

Abstract
This study reports on two individuals with Temple–Baraitser syndrome, manifesting typical hallux and pollex findings, global developmental delay, and seizures. In the five previous cases identified to date, consistent craniofacial and osseous characteristics have been observed. The children described herein exhibit minor differences within this phenotype and are older, highlighting the phenotypic variability and natural history of the clinical and radiographic findings.
Keywords: abnormalities of hallux and pollex, developmental delay disorders, muscular diseases, seizures, Temple–Baraitser syndrome
Introduction
Temple–Baraitser syndrome (TBS, OMIM #611816) was first described in 1991, with the main distinguishing feature being the appearance of the thumbs and great toes (Temple and Baraitser, 1991). Each of the four cases subsequently reported showed consistent findings of severe developmental delay and characteristic craniofacial dysmorphisms and seizures, in addition to the pollex and hallux abnormalities (Gabbett et al., 2008; Jacquinet et al., 2010; Yesil et al., 2014). The thumbs are broad and the great toes are elongated, with the nails either hypoplastic or completely absent. The distal phalangeal bone of these digits exhibit unusual radiographic features, including central lucencies, possible pseudoepiphyses, and missing proximal epiphyses. The two children presented in this report are older and have undergone serial imaging, allowing further characterization of the TBS phenotype.
Clinical reports
Patient 1
She was born at term, weighing 7 pounds and 7 ounces, to an 18-year-old primigravida mother after an uncomplicated pregnancy and delivery. There was no family medical history of neurodevelopmental concerns and the parents were nonconsanguineous. She was first evaluated at 9 months of age because of developmental delay. Her length was 74 cm (95th centile), weight was 8 kg (25th centile), and head circumference was 44 cm (50th centile). Examination showed several dysmorphic features including low anterior hairline, tented vermilion of the upper lip, high palate, and tapering thumbs with near absence of nails on her thumbs and great toes. There was generalized hypotonia. Her development proceeded slowly, with skills such as walking, babbling in consonants, and bringing food to mouth occurring just before 6 years of age. She had one seizure when she was 8 years old. At her last evaluation at 9 years of age, she still had no words and her growth parameters have remained within the normal range. Examination showed a myopathic-appearing face, hypertelorism, a wide nasal bridge, thick alae nasi, slightly low hanging but normal insertion of the columella, full cheeks, everted and thick vermilion of both the upper and lower lips, and a long philtrum (Fig. 1). The thumbs were bulbous distally with near absence of nails. The great toes were longer in appearance and the nails were nearly absent. The remainder of the digits and nails appeared normal. Hand and foot films showed that the distal phalangeal bone of all of the first digits exhibited absence of the secondary ossification center (Fig. 2). The phalangeal bones overall were thin, especially distally, with the exception of the distal phalange of the halluces, which was wider and rounder. She has had a normal brain MRI and single nucleotide polymorphism chromosome microarray.
Fig. 1.
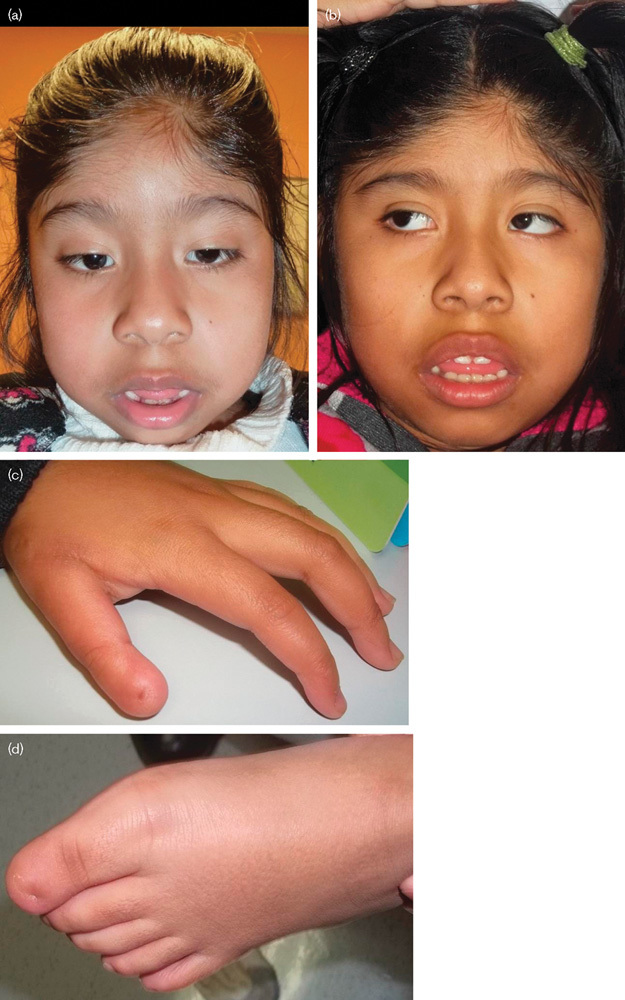
Patient 1 (a) at 5 years of age and (b) at 9 years of age. Note the myopathic-appearing face, low anterior hairline, wide nasal bridge, thick alae nasi, everted and thick vermilion of both the upper and the lower lips. (c) Left hand, (d) left foot; the thumb is bulbous distally and the great toe is elongated; there is near absence of the nail on the first rays only, and the other digits and nails are normal.
Fig. 2.
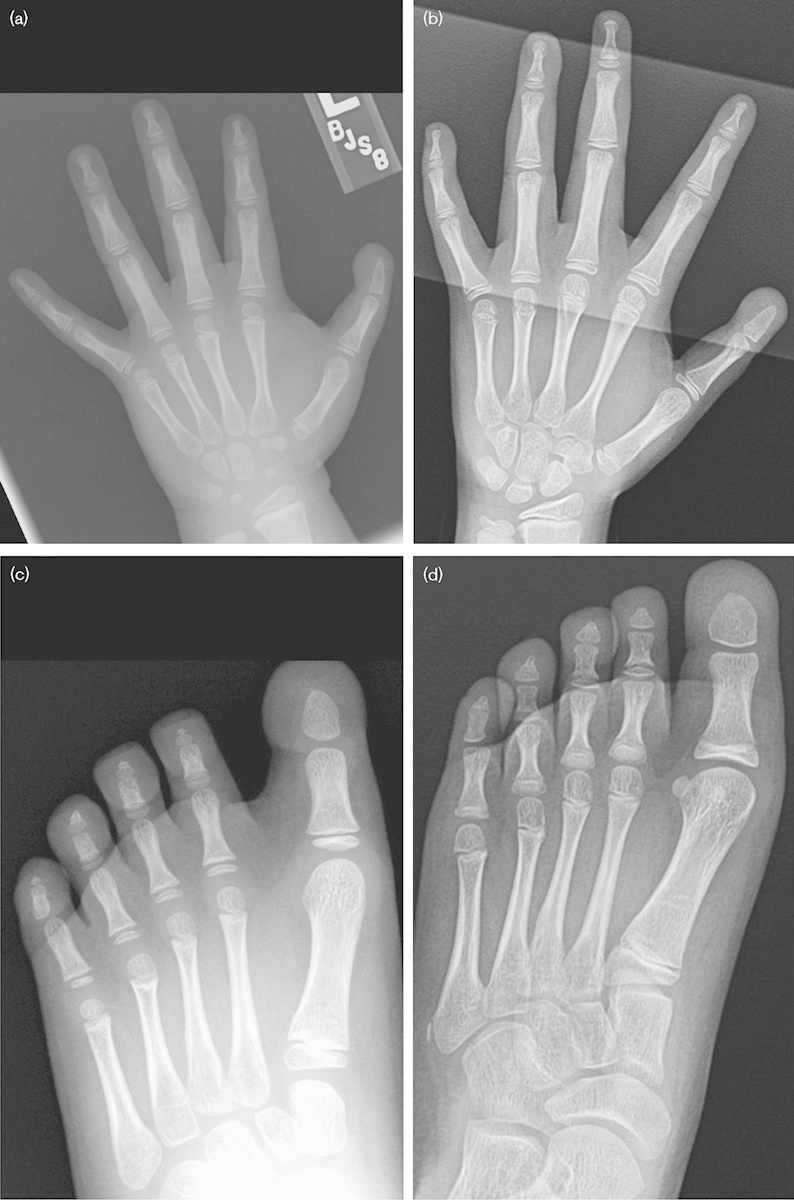
Radiographs of patient 1: (a) left hand at 5 years, (b) left hand at 9 years. Note the abnormal distal phalange of the thumb that is missing the proximal ossification center. (c) Left foot at 5 years, (d) left foot at 9 years; the distal phalange of the great toe has a wide and rounded appearance and lacks the proximal epiphysis.
Patient 2
She was born to a 22-year-old primigravida through a Cesarean section at term because of breech presentation, weighing 7 pounds and 13 ounces. Pregnancy history showed fetal phenytoin exposure (500 mg daily), but otherwise no other exposures nor complications. Family medical history was positive for maternal seizures but otherwise negative for neurodevelopmental concerns, and the parents denied consanguinity. She first presented at 5.5 years of age because of developmental delay and seizures starting at 4.5 years, which were under good control on topiramate and carbamazepine. She had been considered hypotonic since early during infancy, first holding her head up at 8 months, sitting at 12 months, and walking at 3 years and 10/12 months. By 5 years of age she started using single words and was able to feed herself using utensils. Her height was 105 cm (10th centile), weight was 17.6 kg (25th centile), and head circumference was 51 cm (50th centile). Examination showed a high anterior hairline, full cheeks, anteverted nares with a long philtrum, and tented vermilion of the upper lip. All fingers showed smaller nails and hypoplasia of the distal segments, particularly the thumb, and all of her toes had near to true absence of nails. By 7 years of age, she could say two-word phrases. At 9.5 years of age she had a vocabulary of ∼40 words, followed a one-step command, walked up and down stairs, and rode a three-wheeled toy. Up to her most recent evaluation at 14 years, her height and weight have consistently ranged from the 5th to the 10th centile. Dysmorphic features observed at that time included a myopathic-appearing face, a wide and depressed nasal bridge, mild malar flattening, thick alae nasi, full cheeks, high insertion of the columella, tented vermilion of the upper lip, and everted thick vermilion of the lower lip (Fig. 3). Her extremity findings have remained consistent, with hypoplastic and somewhat dystrophic nails on her broad thumbs, nail hypoplasia of her fingers, and longer great toes with essentially absent nails on all of her toes. On radiographs, the distal phalangeal secondary ossification center of the thumbs and great toes were missing, there was a discontinuity in the distal phalange of the left thumb, and the distal phalange of the halluces were wider and rounder than normal (Fig. 4). For all of the remaining digits, the distal phalanges were hypoplastic. Studies performed included a normal brain MRI and a single nucleotide polymorphism microarray, which showed chromosome Xq28 duplication proximal to MECP2. This genomic imbalance was maternally inherited and encompassed seven genes that are not known individually nor collectively to have clinical effects when present in duplicate.
Fig. 3.
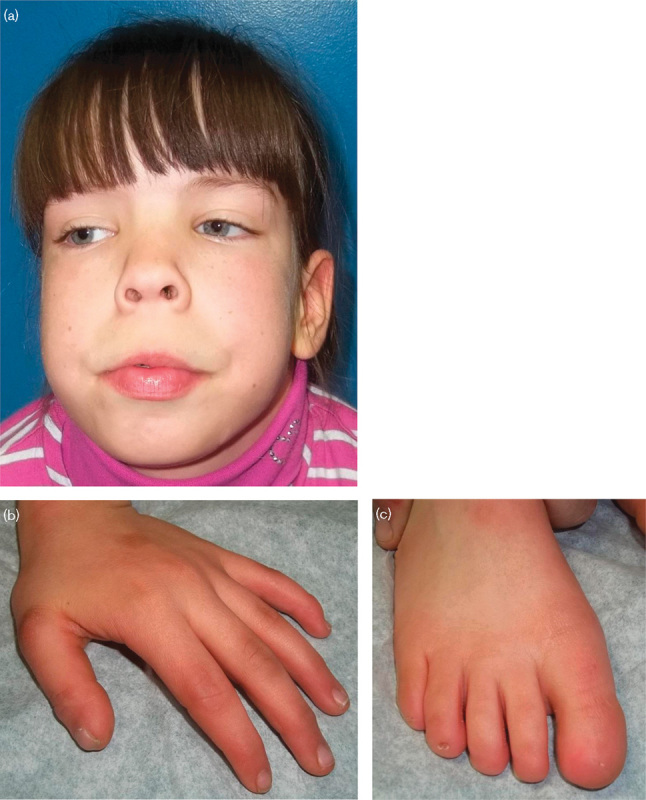
Patient 2 (a) at 14 years of age. Note the myopathic-appearing face, wide and depressed nasal bridge, anteverted nares with thick alae nasi, everted thick vermilion of the lower lip, and long philtrum. (b) Left hand: there is a smaller dystrophic nail on a broad thumb, and the remaining fingernails are hypoplastic. (c) Right foot: note the elongated hallux and near to true absence of toenails.
Fig. 4.
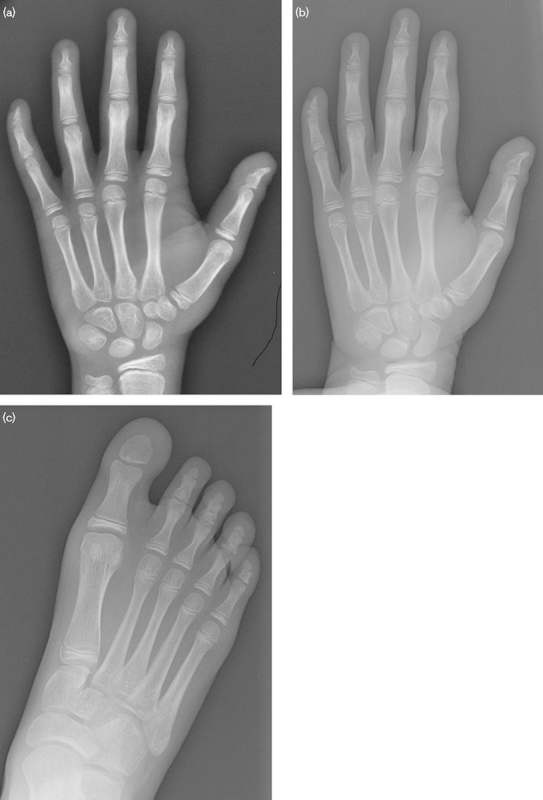
Radiographs of patient 2: (a) left hand at 12 years, (b) left hand at 14 years. Note the discontinuity in the distal phalange of the thumb and absence of the proximal ossification center. The distal phalanges of the other digits are hypoplastic. (c) Right foot at 14 years. The distal phalange of the great toe appears wider and rounder and is missing the proximal epiphysis.
Discussion
Seven cases of TBS, including the two cases in this report, have now been characterized. A remarkably similar set of dysmorphic features of the craniofacies and the extremities has emerged. There is a myopathic-appearing face with full cheeks, a wide and sometimes depressed nasal bridge, and lip findings. The pollex can either be broad or bulbous distally, whereas the hallux is elongated; the nails associated with these digits range from hypoplastic to completely absent. Radiographs of the extremities consistently show that the distal phalangeal bone of the first digits of both the hands and the feet are malformed, whereas the distal phalanges of the remaining digits are typically hypoplastic.
An appropriate set of alternative syndromes to consider has been discussed in previous articles on TBS and include deafness, onychodystrophy, osteodystrophy, mental retardation, seizures [DOOR(S)] syndrome and Lynch–Bushby syndrome. Mutations in TBC1D24 have been recently identified in DOOR(S) syndrome (Campeau et al., 2014), but the absence of deafness, coarse facial features, and neurologic deterioration rule against this diagnosis for the two patients described. In addition, the lack of choreoathetoid movements makes Lynch–Bushby syndrome unlikely (Lynch et al., 1997).
The two children presented herein are older, providing more information about the clinical variability and how the distinctive radiologic findings change over time in TBS. In both patients, skeletal maturity has advanced sufficiently, such that in the distal phalangeal bone of the first digit of all four extremities, the stark absence of the secondary ossification center is readily apparent, similar to that seen in patient 1 in the study by Jacquinet et al. (2010). In addition, the larger bone size accentuates the wide and rounded appearance of the distal phalangeal bone of the halluces. Developmental delay has been reported to be severe, with no language skills in any of the previously described individuals with TBS, with the oldest being 11 year of age (M. Gabbett, personal communication). In this report, patient 1 has the same degree of language delay; however, in contrast, patient 2 by the age of 12 years exhibited language at approximately the 2–3-year-old level.
Additional differences observed in patient 2 are the marked hypoplasia of her fingernails and the near to complete absence of her toenails. None of the other previously published cases of TBS showed this extent of nail involvement. These findings resemble those seen with fetal hydantoin exposure (Jones, 2006), and indeed patient 2 was given this diagnosis initially. Other evidence that could support this diagnosis is her mild growth deficiency and, although the absence of the proximal epiphyseal center of the pollex may be a more distinctive feature of TBS, this finding has also been observed rarely in presumed fetal hydantoin embryopathy (Wood and Young, 1979; Johnson and Goldsmith, 1981). However, the clinical diagnosis of TBS was made because her craniofacial features and hallux appearance more closely matched the observations made previously in other affected individuals with TBS, with this diagnosis validated molecularly upon the recent identification of the causative gene; both patients characterized in this report harbor pathogenic mutations (Simons et al., 2014). As more individuals affected with TBS are identified, whether these differences exhibited by patient 2 represent the influence of in-utero phenytoin exposure, perhaps similar to the case presented by Gabbett et al. (2008) with in-utero carbamazepine exposure, or represent further clinical variation within the TBS phenotype will be clarified.
In summary, two individuals with TBS are presented, with emphasis on the phenotypic variability and natural history. Highlighting the characteristic dysmorphic features, digital findings, and distinctive radiologic appearance of the distal extremities will hopefully allow more individuals to be diagnosed and help expand our understanding of this syndrome.
Acknowledgements
Conflicts of interest
There are no conflicts of interest.
References
- Campeau PM, Kasperaviciute D, Lu JT, Burrage LC, Kim C, Hori M, et al. (2014). The genetic basis of DOORS syndrome: an exome-sequencing study. Lancet Neurol 13:44–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbett MT, Clark RC, McGaughran JM. (2008). A second case of severe mental retardation and absent nails of hallux and pollex (Temple–Baraitser syndrome). Am J Med Genet A 146A:450–452. [DOI] [PubMed] [Google Scholar]
- Jacquinet A, Gérard M, Gabbett MT, Rausin L, Misson JP, Menten B, et al. (2010). Temple–Baraitser syndrome: a rare and possibly unrecognized condition. Am J Med Genet A 152A:2322–2326. [DOI] [PubMed] [Google Scholar]
- Johnson RB, Goldsmith LA. (1981). Dilantin digital defects. J Am Acad Dermatol 5:191–196. [DOI] [PubMed] [Google Scholar]
- Jones KL. Smith’s recognizable patterns of human malformation 6th ed. Philadelphia: WB Saunders; (2006). [Google Scholar]
- Lynch SA, Gardner-Medwin D, Burn J, Bushby KM. (1997). Absent nails, kinesogenic choreoathetosis, epilepsy and developmental delay – a new autosomal dominant disorder? Clin Dysmorphol 6:133–138. [PubMed] [Google Scholar]
- Simons C, Rash LD, Crawford J, Ma L, Cristofori-Armstrong B, Miller D, et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple–Baraitser syndrome and epilepsy. Nat Genet (2014). [Epub ahead of print] doi: 10.1038/ng.3153; PMID - 25420144. [DOI] [PubMed] [Google Scholar]
- Temple IK, Baraitser M. (1991). Severe mental retardation and absent nails of hallux and pollex. Am J Med Genet 41:173–175. [DOI] [PubMed] [Google Scholar]
- Wood BP, Young LW. (1979). Pseudohyperphalangism in fetal Dilantin syndrome. Radiology 131:371–372. [DOI] [PubMed] [Google Scholar]
- Yesil G, Guler S, Yuksel A, Alanay Y. (2014). Report of a patient with Temple–Baraitser syndrome. Am J Med Genet A 164A:848–851. [DOI] [PubMed] [Google Scholar]