Similarly, their etiology is likely to only moderately resemble that of ASD; however, it is surprising that Fragile X syndrome and Rett syndrome, two phenotypically very different disorders, consistently show decreased synaptic density and number in early brain development. To date, no successful studies of iPSC models of idiopathic/non-syndromic autism have been completed. Zeng et al. examined the NRXN1 rare mutation-associated form of autism through a NRXN1 knockdown iPSC model. Their results, in accordance with syndromic autism studies, found disruptions in several neurodevelopmental processes, including synaptic adhesion and neuron differentiation [104]. Finally, it is important to note that the monolayer preps used in many of these studies are limited in terms of their differentiation potential. For example, these studies have not “validated” their differentiation model by showing overall similarity of gene/protein expression to a particular brain region. This may suggest that the preparations have low complexity of neuronal phenotypes, which in turn limits the amount of information they can provide.
In conclusion, disease modeling through iPSCs presents formidable challenges. First and foremost is the difficulty in modeling normal and diseased cell-to-cell connectivity and functional neuronal networks in vitro. Indeed, while excitatory forebrain neurons can be differentiated from iPSC lines, the generation of the large variety of inhibitory forebrain neurons has not yet been achieved, most likely because of their extended period of maturation. Synaptic connections among excitory cells are more akin to reproducing epileptiform activity than functioning as a self-regulated cortical neuronal network. Carefully designed transplantation experiments may be able to overcome, in part, these difficulties. Second, in order to model disorders of multifactorial genetic etiology with a strong epigenetic component, a large number of patients may be needed. Furthermore, we still have limited understanding of the systemic and environmental hormonal influences on epigenetic regulation in normal and abnormal brain development. While iPSCs offer the exceptional promise of identifying disease-specific disruptions in cellular and molecular aspects of brain development, their application to the study of neuropsychiatric disorders is still in its infancy.
Applications of New Technologies to Stem Cell Models
One of the most noticeable advantages of iPSCs over post-mortem brain tissue is availability, as brain cells/tissue can be generated from any individual, thus allowing access to a wider choice of experimental designs. A major unique possibility is to follow individual early stages of brain development, using a combination of molecular and cellular assays at various time points to shed light on the specific developmental trajectory (albeit limited to early stages) and the associated transcriptional regulatory machinery driving it. The “omics revolution,” with the advent of the next generation sequencing at relatively low cost, has opened the way to the simultaneous analysis of whole genome features (i.e., coding and non-coding transcripts, binding sites, open chromatin regions, proteins, metabolites, and so on), allowing a snapshot of the molecular status of the cells. Perturbation experiments can be designed, for instance, by RNA interference to introduce deviations in the developmental trajectory of differentiating hiPSCs from either healthy controls or affected individual to assess the role of specific genes and identify their direct and indirect targets. Also interesting is the possibility of “accurate” genome editing by the recently established CRISPR technology [105-107]. It is now feasible to correct potential disease-causing mutations in iPSCs from disease-affected individuals, or, in the opposite direction, to engineer mutations, in isolation or in combination, in iPSCs from normal controls. These models can then be used for assessing at cellular and molecular levels the resulting phenotype to identify critical nodes (e.g., genes) in the disease network. Work in this direction already has been undertaken in the context of several disorders [105-107].
Drug testing is the next logical step, after the discussed manipulations and analysis strategies have highlighted critical nodes in the disease network.
It should be pointed out that the availability of iPSCs-derived neuronal progenitors or more differentiated neurons is still somewhat limited by the non-negligible reprogramming and differentiation costs, which adds up to any cost associated to genomics analysis. This limits the number of samples that can be analyzed and, consequently, the power for inferring, for instance, transcriptional alterations in patients. In order to (partially) overcome such limitations, our lab exploited the fact that iPSCs can be generated from cells (e.g., fibroblasts) collected from full families encompassing an affected individual, thus allowing statistical tests in a matched pair design, with the pair being the affected individual and its unaffected parent/sibling. This allows, to some extent, to control for the genetic background, and it resulted in a winning approach as we successfully inferred statistically significant differences, at cellular and molecular level, using only four families [unpublished data].
Conclusions and Outlook
Developmental neurobiology has advanced tremendously in the last 25 years, largely through the painstaking analysis of single gene loss and gain of function in several animal models. The genetic revolution has uncovered the mechanisms of many essential processes like the formation of the vertebrate body plan, and the principles by which stem cells differentiate into many cell types. Much still remains to be done. However, as we treasure this body of knowledge, we now realize that to understand the biology of neurodevelopmental biology such as autism, we need a drastic change of approach. Autism and similar complex neurodevelopmental brain disorders are likely to arise as a by-product of the inherent complexity of our brain, as well as subtle inter-individual variations in the processes that build neuronal circuitry. As such, it would be difficult to model these disorders in animals that lack such neuroanatomical and neurophysiological complexity, and inevitably, we will need multiple approaches, including ways to model and manipulate human-specific brain systems. The creation of iPSCs represents a first step in this direction. Necessary future advances will hopefully include the development of organoid systems able to reproduce regions or circuits of the human brain, such as a mini-cortical column circuit with its core of excitatory pyramidal cells and inhibitory neuron component, connected in the appropriate fashion. These are difficult yet not impossible goals, but require inducing in iPSC the appropriate genetic programs generating these cell types in their normal ratio. In the normal brain, such programs do not exist in isolation, but cross-regulate each other, often with reciprocal inhibition. For this reason, the directed differentiation approach may not be successful in recapitulating this complexity.
Another realization is that we need to shift our approach from investigating single genes to investigating how multiple genes interact with one another, needing the creation of tools for the generation and analysis of large datasets. The advent of next generation sequencing, another recent technological advance, is having a transformative effect on the biomedical field. We believe that collaboration and cooperation between scientists at multiple level and across disciplines, including genetics, developmental biology, neurobiology, and bioinformatics, will be essential to understanding the pathways underlying the pathogenesis of autism and lead to rational treatment strategies.
Abbreviations
- ASD
autism spectrum disorder
- iPSCs
induced pluripotent stem cells
- E-I
excitation-inhibition
- CNVs
copy number variations
- GWAS
genome-wide association
- PPI
protein-protein interaction
- FMRP
Fragile X syndrome protein
- hiPSCs
human-induced pluripotent stem cells
- CBMNC
human cord blood mononuclear cell
- UCMSCs
human umbilical cord-derived mesenchymal stem cells
- NSCs
neural stem cells
- NPCs
neural progenitor cells
- MSC
mesenchymal stem cell
- ESC-iPSC
embryonic stem cell/induced pluripotent stem cell
- DTI
diffusion tensor imaging
- CNS
central nervous system
- LM-CNV
lineage manifested copy number variation
- qPCR
quantitative polymerase chain reaction
- SV
structural variation
References
- Prevalence of autism spectrum disorder among children aged 8 years — autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63(2):1–21. [PubMed] [Google Scholar]
- Volkmar FR, McPartland JC. From Kanner to DSM-5: autism as an evolving diagnostic concept. Annu Rev Clin Psychol. 2014;10:193–212. doi: 10.1146/annurev-clinpsy-032813-153710. [DOI] [PubMed] [Google Scholar]
- Santini E, Klann E. Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci Signal. 2014;7(349):re10. doi: 10.1126/scisignal.2005832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: a decade of new twin studies. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(3):255–274. doi: 10.1002/ajmg.b.31159. [DOI] [PubMed] [Google Scholar]
- Ozonoff S, Young GS, Carter A, Messinger D, Yirmiya N, Zwaigenbaum L. et al. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics. 2011;128(3):e488–e495. doi: 10.1542/peds.2010-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders. Annu Rev Genomics Hum Genet. 2013;14:191–213. doi: 10.1146/annurev-genom-091212-153431. [DOI] [PubMed] [Google Scholar]
- Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB. et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46(8):881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl S, Chiang C, Beauchamp RL, Neale BM, Daly MJ, Gusella JF. et al. Lack of association of rare functional variants in TSC1/TSC2 genes with autism spectrum disorder. Mol Autism. 2013;4(1):5. doi: 10.1186/2040-2392-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC. et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34(1):27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP. et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74(3):552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ. et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39(3):319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S. et al. SHANK1 Deletions in Males with Autism Spectrum Disorder. Am J Hum Genet. 2012;90(5):879–887. doi: 10.1016/j.ajhg.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS, Heinrich J, Delorme R, Proepper C, Betancur C, Huguet G. et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet. 2012;8(2):e1002521. doi: 10.1371/journal.pgen.1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S. et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: a gradient of severity in cognitive impairments. PLoS Genet. 2014;10(9):e1004580. doi: 10.1371/journal.pgen.1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F. et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39(1):25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JA, Staal WG, van Daalen E, van Engeland H, Hochstenbach PF, Franke L. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry. 2006;11(1):18–28. doi: 10.1038/sj.mp.4001781. [DOI] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG. et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338(6114):1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J. et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74(2):285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A. et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149(3):525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S. et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ. et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V. et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155(5):1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA. et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155(5):997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S. et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135(3):401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Wang H, Doering LC. Reversing autism by targeting downstream mTOR signaling. Front Cell Neurosci. 2013;7:28. doi: 10.3389/fncel.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455(7215):903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature. 2011;480(7375):63–68. doi: 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14(3):285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A. et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature. 2012;486(7402):256–260. doi: 10.1038/nature11015. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM. et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318(5847):71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro R, Konyukh M, Delorme R, Leblond C, Chaste P, Fauchereau F. et al. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26(8):363–372. doi: 10.1016/j.tig.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Ecker C, Murphy D. Neuroimaging in autism--from basic science to translational research. Nat Rev Neurol. 2014;10(2):82–91. doi: 10.1038/nrneurol.2013.276. [DOI] [PubMed] [Google Scholar]
- Lombardo MV, Chakrabarti B, Bullmore ET, Baron-Cohen S. Specialization of right temporo-parietal junction for mentalizing and its relation to social impairments in autism. Neuroimage. 2011;56(3):1832–1838. doi: 10.1016/j.neuroimage.2011.02.067. [DOI] [PubMed] [Google Scholar]
- Langen M, Leemans A, Johnston P, Ecker C, Daly E, Murphy CM. et al. Fronto-striatal circuitry and inhibitory control in autism: findings from diffusion tensor imaging tractography. Cortex. 2012;48(2):183–193. doi: 10.1016/j.cortex.2011.05.018. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci. 2008;31(3):137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Courchesne E. Abnormal early brain development in autism. Mol Psychiatry. 2002;7 Suppl 2:S21–S23. doi: 10.1038/sj.mp.4001169. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Karns CM, Davis HR, Ziccardi R, Carper RA, Tigue ZD. et al. Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology. 2001;57(2):245–254. doi: 10.1212/wnl.57.2.245. [DOI] [PubMed] [Google Scholar]
- Lainhart JE, Bigler ED, Bocian M, Coon H, Dinh E, Dawson G. et al. Head circumference and height in autism: a study by the Collaborative Program of Excellence in Autism. Am J Med Genet A. 2006;140(21):2257–2274. doi: 10.1002/ajmg.a.31465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks BF, Friedman DD, Shaw DW, Aylward EH, Echelard D, Artru AA. et al. Brain structural abnormalities in young children with autism spectrum disorder. Neurology. 2002;59(2):184–192. doi: 10.1212/wnl.59.2.184. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Carper R, Akshoomoff N. Evidence of brain overgrowth in the first year of life in autism. JAMA. 2003;290(3):337–344. doi: 10.1001/jama.290.3.337. [DOI] [PubMed] [Google Scholar]
- Dawson G, Munson J, Webb SJ, Nalty T, Abbott R, Toth K. Rate of head growth decelerates and symptoms worsen in the second year of life in autism. Biol Psychiatry. 2007;61(4):458–464. doi: 10.1016/j.biopsych.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dementieva YA, Vance DD, Donnelly SL, Elston LA, Wolpert CM, Ravan SA. Accelerated head growth in early development of individuals with autism. Pediatr Neurol. 2005;32(2):102–108. doi: 10.1016/j.pediatrneurol.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Hazlett HC, Poe M, Gerig G, Smith RG, Provenzale J, Ross A. et al. Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Arch Gen Psychiatry. 2005;62(12):1366–1376. doi: 10.1001/archpsyc.62.12.1366. [DOI] [PubMed] [Google Scholar]
- Hazlett HC, Poe MD, Gerig G, Styner M, Chappell C, Smith RG. et al. Early brain overgrowth in autism associated with an increase in cortical surface area before age 2 years. Arch Gen Psychiatry. 2011;68(5):467–476. doi: 10.1001/archgenpsychiatry.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert MR, Ziegler DA, Deutsch CK, O’Brien LM, Lange N, Bakardjiev A. et al. Dissociations of cerebral cortex, subcortical and cerebral white matter volumes in autistic boys. Brain. 2003;126(Pt 5):1182–1192. doi: 10.1093/brain/awg110. [DOI] [PubMed] [Google Scholar]
- Palmen SJ, Hulshoff Pol HE, Kemner C, Schnack HG, Durston S, Lahuis BE. et al. Increased gray-matter volume in medication-naive high-functioning children with autism spectrum disorder. Psychol Med. 2005;35(4):561–570. doi: 10.1017/s0033291704003496. [DOI] [PubMed] [Google Scholar]
- Lotspeich LJ, Kwon H, Schumann CM, Fryer SL, Goodlin-Jones BL, Buonocore MH. et al. Investigation of neuroanatomical differences between autism and Asperger syndrome. Arch Gen Psychiatry. 2004;61(3):291–298. doi: 10.1001/archpsyc.61.3.291. [DOI] [PubMed] [Google Scholar]
- Hazlett HC, Poe MD, Gerig G, Smith RG, Piven J. Cortical gray and white brain tissue volume in adolescents and adults with autism. Biol Psychiatry. 2006;59(1):1–6. doi: 10.1016/j.biopsych.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Carper RA, Courchesne E. Localized enlargement of the frontal cortex in early autism. Biol Psychiatry. 2005;57(2):126–133. doi: 10.1016/j.biopsych.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Herbert MR, Ziegler DA, Makris N, Filipek PA, Kemper TL, Normandin JJ. et al. Localization of white matter volume increase in autism and developmental language disorder. Ann Neurol. 2004;55(4):530–540. doi: 10.1002/ana.20032. [DOI] [PubMed] [Google Scholar]
- Pugliese L, Catani M, Ameis S, Dell’Acqua F, Thiebaut de Schotten M, Murphy C. et al. The anatomy of extended limbic pathways in Asperger syndrome: a preliminary diffusion tensor imaging tractography study. Neuroimage. 2009;47(2):427–434. doi: 10.1016/j.neuroimage.2009.05.014. [DOI] [PubMed] [Google Scholar]
- Shukla DK, Keehn B, Muller RA. Tract-specific analyses of diffusion tensor imaging show widespread white matter compromise in autism spectrum disorder. J Child Psychol Psychiatry. 2011;52(3):286–295. doi: 10.1111/j.1469-7610.2010.02342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshino H, Carpenter PA, Minshew NJ, Cherkassky VL, Keller TA, Just MA. Functional connectivity in an fMRI working memory task in high-functioning autism. Neuroimage. 2005;24(3):810–821. doi: 10.1016/j.neuroimage.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Koshino H, Kana RK, Keller TA, Cherkassky VL, Minshew NJ, Just MA. fMRI investigation of working memory for faces in autism: visual coding and underconnectivity with frontal areas. Cereb Cortex. 2008;18(2):289–300. doi: 10.1093/cercor/bhm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker C, Ronan L, Feng Y, Daly E, Murphy C, Ginestet CE. et al. Intrinsic gray-matter connectivity of the brain in adults with autism spectrum disorder. Proc Natl Acad Sci USA. 2013;110(32):13222–13227. doi: 10.1073/pnas.1221880110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page LA, Daly E, Schmitz N, Simmons A, Toal F, Deeley Q. et al. In vivo 1H-magnetic resonance spectroscopy study of amygdala-hippocampal and parietal regions in autism. Am J Psychiatry. 2006;163(12):2189–2192. doi: 10.1176/appi.ajp.163.12.2189. [DOI] [PubMed] [Google Scholar]
- Mendez MA, Horder J, Myers J, Coghlan S, Stokes P, Erritzoe D. et al. The brain GABA-benzodiazepine receptor alpha-5 subtype in autism spectrum disorder: a pilot [(11)C]Ro15-4513 positron emission tomography study. Neuropharmacology. 2013;68:195–201. doi: 10.1016/j.neuropharm.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Halt AR, Stary JM, Kanodia R, Schulz SC, Realmuto GR. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry. 2002;52(8):805–810. doi: 10.1016/s0006-3223(02)01430-0. [DOI] [PubMed] [Google Scholar]
- Horder J, Lavender T, Mendez MA, O’Gorman R, Daly E, Craig MC. et al. Reduced subcortical glutamate/glutamine in adults with autism spectrum disorders: a [(1)H]MRS study. Transl Psychiatry. 2013;3:e279. doi: 10.1038/tp.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman ML, Kemper TL. Histoanatomic observations of the brain in early infantile autism. Neurology. 1985;35:866–874. doi: 10.1212/wnl.35.6.866. [DOI] [PubMed] [Google Scholar]
- Kemper TL, Bauman ML. The contribution of neuropathologic studies to the understanding of autism. Neurol Clin. 1993;11(1):175–187. [PubMed] [Google Scholar]
- Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum. 2008;7(3):406–416. doi: 10.1007/s12311-008-0043-y. [DOI] [PubMed] [Google Scholar]
- Simms ML, Kemper TL, Timbie CM, Bauman ML, Blatt GJ. Altered posterior cingulate cortical cyctoarchitecture, but normal density of neurons and interneurons in the posterior cingulate cortex and fusiform gyrus in autism. Acta Neuropathol. 2009;118(5):673–684. [Google Scholar]
- Oblak AL, Rosene DL, Kemper TL, Bauman ML, Blatt GJ. Altered posterior cingulate cortical cyctoarchitecture, but normal density of neurons and interneurons in the posterior cingulate cortex and fusiform gyrus in autism. Autism Res. 2011;4(3):200–211. doi: 10.1002/aur.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S. et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med. 2014;370(13):1209–1219. doi: 10.1056/NEJMoa1307491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova MF, El-Baz AS, Kamat SS, Dombroski BA, Khalifa F, Elnakib A. et al. Focal cortical dysplasias in autism spectrum disorders. Acta Neuropathol Commun. 2013;1(1):67. doi: 10.1186/2051-5960-1-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova MF. White matter volume increase and minicolumns in autism. Ann Neurol. 2004;56(3):453, author reply 454. doi: 10.1002/ana.20196. [DOI] [PubMed] [Google Scholar]
- Casanova MF, van Kooten IA, Switala AE, van Engeland H, Heinsen H, Steinbusch HW. et al. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006;112(3):287–303. doi: 10.1007/s00401-006-0085-5. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ. et al. Neuron number and size in prefrontal cortex of children with autism. JAMA. 2011;306(18):2001–2010. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- Lawrence YA, Kemper TL, Bauman ML, Blatt GJ. Parvalbumin-, calbindin-, and calretinin-immunoreactive hippocampal interneuron density in autism. Acta Neurol Scand. 2010;121(2):99–108. doi: 10.1111/j.1600-0404.2009.01234.x. [DOI] [PubMed] [Google Scholar]
- Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J. et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry. 2010;68(4):368–376. doi: 10.1016/j.biopsych.2010.05.024. [DOI] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57(1):67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A. et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron. 2014;83(5):1131–1143. doi: 10.1016/j.neuron.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F. et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26(11):1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J. et al. Generation of human induced pluripotent stem cells from urine samples. Nature Protocols. 2012;7(12):2080–2089. doi: 10.1038/nprot.2012.115. [DOI] [PubMed] [Google Scholar]
- Abyzov A, Mariani J, Palejev D, Zhang Y, Haney MS, Tomasini L. et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492(7429):438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens HE, Mariani J, Coppola G, Vaccarino FM. Neurobiology meets genomic science: the promise of human-induced pluripotent stem cells. Dev Psychopathol. 2012;24(4):1443–1451. doi: 10.1017/S095457941200082X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino FM, Stevens HE, Kocabas A, Palejev D, Szekely A, Grigorenko EL. et al. Induced pluripotent stem cells: A new tool to confront the challenge of neuropsychiatric disorders. Neuropharmacology. 2011;60(7-8):1355–1363. doi: 10.1016/j.neuropharm.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino FM, Urban AE, Stevens HE, Szekely A, Abyzov A, Grigorenko EL. et al. Annual Research Review: The promise of stem cell research for neuropsychiatric disorders. J Child Psychol Psychiatry. 2011;52(4):504–516. doi: 10.1111/j.1469-7610.2010.02348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani J, Simonini MV, Palejev D, Tomasini L, Coppola G, Szekely AM. et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci USA. 2012;109(31):12770–12775. doi: 10.1073/pnas.1202944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas CR, Chen J, Tang Y, Southwell DG, Chalmers N, Vogt D. et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell. 2013;12(5):573–586. doi: 10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espuny-Camacho I, Michelsen KA, Gall D, Linaro D, Hasche A, Bonnefont J. et al. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron. 2013;77(3):440–456. doi: 10.1016/j.neuron.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME. et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nat Biotechnol. 2014;32(8):760–772. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- Siniscalco D, Bradstreet JJ, Sych N, Antonucci N. Perspectives on the use of stem cells for autism treatment. Stem Cells Int. 2013;2013:262438. doi: 10.1155/2013/262438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniscalco D, Sapone A, Cirillo A, Giordano C, Maione S, Antonucci N. Autism spectrum disorders: is mesenchymal stem cell personalized therapy the future? J Biomed Biotechnol. 2012;2012:480289. doi: 10.1155/2012/480289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniscalco D, Bradstreet JJ, Antonucci N. Therapeutic role of hematopoietic stem cells in autism spectrum disorder-related inflammation. Front Immunol. 2013;4:140. doi: 10.3389/fimmu.2013.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Aggarwal S, Heads C. Dysregulated immune system in children with autism: beneficial effects of intravenous immune globulin on autistic characteristics. J Autism Dev Disord. 1996;26(4):439–452. doi: 10.1007/BF02172828. [DOI] [PubMed] [Google Scholar]
- Ashwood P, Wills S, Van de Water J. The immune response in autism: a new frontier for autism research. J Leukoc Biol. 2006;80(1):1–15. doi: 10.1189/jlb.1205707. [DOI] [PubMed] [Google Scholar]
- Jyonouchi H, Sun S, Le H. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. J Neuroimmunol. 2001;120(1-2):170–179. doi: 10.1016/s0165-5728(01)00421-0. [DOI] [PubMed] [Google Scholar]
- Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K. et al. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis. 2008;30(3):303–311. doi: 10.1016/j.nbd.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv YT, Zhang Y, Liu M, Qiuwaxi JN, Ashwood P, Cho SC. et al. Transplantation of human cord blood mononuclear cells and umbilical cord-derived mesenchymal stem cells in autism. J Transl Med. 2013;11:196. doi: 10.1186/1479-5876-11-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y. et al. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell. 2010;143(4):527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AY, Horvath LM, Carrel L, Ellis J. X-chromosome inactivation in rett syndrome human induced pluripotent stem cells. Front Psychiatry. 2012;3:24. doi: 10.3389/fpsyt.2012.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananiev G, Williams EC, Li H, Chang Q. Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS One. 2011;6(9):e25255. doi: 10.1371/journal.pone.0025255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L. et al. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 2011;6(10):e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Koscielska KA, Cao Z, Hulsizer S, Grace N, Mitchell G. et al. Signaling defects in iPSC-derived fragile X premutation neurons. Hum Mol Genet. 2012;21(17):3795–3805. doi: 10.1093/hmg/dds207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM. et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17(12):1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Zhang P, Shi L, Yamamoto V, Lu W, Wang K. Functional impacts of NRXN1 knockdown on neurodevelopment in stem cell models. PLoS One. 2013;8(3):e59685. doi: 10.1371/journal.pone.0059685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T. et al. Precise Correction of the Dystrophin Gene in Duchenne Muscular Dystrophy Patient Induced Pluripotent Stem Cells by TALEN and CRISPR-Cas9. Stem Cell Reports. 2015;4(1):143–154. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Suzuki K, Kim NY, Liu GH, Izpisua Belmonte JC. A cut above the rest: targeted genome editing technologies in human pluripotent stem cells. J Biol Chem. 2014;289(8):4594–4599. doi: 10.1074/jbc.R113.488247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Abalde-Atristain L, He C, Brodsky BR, Braunstein EM, Chaudhari P. et al. Efficient and Allele-Specific Genome Editing of Disease Loci in Human iPSCs. Mol Ther. 2014 Nov 24; doi: 10.1038/mt.2014.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera M, Mirotsou M. Stem cells: potential and challenges for kidney repair. Am J Physiol Renal Physiol. 2014;3-6(1):F12–F23. doi: 10.1152/ajprenal.00238.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]