

Abstract
The human immunodeficiency virus (HIV) envelope protein gp120 promotes neuronal injury which is believed to contribute to HIV-associated neurocognitive disorders. Therefore, blocking the neurotoxic effect of gp120 may lead to alternative strategies to reduce the neurotoxic effect of HIV. In vitro, the neurotoxic effect of M-tropic gp120BaL is reduced by the chemokine CCL5, the natural ligand of CCR5 receptors. To determine whether CCL5 reduces the toxic effect of gp120BaL in vivo, animals were intrastriatally injected with lentiviral vectors overexpressing CCL5 prior to an intrastriatal injection of gp120BaL (400 ng). Neuronal injury was determined by silver staining, cleaved caspase-3 and TUNEL. Overexpression of CCL5 decreased gp120-mediated neuronal injury. CCL5 expression can be up-regulated by chronic morphine. Therefore, we examined whether morphine reduces the neurotoxic effect of gp120BaL. Rats stereotaxically injected with gp120BaL into the striatum received saline or chronic morphine for five days (10 mg/kg escalating to 30 mg/kg twice a day). Morphine-treated rats showed a decrease in all markers used to determine neuronal degeneration compared to saline-treated rats. The neuroprotective effect of morphine was significantly attenuated by expressing CCL5 shRNA. Our results suggest that compounds that increase the endogenous production of CCL5 may be used to reduce the pathogenesis of HIV–associated neurocognitive disorders.
Keywords: caspase-3, CCL5-lentivirus, IL-1β, morphine withdrawal, neurodegeneration, neuroprotection
1. Introduction
HIV infection of glial cells is one of the leading causes for the development of HIV-associated neurocognitive disorder (HAND) (Gonzalez-Scarano and Martin-Garcia, 2005). HAND symptoms may include asymptomatic neurocognitive impairment, minor cognitive disorders or, in its more severe form, profound motor and behavioral/psychosocial abnormalities (McArthur et al., 2005). In addition, despite the use of combined retroviral therapy, near 50% of HIV subjects remain neurocognitively impaired (Ellis et al., 2010). Neuropathological correlates of post-mortem brains point at synaptic simplification and neuronal loss in cortical and subcortical structures as the major causes of the neurological complications (Ellis et al., 2007). Thus, new adjunct therapies must be developed to provide a more efficient treatment of HIV neurotoxicity that prevents the loss of neurons.
The strain of HIV that is acquired and propagated during primary infection is the so called M-tropic strain because it infects primarily macrophages by utilizing the chemokine receptor CCR5 as co-receptor for entry (Deng et al., 1996; Gorry et al., 2001). However, HIV replicates in microglia and therefore is found in the brain of individuals with neurocognitive disorders (Peters et al., 2004; Smit et al., 2001). CCR5 is also expressed by neuronal cells and can induce neuronal apoptosis upon activation by the HIV envelope protein gp120 (Bachis et al., 2010; Kaul et al., 2007; Maung et al., 2014). Because neuronal apoptosis has been described in the brain of HIV subjects (Garden et al., 2002; Gelbard and Epstein, 1995), various strains of gp120 have also been used in rodents (Bachis et al., 2010; Bansal et al., 2000; Toggas et al., 1994) to reproduce neuronal loss observed in HAND subjects (Masliah et al., 1996).
Infection (Cocchi et al., 1995) and neurotoxicity (Bachis et al., 2009; Kaul et al., 2007) from M-tropic strains of HIV can be prevented by the chemokine CCL5, a CCR5 natural ligand. In addition, it has been shown that HIV-infected individuals with higher cerebrospinal fluid concentrations of CCL5 perform better on neuropsychological measures than those with low or undetectable levels (Letendre et al., 1999; Sozzani et al., 1997). CCL5, which was originally discovered for its ability to control immune cell migration to sites of injury, is constitutively expressed in non-pathological states in the brain and exerts neuroprotective activities in vitro. For instance, CCL5 promotes survival of human astrocytes (Bakhiet et al., 2001), inhibits the neurotoxic effect of amyloid-β peptide (Ignatov et al., 2006), a hallmark of Alzheimer's disease (Stokin et al., 2005), as well as glutamate toxicity (Bruno et al., 2000). Importantly, CCL5 has been shown to exhibit a neuroprotective profile against various HIV proteins including gp120 and Tat (Brenneman et al., 2002; Kaul and Lipton, 1999; Meucci et al., 1998; Rozzi et al., 2014). Therefore, CCL5 could be a valid adjunct therapy to prevent neurodegeneration caused by HIV. However, the neuroprotective properties of CCL5 need to be validated in vivo.
CCL5 is up-regulated by chronic morphine in the pre-frontal cortex and striatum (Campbell et al., 2013). In this study, we compare the effect of exogenously delivered CCL5 with that of endogenously produced by morphine on M-tropic gp120-mediated neuronal degeneration to determine whether CCL5 is neuroprotective in vivo. We show that CCL5, either delivered into the brain or induced by morphine inhibits gp120-mediated neuronal injury.
2. Materials and Methods
2.1. Transduction of HEK293FT
Human embryonic kidney cells line HEK293FT (Invitrogen, Grand Island, NY) were grown in high glucose Dulbecco's Modified Eagle Medium (DMEM) (MediaTech, Herdon, VA) supplemented with 10% fetal bovine serum (FBS) and 50U/ml penicillin, 50U/ml streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Cells were transfected with selected cDNA and lipoD293™ in Vitro DNA Transfection Reagent at a 3:1 ratio respectively. After 24 hr cell lysates were collected using 1× RIPA buffer with protease/phosphatase inhibitor (Thermo Scientific, Pittsburg, PA).
2.2 Lentiviral preparation
A recombinant lentivirus expressing CCL5-FLAG was generated by subcloning rat CCL5 cDNA (Origene, Rockville, MD) into the multiple cloning site of the pCDH vector (System Biosciences, Mountain View, CA). This vector also expresses green fluorescent protein (GFP) under the independent CMV promoter. CCL5-FLAG tag cDNA was created by polymerase chain reaction (PCR) using the primers described in Table 1. The resulting product was purified and ligated into the pCDH vector using Nhel and Notl restriction enzymes (Thermo Scientific). The orientation of CCL5-FLAG tag was examined by DNA sequencing. CCL5 shRNA constructs were generated by cloning CCL5 shRNA sequences into pGreenPuro™ shRNA cloning vector (System Biosciences). Three CCL5 shRNA's were created using the sequences described in Table 1 and were specific for rat CCL5. The sequences were replicated using PCR and ligated into the pGreenPuro™ lentiviral vector using BamHI and EcoRI (Thermo Scientific). All vectors were packaged into pseudoviral particles using an established protocol (Tiscornia et al., 2006) with minor modifications. In brief, HEK293FT cells were transfected with the expression construct (300 μg), packaging plasmids Gag (196 μg) and Rev (76 μg), and envelope vector VSV-G (106 μg) using calcium chloride. Media was collected and pseudovirus particles were concentrated by ultracentrifugation. Concentrated virus was further purified using a 20% sucrose gradient for in vivo grade product. Titer of lentiviral preparation was determined using Lenti-X™ p24 Rapid Titer Kit (Clontech, Mountain View, CA). Typical titer acquired is 4×108 TU/ml.
Table 1. List of primers.
| Type | Forward | Reverse |
|---|---|---|
| CCL5 subclone | 5′-GGGGCTAGCATGAAGATCTCCACA GCTGCA-3′ | 5′-GGGCGGCCGCCTACTTATCGTCGTCATC CTTGTAATCGCTCATCTCCAAATAGTTGAT-3′ |
| CCL5 shRNA-1 | 5′-Phos-CCAAGTGCTCCAACCTTGC AGTCGTCTTTGCTTCTTGTCAGAC AAAGACGACTGCAAGGTTGGAGCACT TGTTTTTG-3′ | 5′-ATTCAAAAACAAGTGCTCCAACCTT GCAGTCGTCTTTGTCTGACAAGAAGCAAAGAC GACTGCAAGGTTGGAGCACTTGG-3′ |
| CCL5 shRNA-2 | 5′-Phos-GATCCAACCTTGCAGTCGTCT TTGTCTTCCTCTCAGAACAAAGACGAC TGCAAGGTTTTTTTG-3′ | 5′-AATTCAAAAAAACCTTGCAGTCGTCTT TGTTCTGACAGGAAGACAAAGACGACTGCAAG GTTG-3′ |
| CCL5 shRNA-3 | 5′-Phos-GATCCGAAGTGGGTTCAAG AATACACTTCTTGTCAGATGTATTCTT GAACCCACTTCTTTTTG-3′ | 5′-AATTCAAAAAGAAGTGGGTTCAAGAA TACATCTGACAGGAAGTGTATTCTTGAACCCA CTTCG-3′ |
CCL5 shRNA-3 was used for the in vivo studies
2.3. Primary cultures
Primary cortical neuronal cultures and astrocytes were prepared as previously described (Avdoshina et al., 2010; Bachis et al., 2012). In brief, cortices were dissected from embryonic (E17-18) Sprague-Dawley rats (Charles River, Germantown, MD). Cortices were cleaned of blood vessels in Krebs-ringer bicarbonate buffer containing 0.3% bovine bovine serum albumin (BSA), and then dissociated using 1,800 U/ml trypsin at 37°C for 30 min. Trypsin was inactivated using soybean trypsin inhibitor/DNase (Sigma) and cells were centrifuged through a 4% BSA layer to form a pellet. Cell pellet was resuspended in Neurobasal medium containing 2% B-27 supplement, 25 mM glutamate, 0.5 mM L-glutamine and 1% antibiotic-antimycotic solution (Invitrogen, Grand Island, NY). Cells were seeded at a density of 0.5 × 106 cells per ml and grown in 95% air and 5% CO2 at 37°C for 7 days.
Astrocytes were prepared from the cerebral cortex of 1-2 day old rats. Cortices were cleaned of blood vessels and mechanically dissociated. Cells were seeded on 75 cm2 culture flasks and grown in DMEM medium containing 10% FBS and 2% antibiotic-antimycotic in 95% air and 5% CO2 at 37°C. After 6 days in culture, flasks were shaken for 4 days to remove microglia and oligodendrocytes. Resulting astrocyte culture was trypsinized and re-seeded on poly-L-lysine coated plates with DMEM media.
Cortical and astrocyte cultures were infected with prepared pseudoviral particles at 100 multiplicity of infection (MOI) and 10 MOI respectively. Cells were shaken every 15 min for 1 hr post infection, and then washed with warmed phosphate buffer and new conditioned medium was added.
2.4. Experimental procedures
Three-month-old male Sprague-Dawley rats (Charles River) were housed under standard conditions, two per cage, with food and water available ad libitum, and were maintained on a 12-hr light/dark cycle for the duration of the treatment protocols. All studies were carried out following the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health and approved by Georgetown University Animal Care and Use Committee. Efforts were made to minimize animal suffering and to reduce the number of animal used.
Gp120BaL or prepared lentiviruses were injected into the rat striatum using established protocols (Bachis et al., 2010; Nosheny et al., 2006). In brief, animals were anaesthetized with a combination of ketamine and xylazine (80 and 10 mg/kg respectively, i.p.) (Henry Schein, Dublin, OH) and mounted on a Kopf stereotaxic frame. A burr hole was drilled into the skull and a cannula was lowered into the forebrain and appropriate compounds (e.g. gp120BaL, pCDH-CCL5) were injected via polyethylene tubing and a 50 μl Hamilton syringe. The injection site, determined by stereotaxic guidance and expressed as coordinates from bregma, was anterior-posterior +0.7mm, mediolateral ±3mm, dorsoventral -6mm (-6.5mm for gp120BaL), according to (Paxinos and Watson, 1998). For all microinjection studies, the needle remained in place at the injection site for an additional 4 min after the injection. For gp120BaL (NIH AIDS Research Reference Reagent Program) microinjection, 4 μl (100 ng/μl concentration in 0.1% BSA) of gp120BaL or boiled gp120BaL were microinjected at a rate of 0.5 μl/min. For lentiviral experiments, rats were injected with 2 μl (Biological titer 4×108 TU/ml) of pCDH Empty vector, pCDH CCL5, scramble shRNA, or CCL5 shRNA-3 at a rate of 0.2 μl/min.
2.4.1. Animal treatment
A group of rats was treated, two days after surgery, with either chronic morphine or morphine withdrawal paradigms as previously described (Campbell et al., 2013). In brief, rats were treated twice a day with escalating doses of morphine for 5 days (10 mg/kg s.c. for 2 days and then escalating up to 30 mg/kg, s.c. from day 3). Animals were perfused and brains harvested 2 hr after the last injection. For morphine withdrawal, animals were treated with escalating doses of morphine for 5 days. At the fifth day morphine treatment was terminated, and animals were allowed to go through withdrawal for 60 hr, after which, animals were perfused and brains harvested for immunohistochemistry. Morphine sulfate was received from the National Institutes of Drug Abuse, (NIH, Division of Neuroscience & Behavioral Research, Research Triangle Park, NC). Morphine was diluted in isotonic and filtered sterilized saline.
After treatment regimens animals were anesthetized with ketamine/xylazine and intracardially perfused with phosphate buffer saline pH 7.4. Whole brains were harvested and post fixed in 4% paraformaldehyde for 24 hr for immunohistochemistry.
2.5. Immunohistochemistry
Fixed brains were transferred to 30% sucrose and sectioned at 20 μm by a sliding microtome (Microm International, Heidelberg, Germany). Sections were blocked in phosphate saline-Blocking Buffer (1% BSA, 0.2% milk, 0.3% Triton) for 30 minutes prior to primary antibody. Primary antibody dilutions were as follows: cleaved caspase-3 (1:10000, Sigma, St. Louis, MO), NeuN (1:200, Cell Signaling, Danvers, MA), ANTI-FLAG® M2 (1:200, Sigma), CCL5 (1:200, Sigma). Sections were incubated in primary antibodies overnight at 4°C, and then incubated with correspondent secondary fluorescent antibodies (1:1000, Alexaflour®, Invitrogen) and analyzed with a Nikon Eclipse Ni microscope at 450 nm, 594 nm or 670 nm wavelengths. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) labeling was performed according to the manufacturer's instructions (Abcam, Cambridge, MA) as previously described (Bachis et al., 2010; Nosheny et al., 2006).
2.5.1 Silver staining
Degenerating neurons and processes were detected using the FD Neurosilver™ Kit II (FD Neurotechnologies, Inc., Columbia, MD) according to the manufacturer's instructions.
2.6. Quantification of neuronal injury
Neuronal injury (silver staining, cleaved caspase-3 and TUNEL) was quantified by ImageJ Imaging software (National Institute of Health, Bethesda, MD). At least 10 striatal sections (one every 100 μm) rostral and caudal to the injection site per animal were used (∼2.5 mm total length of the striatum). The area of the needle tract (∼100 μm) was omitted. Silver stained tissue was color thresholded using ImageJ and further grayscaled to separate positively stained pixels from non-stained. The pixel amount was quantified as percent of silver stained pixels in the region of interest (% of Area). Cleaved caspase-3 or TUNEL positive cells were counted using a 20× objective. Cleaved caspase-3 or TUNEL positive cells were counted only based on staining, not shape or size. Size and shape of the lateral ventricle were examined to ensure that sections from identical coronal planes were used for each animal.
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
Two weeks after injection of pCDH-CCL5 or CCL5 shRNA, the striatum was dissected and homogenized in 1× RIPA buffer with protease/phosphatase inhibitor. Analysis of CCL5 or interleukin-1 beta (IL-1β) levels was carried out using the DuoSet ELISA Development System Kits (R&D), according to the manufacturer's instructions and as previously described (Campbell et al., 2013).
2.8. Statistical Analysis
Data were evaluated by ANOVA with multiple comparisons by Tukey's test (Sigma Plot®, Systat Software Inc., Chicago, IL). Data were considered statistically significant if a p-values was <0.05.
3. Results
3.1. CCL5 reduces the neurotoxic effect of gp120BaL
To examine whether CCL5 is neuroprotective in vivo we used a lentivirus expressing CCL5. We subcloned the CCL5 sequence including a FLAG-tag into a mammalian lentiviral vector (pCDH) expressing green fluorescent protein (GFP). We first confirmed CCL5 protein expression by Western blot after transfecting either pCDH empty vector or pCDH CCL5 into HEK293FT cells. A prominent and abundant 8kDa band was detected when the blot was probed with a CCL5 specific antibody (Fig. 1A). Lentivirus was then prepared and tested for production of CCL5 in rat cortical neurons. Cortical neurons were used because they do not release CCL5 under basal conditions (Avdoshina et al., 2010). We observed a time-dependent accumulation of CCL5 in the medium (Fig. 1B) by 24 and 48 hr post infection, suggesting that viral CCL5 is released.
Figure 1. Preparation and testing of a pCDH-CCL5 pseudovirus.
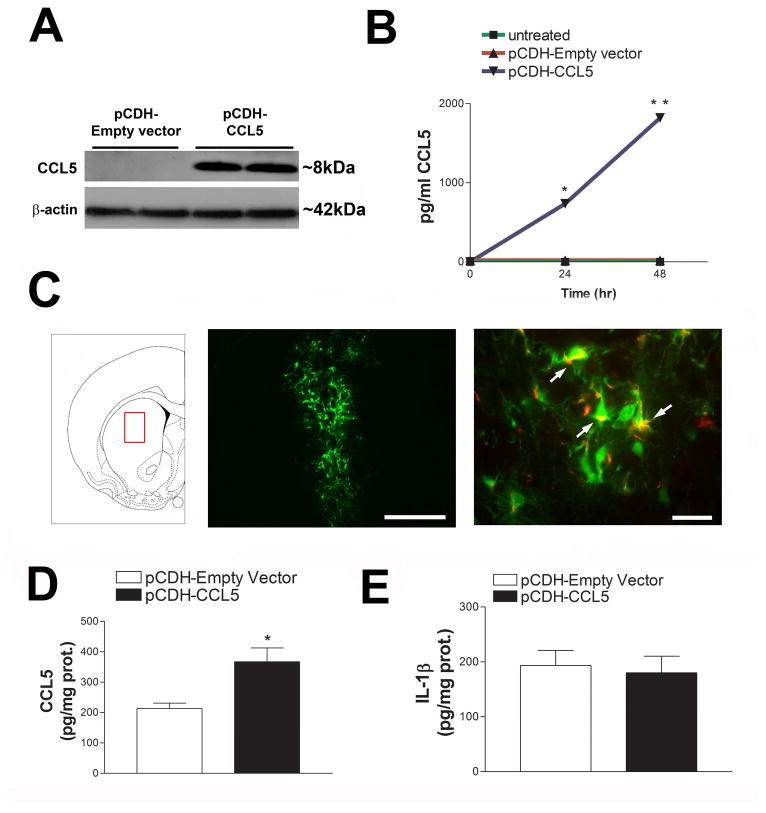
(A) pCDH-Empty vector or pCDH-CCL5 were transfected into HEK293FT cells using Lipo293 for 24 hr. Representative Western blot analysis of cell lysates using a CCL5 specific antibody. (B) Pseudovirus particles were created using established methods and primary neuronal cultures were infected with either pCDH-Empty vector or pCDH-CCL5 (100 MOI). Media was collected from the cells 24 and 48 hr post infection and CCL5 levels were measured by ELISA. Data are ± SEM (n=3 separate experiments), *p< 0.001 vs untreated and pCDH-Empty vector, **p< 0.001 vs pCDH-CCL5 at 24 hr. (C) Left panel: pCDH-CCL5 pseudovirus was injected into the rat striatum (AP = +0.7mm, ML = +3.0mm, DV = -6.0mm). Middle panel: example of viral infection detected by GFP. Bar=500 μm. Right panel. Protein production by pCDH-CCL5 infection was visualized using the anti-FLAG antibody (red). Arrows indicate CCL5-FLAG production (yellow= overlay). Bar= 20 μm. Levels of CCL5 (D) and IL-1β (E) were measured in the striatum by ELISA two weeks after the injection of pCDH-Empty vector or pCDH-CCL5 pseudovirus. Data are expressed as mean ± SEM (n = 5 per group), *p< 0.05.
We next determined the production of CCL5 in vivo by injecting pCDH empty vector or pCDH CCL5 (2 μl of 4×108 TU/ml) into the rat striatum (Fig. 1C left panel). This brain area was selected because it is sensitive to the toxic effect of gp120 (Bachis et al., 2010). After two weeks, rats were sacrificed and brains were prepared for immunohistochemistry. The expression of lenti-derived CCL5 was detected in several sections up to 2.5 mm rostral and caudal to the injection site (Fig. 1C, middle panel). Cells were both GFP and FLAG positive (Fig. 1C, right panel), suggesting expression of viral CCL5. ELISA was then used to quantify the amount of CCL5 in the striatum. Rats injected with pCDH-CCL5 exhibited higher levels of CCL5 than those injected with pCDH empty vector (Fig. 1D). Because lentiviral vectors may induce an inflammatory response (Abordo-Adesida et al., 2005), we also measured the levels of the pro-inflammatory cytokine IL-1β. We observed no significant changes in this cytokine by the CCL5 vector (Fig. 1E).
To test the effectiveness of CCL5 neuroprotection in vivo, animals were injected with either pCDH-empty vector or pCDH-CCL5 into the striatum. After two weeks, animals were injected with boiled gp120BaL (control) or gp120BaL (400 ng) and sacrificed 7 day later. Brains were then processed for silver stain, which detects degenerating neurons and axons (Shitaka et al., 2011; Tenkova and Goldberg, 2007). Control animals (empty vector + boiled gp120) did not exhibit silver staining (Figs. 2A and M). In contrast, in gp120-treated rats receiving the empty vector we noted a characteristic silver-impregnated degenerated synaptic terminals and fibers (Fig. 2G) suggesting axonal degeneration. No silver staining was observed in rats injected with pCDH-CCL5 and boiled gp120 (Figs. 2D and M), but overexpression of CCL5 significantly decreased silver-labeled neuronal processes evoked by gp120 (Figs. 2J and M). (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007) (Shitaka et al., 2011; Tenkova and Goldberg, 2007)
Figure 2. pCDH-CCL5 decreases gp120BaL-induced neuronal injury in the rat striatum.
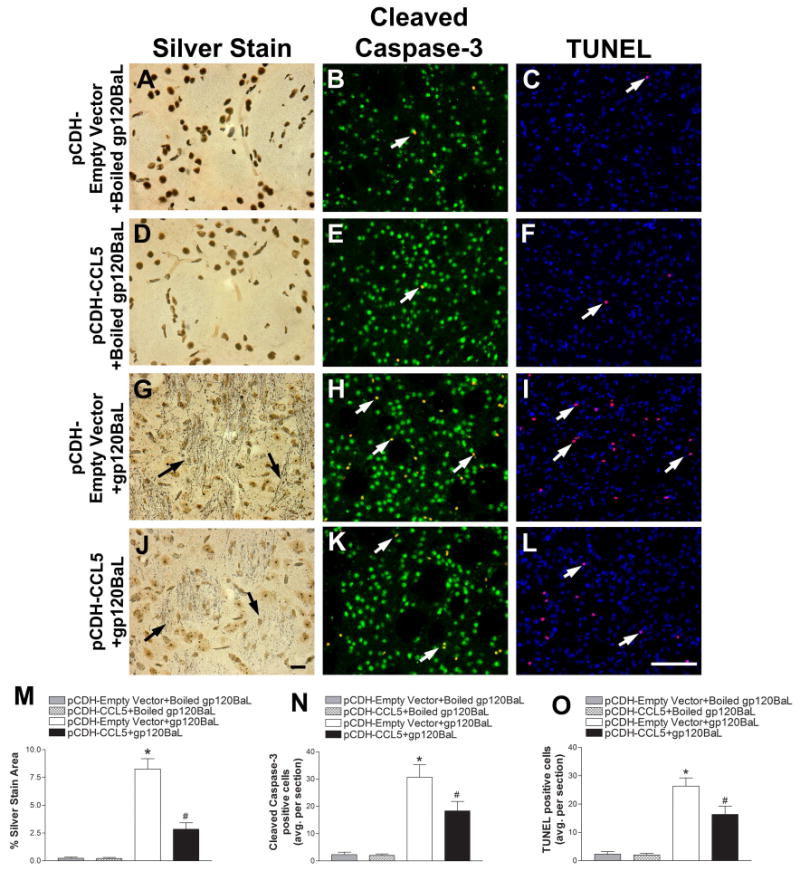
pCDH-Empty vector or pCDH-CCL5 packaged into pseudoviruses were injected into the rat striatum two weeks prior to gp120BaL or boiled gp120BaL. Serial sections were then processed for silver staining, cleaved caspase-3 and NeuN (green), and TUNEL and DAPI (blue) five days after gp120BaL injection. Representative images of striata from pCDH-Empty vector + boiled gp120BaL (A, B, C), pCDH-CCL5 + boiled gp120BaL (D, E, F), pCDH-Empty vector + gp120BaL (G, H, I) and pCDH-CCL5 + gp120BaL (J, K, L) - treated rats. Black arrows indicate silver impregnated, degenerating neuronal processes. White arrows indicate either cleaved caspase-3 positive neurons (yellow, green + red) or TUNEL (purple, blue + red) positive cells. Scale bar in J = 10 μm, L = 100 μm. Silver staining, cleaved caspase-3 and TUNEL positive cells, respectively were quantified by ImageJ in serial sections of treated animals (M, N, O). Data are the mean ± SEM (n = rats 6 per group). *p< 0.01 vs boiled gp120BaL. #p< 0.05 vs pCDH-empty vector+gp120BaL.
To confirm the neurotoxic effect of gp120 and the neuroprotective property of CCL5, striatal sectioned were also analyzed for cleaved caspase-3 and the neuronal marker NeuN, and TUNEL to detect apoptotic cells. An average of 3 cells both NeuN and caspase-3 positive per section were observed in control (Figs. 2B and N) and pCDH-CCL5 (Figs. 2E and N) animals. A similar scenario was obtained by using TUNEL (Figs. 2C, F and O). Gp120 treatment elicited a significant increase in caspase-3 positive neurons (Figs. 2H and N) as well as TUNEL positive cells (Figs. 2I and O). CCL5 vector significantly reduced the number of activated caspase-3 (Figs. 2K and N) as well as TUNEL positive cells (Figs. 2L and O) when compared to rats receiving empty vector prior to gp120.
3.2. Morphine and morphine withdrawal have the opposite effect on gp120BaL mediated neuronal injury
A previous study (Campbell et al., 2013) has shown that rats treated with a morphine regimen that induces tolerance and dependence to morphine exhibited increased striatal levels of CCL5. The opposite was true in rats undergoing morphine withdrawal. Thus, chronic morphine might prevent or limit gp120-mediated neuronal injury whereas withdrawal may exacerbate the toxic effect of gp120. To test these hypotheses, rats were injected with heat inactivated gp120BaL or gp120BaL (400 ng) into the striatum. Two days later, rats were treated for 5 days with saline or chronic escalating doses of morphine that increase striatal CCL5 (Campbell et al., 2013). Rats were perfused 2 hr after the last injection. A group of rats was allowed to undergo spontaneous withdrawal for 60 hr by the cessation of morphine treatment. The striatum was sectioned and processed for silver staining, activated caspase-3 and TUNEL. Morphine was able to reduce gp120BaL-induced neuronal injury. Indeed, the number and intensity of fibers positive for silver stain (Figs. 3D and J) was significantly lower in gp120 + morphine than gp120+ saline-treated animals (Figs. 3A and J). The opposite was obtained in animals undergoing withdrawal. In fact, an increased in the area and intensity of silver stain (Fig. 3G and J) was observed in gp120BaL-treated rats undergoing spontaneous withdrawal when compared to saline + gp120BaL or chronic morphine + gp120.
Figure 3. Chronic morphine and morphine withdrawal differentially affect gp120BaL-induced neuronal injury.
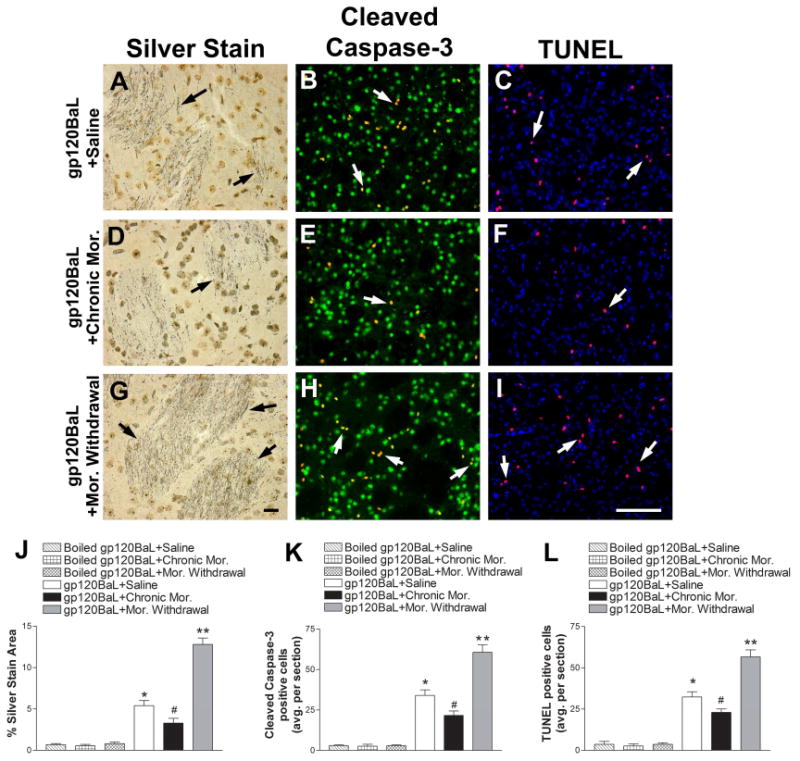
Rats were injected with gp120BaL (400 ng) into the striatum two days prior to receiving saline or escalating doses (see Materials and Methods) of morphine (Mor.) over the course of five days. Another group of animals were underwent spontaneous morphine withdrawal. Boiled gp120 was used as a control. Animals were perfused and neuronal injury was examined in serial striatal sections by silver staining, cleaved caspase-3 (red) and NeuN (green), and TUNEL (red) and DAPI (blue). Representative images from striata of gp120BaL + saline (A, B, C), gp120BaL + chronic mor. (D, E, F), and gp120BaL + mor. withdrawal (G, H, I) - treated animals. Black arrows indicate silver impregnated, degenerating neuronal processes. White arrows indicate either cleaved caspase-3 positive neurons or TUNEL positive cells. Scale bar in G = 10 μm, I = 100 μm. J, K, L. Silver staining, cleaved caspase-3 (yellow) and TUNEL (purple) positive cells, respectively were quantified by ImageJ in serial sections. Data are the mean ± SEM (n = 6 rats per group). *p< 0.01 vs boiled gp120BaL, #p< 0.05 vs saline + gp120BaL, **p<0.01 vs saline + gp120BaL.
The “neuroprotective” and “neurotoxic” effect of morphine and morphine withdrawal was confirmed by determining the number of activated caspase-3 and TUNEL positive cells. In fact, we observed that gp120-treated rats receiving chronic morphine exhibited fewer neurons positive for cleaved caspase-3 (Figs. 3E and K) or TUNEL (Figs. 3F and L) whereas withdrawal increased this number (Figs. 3H and I) compared to gp120 + saline treated animals (Figs. 3K and L). Thus, it appears that chronic morphine and morphine withdrawal have opposite effects on gp120BaL-mediated neuronal degeneration.
3.3. CCL5 shRNA prevents morphine-mediated neuroprotection
To further support the hypothesis that morphine may reduce the toxic effect of gp120 through CCL5, we created a CCL5 shRNA sequence that could block CCL5 synthesis in vivo. Three 9-11 base pair sequences were chosen and cloned into a pGreenPuro™ shRNA cloning vector along with a scramble shRNA control. We then transfected HEK293FT cells with our pCDH CCL5 construct alone or in combination with either scramble shRNA, CCL5 shRNA-1, CCL5 shRNA-2, or CCL5 shRNA-3. By Western blot analysis, we observe no effect in CCL5 expression with scramble shRNA, but a reduction with CCL5 shRNA-1, and CCL5 shRNA-2 (Fig. 4A). CCL5 shRNA-3 sequence promoted a complete CCL5 silencing (Fig. 4A). Therefore, we used CCL5 shRNA-3. We first tested the efficacy of CCL5 shRNA-3 in reducing CCL5 synthesis in primary cultures of astrocytes. Astrocytes were chosen because of their ability to induce CCL5 synthesis and release after morphine exposure (Avdoshina et al., 2010). Twenty-four hr following infection, astrocytes were exposed to 10 μM morphine and the media was collected at 12, 24, and 48 hr. We observed a time dependent increase in CCL5 release into the media by both morphine stimulated astrocytes and morphine stimulated + scramble shRNA-treated cells. However, CCL5 shRNA-3 completely abolished morphine stimulated CCL5 release (Fig. 4B).
Figure 4. Characterization of CCL5 shRNA lentivirus for in vivo silencing.
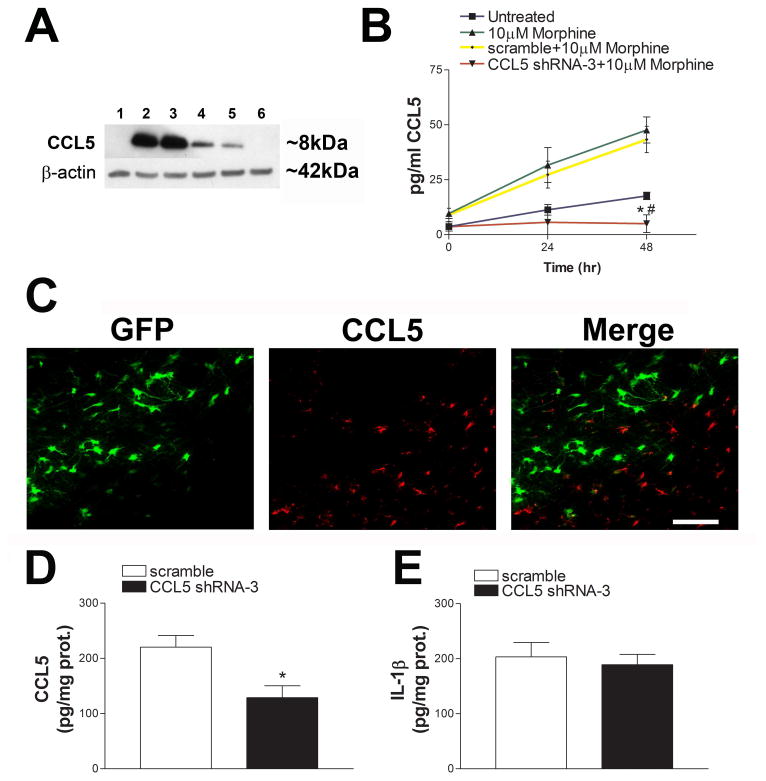
(A) HEK293FT cells were transfected with plasmids containing 1 = no treatment, 2 = pCDH CCL5, 3 = pCDH CCL5 + scramble shRNA, 4 = pCDH CCL5 + CCL5 shRNA-1, 5 = pCDH CCL5 + CCL5 shRNA-2, 6 = pCDH CCL5 + CCL5 shRNA-3. 24 hr later lysates were prepared and analyzed by Western blot. Please note that CCL5 shRNA-3 exhibited the highest amount of knockdown and was used for the subsequent experiments. (B) Primary rat astrocytes were infected with pseudovirus particles carrying either scramble shRNA or CCL5 shRNA-3 for 24 hr prior to stimulation with 10 μM morphine. Media was collected 24 and 48 hr following morphine stimulation and CCL5 levels were measured by ELISA. Data are expressed as the mean ± SEM (n=3 separate experiments), *p< 0.05 vs untreated, #p <0.001 vs 10μM morphine and 10 μM morphine + scramble shRNA. (C) Left panel. Example of CCL5 shRNA-3 infection (green). Middle panel. Representative image showing cells positive for endogenous CCL5 (red). Right panel. Image shows that shRNA-3 infected cells are CCL5 negative. Scramble shRNA or CCL5 shRNA-3 pseudovirus was injected into the rat striatum and levels of CCL5 (D) and IL-1β (E) were measured by ELISA two weeks later. Data are expressed as the mean ± SEM (n = 5 per group), *p< 0.05.
CCL5 shRNA-3 was then injected into the rat striatum. After a two week period, animals were perfused and brains were prepared for the determination of CCL5 by immunohistochemistry and ELISA. Histological analysis of CCL5 immunoreactivity revealed that cells positive for GFP exhibited no positivity for CCL5 (Fig. 4C). Analysis of lysates by ELISA confirmed a significant decrease in CCL5 levels in rats treated with CCL5 shRNA-3 compared to scramble shRNA (Fig. 4D). To establish whether shRNA promotes an inflammatory response we determined the levels of IL-1β. We observed no differences in IL-1β levels between scramble shRNA and shRNA-3 infected animals (Fig. 4E).
CCL5 shRNA-3 or scramble shRNA was then injected into the rat striatum. Two weeks later, gp120BaL was infused into the striatum and animals underwent either saline treatment or chronic morphine. At the completion of the treatments, animals were perfused and brains were sectioned and prepared for silver staining, cleaved caspase-3 and TUNEL. Scramble shRNA did not prevent the protective effect of chronic morphine, as there continued to be a decrease in silver-labeled neurons (Figs. 5A and G), cleaved caspase-3 (Figs. 5B and H) positive neurons and TUNEL (Figs. 5C and I) positive cells. However, CCL5 shRNA-3 infection reduced the protective effect of chronic morphine as detected by an increase in all three parameters of neuronal injury (Fig. 5). Overall our data suggests that the increase in CCL5 contributes to the neuroprotective activity of morphine.
Figure 5. CCL5 shRNA-3 decreases morphine-mediated neuroprotection against gp120BaL.
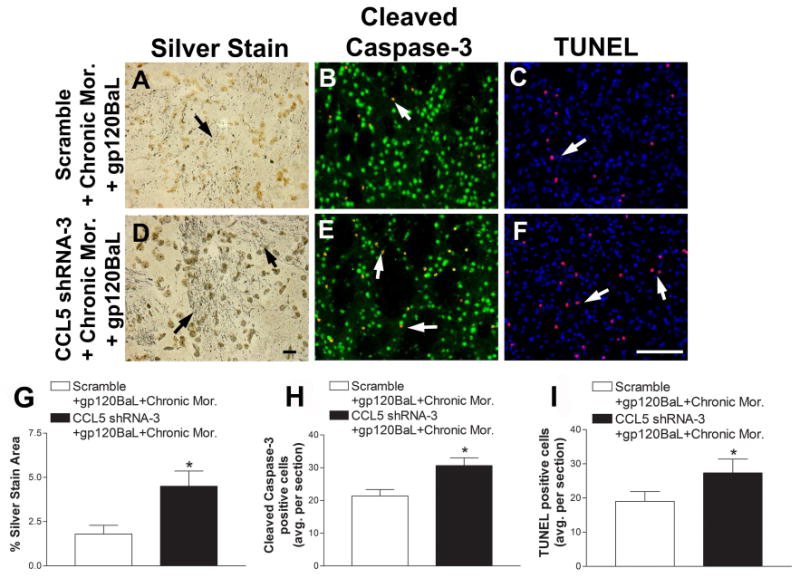
Scramble shRNA or CCL5 shRNA-3 packaged into pseudoviruses were injected into the rat striatum two weeks prior to intrastriatal injection of gp120BaL (400 ng). Two days later, rats were treated with chronic morphine (Mor). Serial striatal sections were then processed for (A, D) silver stain, (B, E) cleaved caspase-3 and NeuN (green), and (C, F) TUNEL and DAPI (blue). A, B and C are representative striatal images of scramble shRNA + chronic morphine + gp120BaL treated rats. D, E and F are representative striatal images of CCL5 shRNA-3 + chronic morphine + gp120BaL–treated animals. Black arrows indicate silver impregnated, degenerating neuronal processes. White arrows indicate either cleaved caspase-3 positive neurons (yellow, red + green) or TUNEL positive cells (purple, blue + red). Scale bar in D = 10 μm, F = 100 μm. Silver stained area (G), cleaved caspase-3 positive neurons (H) and TUNEL (I) positive cells were quantified by ImageJ in serial sections. Data are the mean ± SEM (n = 6 rats per group). *p< 0.05 vs scramble shRNA + chronic morphine + gp120BaL.
4. Discussion
Synaptic simplification and injury are pathological features observed in HAND (Ellis et al., 2007; Masliah et al., 1997). The brain is infected predominantly by the M-tropic strain of HIV (Gorry et al., 2001) and, in rodents, the M-tropic strain of gp120 causes neuronal cell loss (Bachis et al., 2010). Thus, discovering ways of preventing the neurotoxic effect of gp120BaL may reduce HAND. In this work we have shown that CCL5 either delivered by a lentiviral vector or induced pharmacologically by chronic morphine inhibits the neurotoxic action of M-tropic gp120.
CCL5 is a promiscuous ligand that binds to different receptors; however, the main receptor for CCL5 is CCR5 (Rossi and Zlotnik, 2000). In the adult brain, non-neuronal cells such as microglia, astrocytes and oligodendrocytes express this receptor (He et al., 1997; Klein et al., 1999; van der Meer et al., 2000). Nevertheless, CCR5 is also expressed by neurons, although this expression varies between species, methods of detection, and type of cultures (Avdoshina et al., 2011; Klein et al., 1999; Rottman et al., 1997; van der Meer et al., 2000). CCR5 is also the main receptor that mediates the neurotoxic effect of M-tropic gp120. In fact, CCR5 preferring gp120s fail to induce neuronal loss in cultures obtained from CCR5-deficient mice (Kaul et al., 2007). Moreover, two ligands of CCR5, CCL5 and CCL4, block the neurotoxic effect of gp120 (Kaul et al., 2007) suggesting that CCL5 may prevent/displace gp120BaL from binding to neuronal CCR5. However, CCL5 has also been shown to be protective against numerous neurotoxic insults including N-methyl-D-aspartic acid (Bruno et al., 2000), β-amyloid (Tripathy et al., 2010), and glutamate (Brenneman et al., 2002; Lin et al., 2011) as well as other HIV proteins (Brenneman et al., 2002; Kaul et al., 2007; Rozzi et al., 2014). Moreover, CCL5 enhances neurite formation and dendritic arborization (Chou et al., 2008), two hallmarks shared by neurotrophic factors. These properties suggest an additional mechanism of action of CCL5, which may involve a neuroprotective signal transduction. Indeed, CCL5 activates the neuroprotective pathway Akt/protein kinase B (Kaul et al., 2007). Thus, CCL5 may have a dual effect, acting as a ligand “displacing” gp120 from CCR5 as well as naturally occurring synaptic promoting agent.
In this work, we were able to show that while pCDH-CCL5 promoted a significant protection against gp120BaL, it did not completely abolish the HIV protein toxicity. There are several explanations for the partial neuroprotection. We have used a viral titer that produced a two-fold increase in CCL5 levels to avoid side effect of overexpressing CCL5. Thus, this amount of CCL5 may not be sufficient to confer complete neuroprotection. This suggestion is supported by in vitro data showing that the neuroprotective concentrations of CCL5 are in the nanomolar range. In addition, while CCL5 blocks gp120BaL from binding to CCR5, it may not block the production of toxic cytokines, such as tumor necrosis factor-α (TNFα), that promote neuronal apoptosis independently from CCR5 activation. Lastly, CCR5 may be recycled back to the membrane surface despite the presence of CCL5 and therefore be sensitive to further ligand interactions (Signoret et al., 2000). Future studies are needed to elucidate these mechanisms.
Previous in vitro studies suggested that morphine rescues neurons against gp120BaL through the release of CCL5 from astrocytes (Avdoshina et al., 2010). Therefore, we determined if this effect would be reproducible in vivo, by using a chronic morphine treatment previously described to increase levels of CCL5 in the rat striatum (Campbell et al., 2013). We found that morphine decreases the number of degenerating neurons caused by a microinjection of gp120BaL in the rat striatum. Moreover, decreasing the endogenous levels of CCL5 through a lentiviral shRNA significantly reduced the neuroprotective effect of morphine, supporting previous data that CCL5 mediates the neuroprotective activity of morphine. We could not confirm this hypothesis with specific antagonists of CCL5 receptors because CCL5 is a promiscuous ligand that binds to four different G-protein coupled receptors, CCR1, 3 and 5 (Rossi and Zlotnik, 2000), and GPR75 (Ignatov et al., 2006). There are no selective antagonists for these receptors that can be used to block CCL5 activity in vivo. Nevertheless, our data showing that CCL5 shRNA abolishes the neuroprotective effect of morphine support a role of CCL5 in the neuroprotective effect of morphine against M-tropic viral proteins.
The abuse of opioids is commonly associated with a withdrawal effect. Morphine withdrawal can lead to an increase in pro-inflammatory cytokines produced by glial cells in the brain (Hutchinson et al., 2009). These properties are comparable to the damaging effects produced by gp120BaL alone, which includes not only direct neurotoxicity through the CCR5 receptor (Kaul et al., 2007), but enhanced levels of pro-inflammatory cytokines such as TNF-α (Bachis et al., 2010). Indeed, in this study, we have observed that morphine withdrawal enhances the neurotoxic properties of gp120BaL. This effect may correlates with a decrease in CCL5. However, a reduction in CCL5 alone may not be the only mechanism that can account for the neurotoxic effect of withdrawal. For instance, morphine withdrawal has been shown to increase the number of microglia cells as well as pro-inflammatory cytokines such as IL-1β and TNF-α (Campbell et al., 2013) that promote caspase-8 and -3 dependent apoptosis (Emeterio et al., 2006). Opioid withdrawal also increases the levels of corticosteroids (Martínez-Laorden et al., 2012) which are known to enhance gp120 toxicity (Brooke and Sapolsky, 2002). Moreover, a recent study suggested that opioid withdrawal may accelerate progression to HIV-associated neurocognitive disorders by increasing ceramide (Bandaru et al., 2011), a sphingolipid that negatively regulates synaptic function. Thus, withdrawal may create a toxic environment composed of microglia proliferation, pro-inflammatory cytokines, elevated glucocorticoids and ceramide, which synergizes with HIV and its viral proteins to exacerbate neuronal damage.
Although the scope of this work was to establish the neuroprotective effect of CCL5 in vivo, our results may support new investigations on whether opioids such as methadone and buprenorphine can have protective effects against HIV neurotoxicity. These drugs are long lasting opioids, which are commonly used as a maintenance therapy and are shown to diminish withdrawal effects and relapse into intermittent drug use (Schaub et al., 2010). Because of these findings, we may hypothesize that opioid maintenance therapy may be beneficial in suppressing progression to HAND. This particular facet should be explored in future research, as it has direct clinical significance.
We show that the HIV protein gp120BaL causes synaptic degeneration in vivo.
morphine abuse and withdrawal differentially affect gp120 neurotoxic effect
CCL5 plays a role in the neuroprotective effect of morphine in vivo.
Acknowledgments
The authors would like to acknowledge support from HHS grants DA026174, NS079172 and F31DA032282. Special thanks to Mrs. Maia Parsadanian and Ms. Allyssia Boelk for technical assistance. The authors wish to thank the National Institute of Health, National Institute of Drug Abuse, Division of Neuroscience & Behavioral Research for morphine and NIH AIDS Research Reference Reagent Program for gp120BaL.
Abbreviations
- BSA
bovine serum albumin
- DAPI
diamidino-2-phenylindole
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- HIV
human immunodeficiency virus
- HAND
HIV-associated neurocognitive disorder
- IL-1β
interleukin-1 beta
- NeuN
Neuronal Nuclei
- PCR
polymerase chain reaction
- TNFα
tumor necrosis factor-α
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abordo-Adesida E, Follenzi A, Barcia C, Sciascia S, Castro MG, Naldini L, Lowenstein PR. Stability of lentiviral vector-mediated transgene expression in the brain in the presence of systemic antivector immune responses. Hum Gene Ther. 2005;16:741–751. doi: 10.1089/hum.2005.16.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdoshina V, Becker J, Campbell L, Parsadanian M, Mhyre T, Tessarollo L, Mocchetti I. Neurotrophins modulate the expression of chemokine receptors in the brain. J Neurovirol. 2011;17:58–62. doi: 10.1007/s13365-010-0004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdoshina V, Biggio F, Palchik G, Campbell LA, Mocchetti I. Morphine induces the release of CCL5 from astrocytes: potential neuroprotective mechanism against the HIV protein gp120. Glia. 2010;58:1630–1639. doi: 10.1002/glia.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachis A, Avdoshina V, Zecca L, Parsadanian M, Mocchetti I. Human immunodeficiency virus type 1 alters brain-derived neurotrophic factor processing in neurons. J Neurosci. 2012;32:9477–9484. doi: 10.1523/JNEUROSCI.0865-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachis A, Biggio F, Major EO, Mocchetti I. M- and T-tropic HIVs promote apoptosis in rat neurons. J Neuroimmune Pharmacol. 2009;4:150–160. doi: 10.1007/s11481-008-9141-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachis A, Cruz MI, Mocchetti I. M-tropic HIV envelope protein gp120 exhibits a different neuropathological profile than T-tropic gp120 in rat striatum. Eur J Neurosci. 2010;32:570–578. doi: 10.1111/j.1460-9568.2010.07325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhiet M, Tjernlund A, Mousa A, Gad A, Stromblad S, Kuziel WA, Seiger A, Andersson J. RANTES promotes growth and survival of human first-trimester forebrain astrocytes. Nat Cell Biol. 2001;3:150–157. doi: 10.1038/35055057. [DOI] [PubMed] [Google Scholar]
- Bandaru VV, Patel N, Ewaleifoh O, Haughey NJ. A failure to normalize biochemical and metabolic insults during morphine withdrawal disrupts synaptic repair in mice transgenic for HIV-gp120. J Neuroimmune Pharmacol. 2011;6:640–649. doi: 10.1007/s11481-011-9289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM. Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res. 2000;879:42–49. doi: 10.1016/s0006-8993(00)02725-6. [DOI] [PubMed] [Google Scholar]
- Brenneman DE, Hauser JM, Spong C, Phillips TM. Chemokine release is associated with the protective action of PACAP-38 against HIV envelope protein neurotoxicity. Neuropeptides. 2002;36:271–280. doi: 10.1016/s0143-4179(02)00045-8. [DOI] [PubMed] [Google Scholar]
- Brooke SM, Sapolsky RM. Glucocorticoid exacerbation of gp120 neurotoxicity: role of microglia. Exp Neurol. 2002;177:151–158. doi: 10.1006/exnr.2002.7956. [DOI] [PubMed] [Google Scholar]
- Bruno V, Copani A, Besong G, Scoto G, Nicoletti F. Neuroprotective activity of chemokines against N-methyl-D-aspartate or beta-amyloid-induced toxicity in culture. Eur J Pharmacol. 2000;399:117–121. doi: 10.1016/s0014-2999(00)00367-8. [DOI] [PubMed] [Google Scholar]
- Campbell LA, Avdoshina V, Rozzi S, Mocchetti I. CCL5 and cytokine expression in the rat brain: differential modulation by chronic morphine and morphine withdrawal. Brain Behav Immun. 2013;34:130–140. doi: 10.1016/j.bbi.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SY, Weng JY, HL L, Liao F, Sun SH, Tu PH, Dickson DW, Chern Y. Expanded-polyglutamine huntingtin protein supresses the secretion and production of a chemokine (CCL5/RANTES) by astrocytes. J Neurosci. 2008 Mar 26;28:3277–3290. doi: 10.1523/JNEUROSCI.0116-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Rosario D, Clifford DB, McArthur JC, Simpson D, Alexander T, Gelman BB, Vaida F, Collier A, Marra CM, Ances B, Atkinson JH, Dworkin RH, Morgello S, Grant I, Group CS. Continued high prevalence and adverse clinical impact of human immunodeficiency virus-associated sensory neuropathy in the era of combination antiretroviral therapy: the CHARTER Study. Arch Neurol. 2010;67:552–558. doi: 10.1001/archneurol.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emeterio EP, Tramullas M, Hurle MA. Modulation of apoptosis in the mouse brain after morphine treatments and morphine withdrawal. J Neurosci Res. 2006;83:1352–1361. doi: 10.1002/jnr.20812. [DOI] [PubMed] [Google Scholar]
- Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D'Emilia DM, Friedlander RM, Yuan J, Masliah E, Lipton SA. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J Neurosci. 2002;22:4015–4024. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbard HA, Epstein LG. HIV-1 encephalopathy in children. Curr Opin Pediatr. 1995;7:655–662. doi: 10.1097/00008480-199512000-00005. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gorry PR, Bristol G, Zack JA, Ritola K, Swanstrom R, Birch CJ, Bell JE, Bannert N, Crawford K, Wang H, Schols D, De Clercq E, Kunstman K, Wolinsky SM, Gabuzda D. Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. J Virol. 2001;75:10073–10089. doi: 10.1128/JVI.75.21.10073-10089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, Busciglio J, Yang X, Hofmann W, Newman W, Mackay CR, Sodroski J, Gabuzda D. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. 1997;385:645–649. doi: 10.1038/385645a0. [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Lewis SS, Coats BD, Skyba DA, Crysdale NY, Berkelhammer DL, Brzeski A, Northcutt A, Vietz CM, Judd CM, Maier SF, Watkins LR, Johnson KW. Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast) Brain Behav Immun. 2009;23:240–250. doi: 10.1016/j.bbi.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatov A, Robert J, Gregory-Evans C, Schaller HC. RANTES stimulates Ca2+ mobilization and inositol trisphosphate (IP3) formation in cells transfected with G protein-coupled receptor 75. Br J Pharmacol. 2006;149:490–497. doi: 10.1038/sj.bjp.0706909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Ma Q, Medders KE, Desai MK, Lipton SA. HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ. 2007;14:296–305. doi: 10.1038/sj.cdd.4402006. [DOI] [PubMed] [Google Scholar]
- Klein RS, Williams KC, Alvarez-Hernandez X, Westmoreland S, Force T, Lackner AA, Luster AD. Chemokine receptor expression and signaling in macaque and human fetal neurons and astrocytes: implications for the neuropathogenesis of AIDS. J Immunol. 1999;163:1636–1646. [PubMed] [Google Scholar]
- Letendre SL, Lanier ER, McCutchan JA. Cerebrospinal fluid beta chemokine concentrations in neurocognitively impaired individuals infected with human immunodeficiency virus type 1. J Infect Dis. 1999;180:310–319. doi: 10.1086/314866. [DOI] [PubMed] [Google Scholar]
- Lin MS, Hung KS, Chiu WT, Sun YY, Tsai SH, Lin JW, Lee YH. Curcumin enhances neuronal survival in N-methyl-d-aspartic acid toxicity by inducing RANTES expression in astrocytes via PI-3K and MAPK signaling pathways. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:931–938. doi: 10.1016/j.pnpbp.2010.12.022. [DOI] [PubMed] [Google Scholar]
- Martínez-Laorden E, Hurle MA, Milanés MV, Laorden ML, Almela P. Morphine Withdrawal Activates Hypothalamic-Pituitary-Adrenal Axis and Heat Shock Protein 27 in the Left Ventricle: The Role of Extracellular Signal-Regulated Kinase. Journal of Pharmacology and Experimental Therapeutics. 2012;342:665–675. doi: 10.1124/jpet.112.193581. [DOI] [PubMed] [Google Scholar]
- Masliah E, Ge N, Mucke L. Pathogenesis of HIV-1 associated neurodegeneration. Crit Rev Neurobiol. 1996;10:57–67. doi: 10.1615/critrevneurobiol.v10.i1.30. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42:963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- Maung R, Hoefer MM, Sanchez AB, Sejbuk NE, Medders KE, Desai MK, Catalan IC, Dowling CC, de Rozieres CM, Garden GA, Russo R, Roberts AJ, Williams R, Kaul M. CCR5 knockout prevents neuronal injury and behavioral impairment induced in a transgenic mouse model by a CXCR4-using HIV-1 glycoprotein 120. J Immunol. 2014;193:1895–1910. doi: 10.4049/jimmunol.1302915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543–555. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosheny RL, Bachis A, Aden SA, De Bernardi MA, Mocchetti I. Intrastriatal administration of human immunodeficiency virus-1 glycoprotein 120 reduces glial cell-line derived neurotrophic factor levels and causes apoptosis in the substantia nigra. J Neurobiol. 2006;66:1311–1321. doi: 10.1002/neu.20288. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego (CA): Academic Press; 1998. [Google Scholar]
- Peters P, Bhattacharya J, Hibbitts S, dittmar MT, Simmons G, Bell J, Simmonds P, Clapham PR. Biological analysis of human immunodeficiency virus type 1 R5 envelopes amplified from brain and LN tissues of AIDS patients with neuropathology reveals two distinct tropism phenotypes and identifies envelopes in the brain that confer an enhanced tropism and fusigenicity for macrophages. J Virol. 2004;78:6915–6926. doi: 10.1128/JVI.78.13.6915-6926.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- Rottman JB, Ganley KP, Williams K, Wu L, Mackay CR, Ringler DJ. Cellular localization of the chemokine receptor CCR5. Correlation to cellular targets of HIV-1 infection. Am J Pathol. 1997;151:1341–1351. [PMC free article] [PubMed] [Google Scholar]
- Rozzi SJ, Borelli G, Ryan K, Steiner JP, Reglodi D, Mocchetti I, Avdoshina V. PACAP27 is Protective Against Tat-Induced Neurotoxicity. J Mol Neurosci. 2014 doi: 10.1007/s12031-014-0273-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub M, Chtenguelov V, Subata E, Weiler G, Uchtenhagen A. Feasibility of buprenorphine and methadone maintenance programmes among users of home made opioids in Ukraine. Int J Drug Policy. 2010;21:229–233. doi: 10.1016/j.drugpo.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Shitaka Y, Tran HT, Bennett RE, Sanchez L, Levy MA, Dikranian K, Brody DL. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol. 2011;70:551–567. doi: 10.1097/NEN.0b013e31821f891f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signoret N, Pelchen-Matthews A, Mack M, Proudfoot AE, Marsh M. Endocytosis and recycling of the HIV coreceptor CCR5. J Cell Biol. 2000;151:1281–1294. doi: 10.1083/jcb.151.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit TK, Wang B, Ng T, Osborne R, Brew B, Saksena N. Varied tropism of HIV-1 isolates derived from different regions of adult brain cortex discriminate between patients with and without AIDS dementia complex (ADC): evidence for neurotropic HIV variants. Virology. 2001 Jan 20;:509–526. doi: 10.1006/viro.2000.0681. [DOI] [PubMed] [Google Scholar]
- Sozzani S, Introna M, Bernasconi S, Polentarutti N, Cinque P, Poli G, Sica A, Mantovani A. MCP-1 and CCR2 in HIV infection: regulation of agonist and receptor expression. J Leukoc Biol. 1997;62:30–33. doi: 10.1002/jlb.62.1.30. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Tenkova TI, Goldberg MP. A modified silver technique (de Olmos stain) for assessment of neuronal and axonal degeneration. Methods Mol Biol. 2007;399:31–39. doi: 10.1007/978-1-59745-504-6_3. [DOI] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- Tripathy D, Thirumangalakudi L, Grammas P. RANTES upregulation in the Alzheimer's disease brain: a possible neuroprotective role. Neurobiol Aging. 2010;31:8–16. doi: 10.1016/j.neurobiolaging.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer P, Ulrich AM, Gonzalez-Scarano F, Lavi E. Immunohistochemical analysis of CCR2, CCR3, CCR5, and CXCR4 in the human brain: potential mechanisms for HIV dementia. Exp Mol Pathol. 2000;69:192–201. doi: 10.1006/exmp.2000.2336. [DOI] [PubMed] [Google Scholar]