

Abstract
MTL-homozygous (a/a or α/α) white cells form a complex sexual biofilm that exhibits the same architecture as that of MTL-heterozygous (a/α) pathogenic biofilms. However, the former is regulated by the mitogen-activated protein (MAP) kinase pathway, while the latter is regulated by the Ras1/cyclic AMP (cAMP) pathway. We previously demonstrated that in the formation of an MTL-homozygous, mature (48 h) sexual biofilm in RPMI 1640 medium, the MAP kinase pathway targets Tec1 rather than Cph1, the latter of which is the target of the same pathway, but for the opaque cell mating response. Here we continued our analysis of the role of Tec1 by comparing the effects of deleting TEC1 on initial adhesion to silicone elastomer, high-resolution confocal microscopy assessments of the stages and cellular phenotypes during the 48 h of biofilm development, human white cell penetration, and biofilm fragility. We show that although Tec1 plays only a minor role in initial adhesion to the silicone elastomer, it does play a major role in the growth of the basal yeast cell polylayer, vertical extension of hyphae and matrix deposition in the upper portion of the biofilm, final biofilm thickness, penetrability of human white blood cells, and final biofilm integrity (i.e., resistance to fluid flow). These results provide a more detailed description of normal biofilm development and architecture and confirm the central role played by the transcription factor Tec1 in the biofilm model employed here.
INTRODUCTION
Candida albicans forms biofilms with different functional characteristics, depending upon the configuration of the MTL locus (1–4). After 48 h in pH-stabilized RPMI 1640 medium at 37°C in air, a/α cells form a biofilm on silicone elastomer that is approximately 100 μm thick and composed of a basal yeast cell polylayer (∼20 μm thick) and an extensive upper layer of vertically oriented hyphae (∼80 μm thick) that are uniformly distributed and embedded in a dense extracellular matrix. These a/α biofilms are firmly attached to the silicone elastomer substratum, highly resistant to penetration by phagocytic human white blood cells, resistant to drugs, such as fluconazole, and impermeable to low- and high-molecular-weight molecules (5). Under exactly the same conditions, a/a and α/α cells in the white phase of the white-opaque transition (6) form biofilms that have the same architecture and integrity as those of a/α biofilms but differ in that they are readily penetrated by human phagocytic white blood cells, susceptible to fluconazole, and permeable to low- and high-molecular-weight molecules (1, 5). MTL-heterozygous and MTL-homozygous biofilms also differ in the capacity to support mating (2, 7). MTL-homozygous biofilms support mating of seeded minority opaque cells at 10 to over 100 times the frequency of that of MTL-heterozygous biofilms (7). Because of the differences in the latter pathogenic and mating characteristics, biofilms formed by a/α cells, which represent the predominant MTL genotype colonizing hosts (8–12), were deemed “pathogenic,” while those formed by a/a and α/α cells were termed “sexual” (5, 13).
Mutational studies revealed that pathogenic a/α and sexual a/a or α/α biofilms formed in the model we use, which was pioneered by Douglas and coworkers (14–16), are regulated by different signal transduction pathways (4, 17–21). The formation of a pathogenic a/α biofilm is regulated by the Ras1/cyclic AMP (cAMP) pathway, which targets a transcription factor pathway that includes the cascade Efg1 → Tec1 → Bcr1 (5, 13). The formation of a sexual white cell biofilm, in contrast, is regulated by the pheromone receptor, trimeric G protein complex, and mitogen-activated protein (MAP) kinase pathways, which also target Tec1 and, in turn, a downstream transcription factor that has yet to be identified (5, 22). Because our mutational analyses suggested that the signal, receptor, trimeric G protein complex, and MAP kinase cascades regulating formation of the sexual biofilm pathway are identical to the components of the opaque cell pheromone response pathway but target different transcription factors (Tec1 in white cells and Cph1 in opaque cells), we proposed a working hypothesis for the evolution of the white cell biofilm in which the entire upper portion is derived intact from the highly conserved pheromone response pathway for mating, but instead of Cph1, the major targeted transcription factor in the mating response by opaque cells, Tec1 is the major targeted transcription factor in sexual biofilm formation (5, 13, 23, 24). Since sexual biofilms facilitate mating of minority opaque cells (7), the hypothesis provided a possible reason for why the two cell-type-specific responses employ the same signal and signal response pathway. Using the same signal provides a means of coordinating the two intertwined processes of mating by minority opaque cells and sexual biofilm formation in majority white cells. And since the white cell biofilm response and the opaque cell mating response are phenotypically different, the hypothesis provided a reason for why the pathway targets different transcription factors for different phenotypic outcomes. Recently, our conclusion that the MAP kinase pathway targets Tec1 in the regulation of a sexual biofilm was challenged by Lin et al. (25), who concluded that the MAP kinase pathway actually targets Cph1 in the regulation of MTL-homozygous biofilm formation. However, the conditions used by Lin et al. (25) and the resulting biofilm preparation were dramatically different and not comparable to ours, as we demonstrated in a recent study (1).
Here we extended our analysis of the role of Tec1 in biofilm formation by comparing the temporal steps in the developmental program, the cellular phenotypes in the two main regions of the final biofilm, the final general architecture, the penetrability by human white phagocytic cells, and the integrity of the biofilms formed by the parent strain a/a P37005, two independently generated a/a cph1Δ/cph1Δ mutant strains, two independently generated a/a tec1Δ/tec1Δ mutant strains, and complemented mutant a/a tec1Δ/TEC1c strains, using the Douglas model (14–16). The majority of planktonic cells of the a/a P37005 parent strain adhered to the silicone elastomer within 1.5 h and then formed a uniformly distributed basal yeast-phase cell polylayer of approximately 10 cells thick (∼20 μm) within the next 5 h. Cells at the surface of the polylayer then formed hyphae that extended vertically and relatively equidistant from one another and were engulfed in a cell-free extracellular matrix deposited as they grew. The final thickness of the upper hypha-matrix layer was approximately 80 μm. The upper layer achieved half of its thickness after 16 h. After 48 h, the thickness of the upper layer was close to the maximum, and the deposited matrix decreased with height, such that hyphal ends at the biofilm surface were relatively free of encapsulating matrix and were bent (26). Adhesion to the silicone elastomer and the formation of biofilms by the a/a cph1Δ/cph1Δ mutants were highly similar to those of the parental strain, but this was not the case for the a/a tec1Δ/tec1Δ mutants. Mutant a/a tec1Δ/tec1Δ cells also adhered to silicone elastomer, but adherence was slightly reduced, and the remaining developmental program was even more defective. Cells of the a/a tec1Δ/tec1Δ strains formed a thinner, nonuniform basal layer of yeast cells and then extended shorter hyphae in random directions rather than in a vertical, uniform array, resulting in intertwined hyphae. The final thickness of a/a tec1Δ/tec1Δ biofilms was approximately half that of wild-type biofilms, with a less dense matrix. The aberrant a/a tec1Δ/tec1Δ biofilms were also far more penetrable by human white blood cells and were highly fragile compared to wild-type biofilms, releasing from the silicone elastomer surface and fragmenting when swirled. This study not only provides a more detailed temporal description of the steps in the complex developmental program of biofilm formation of wild-type a/a cells in the model employed (14, 15) but also demonstrates that Tec1 plays a critical role in the entire developmental program.
MATERIALS AND METHODS
Strains.
Strain P37005, a natural isolate from a bloodstream infection with the MTL genotype a/a (27), was used to generate two independent a/a cph1Δ/cph1Δ deletion mutants, (A) and (B), and two independent a/a tec1Δ/tec1Δ deletion mutants, (A) and (B), as previously described (3, 4). Complementation strains a/a tec1Δ/TEC1c (A) and (B) were generated from the homozygous mutants. The deletion mutants were generated using the recyclable flipper cassette pSFS2A (28), which contains a dominant nourseothricin resistance marker (CaSAT1). The plasmid was a gift from Joachim Morschaüser of the University of Würzburg. Deletion cassettes I and II were generated by use of a two-step disruption strategy. For each gene, two mutants were confirmed by PCR and Southern analysis as homozygous. Both a/a tec1Δ/tec1Δ (A) and (B) were complemented by integrating wild-type TEC1 back into its native locus, placing it under the control of its own promoter. To generate the plasmid pTEC1c, used for complementation of a/a tec1Δ/tec1Δ (A) and (B), the CaSAT1 gene in the plasmid pNIMI (29) was flanked with the promoter and coding sequences of TEC1 and the 3′ region of TEC1, which were amplified by PCR with the primer pairs TEC1-F1/-R1 and TEC1-F2/-R2, respectively (see Table S1 in the supplemental material for a list of all primers used in this study). The genomic DNA of the SC5314 strain served as the template for PCR amplification of the TEC1 gene. All sequences in the plasmid were verified by DNA sequencing. The ApaI-SacII-digested DNA fragment obtained from the plasmid pTEC1C was integrated into one of the tec1Δ alleles of each a/a tec1Δ/tec1Δ mutant strain. All strains were maintained at 25°C on agar containing Lee's medium (30) supplemented with arginine and zinc, with glucose as the carbon source (sLee's-glucose agar) (31). This medium contained phloxine B (5 μg/ml), which stains opaque cells and colonies red (32). The phenotypes of white- and opaque-phase cells from respective colonies were verified as >99% homogeneous at the cellular level by microscopy prior to use.
Mutant validation.
Total genomic DNAs from parental strain a/a P37005, mutant strain a/a cph1Δ/cph1Δ clones (A) and (B), and a/a tec1Δ/tec1Δ clones (A) and (B) were amplified by PCR, using the CPH1-specific primer pair P1 and P2 and the TEC1-specific primer pair P3 and P4 (see Table S1 in the supplemental material). Validation of the complemented a/a TEC1c strains employed the primer pair TEC1-SR/CASAT1-F (see Table S1). The PCR products were resolved in Tris-borate-EDTA (TBE)–agarose gels and purified using a Qiagen QIAquick gel extraction kit. The purified DNA bands were sequenced with the same CPH1- and TEC1-specific primer pairs used for amplification. The nucleotide sequences were compared with the Candida Genome Database to determine the deleted regions in the mutants. The deletions are noted in the putative amino acid sequences deduced from the DNA sequences (see Fig. 1C and D). The complemented strains were similarly verified by PCR.
FIG 1.

Verification of a/a cph1Δ/cph1Δ and a/a tec1Δ/tec1Δ deletion mutants by PCR amplification and DNA sequencing. (A) Primer sites for amplification and predicted deletion regions. (B) PCR amplification products of wild-type and mutant strains. (C and D) Deleted regions of proteins revealed by DNA sequencing. The same respective sequencing data were obtained for the two independent deletion derivatives of CPH1 and those of TEC1. The deduced deletions in the final proteins are presented.
Medium used for biofilm development.
RPMI 1640 medium (Life Technologies) was modified with the buffer morpholinepropanesulfonic acid (MOPS) (16) to stabilize the pH at 7.0. In this report, this buffered RPMI 1640 medium is simply referred to as RPMI 1640.
Preparation of cells for biofilm development.
Cells from 5-day-old colonies were grown in suspension cultures in supplemented Lee's medium grown to stationary phase (48 h) in flasks rotated in air at 25°C and 200 rpm. At 48 h, the cells, which were over 95% unbudded singlets, were pelleted, washed in RPMI 1640 medium, and counted in a hemocytometer.
Preparation of silicone elastomer discs.
Biofilms were developed on silicone elastomers in the wells of cluster dishes in RPMI 1640 medium. Silicone elastomer discs were cut from sterile silicone elastomer sheets that were 0.04 in. thick (Bentec Medical, Woodland, CA), using a 10-mm biopsy punch (Acu-Punch; Acuderm, Inc., Ft. Lauderdale, FL). The discs were washed and sterilized as previously described (1), placed in a 24-well cluster dish (Costar; Corning, Inc.), and incubated overnight in 2 ml of RPMI 1640 medium at 30°C.
Biofilm development.
The incubation medium in which the silicone elastomer discs were incubated was replaced with 2 ml of fresh RPMI 1640 medium containing 2 × 107 stationary-phase cells. The cells were allowed to adhere without agitation for 90 min at 37°C, unless otherwise noted. After adhesion, the discs were removed and gently rinsed with Dulbecco's modified phosphate-buffered saline (D-PBS), without the cations Ca2+ and Mg2+. The discs were then transferred to a 12-well cluster dish containing 2.5 ml of fresh RPMI 1640 medium. The discs were incubated at 37°C, unless otherwise noted, in a humidified incubator on a platform rocker with a 60° total deflection every 7.5 s over a 48-h period.
Cell adhesion to silicone elastomer discs.
The number of cells adhering to the silicone elastomer discs was quantitated following the initial 90-min adhesion step of biofilm development (see above), using two methods. In the first method, discs supporting adherent cells after 90 min in RPMI 1640 medium at 37°C without agitation were removed, gently rinsed in D-PBS, and placed in 0.05% trypsin-EDTA solution (Invitrogen-Life Technologies) to release adherent cells from the disc substrate. Cells released were counted in a hemocytometer. White- and opaque-phase cells were distinguished by size and morphology (6, 32) and counted. Discs were microscopically evaluated for the total release of cells caused by the trypsin-EDTA solution. In the second method, we transferred the disc to fresh medium at 90 min and then fixed the cells and stained them for nuclei with Syto 9 (Invitrogen-Life Technologies), using the method of Zhao et al. (33). The number of nuclei per microscope field (60× objective) was counted using a laser scanning confocal microscope (LSCM).
Assessing biofilm thickness and architecture.
After 48 h, biofilms were fixed with 2% (vol/vol) paraformaldehyde, rinsed with D-PBS, and stained with calcofluor M2R fluorescent white (Sigma-Aldrich, St. Louis, MO) for cell wall chitin and the extracellular matrix, as previously described (1). Biofilm thickness and architecture were analyzed as described previously, with minor changes. Briefly, imaging was acquired using a Bio-Rad 2100 multiphoton LSCM equipped with a Mai-Tai infrared (IR) laser. Calcofluor white was excited with the IR laser at 780 nm, and emission was captured at 460 nm. Z-series scans were collected using LaserSharp software (Bio-Rad). Z-stack images were gathered at a high resolution, with a step interval of 0.25 μm through 125 μm in the z axis, for a total of 500 images. The images were then processed using Imaris 3D Image Processing & Analysis software (Bitplane, Zurich, Switzerland). Slices were stacked and the image rotated so that a side view was obtained. For internal analysis of hypha orientation and distribution, a 20-μm stack, comprised of 80 individual scans from the center of a biofilm at 48 h, was imaged from the top. This provided information on the architecture (cross sections, vertical orientation, and distribution) in the middle of a mature biofilm. To assess the intensity of matrix staining, the laser intensity was increased uniformly for compared biofilms.
Biofilm penetrability.
Biofilm penetrability by human white blood cells was assessed by overlaying mature 48-h biofilms with green fluorescent protein (GFP)-labeled HL-60 cells (34, 35) and measuring the distance penetrated after 90 min, using methods previously published in detail (1). The extent of cell penetration was determined by LSCM, with GFP excitation at 488 nm and emission captured at 515 nm.
Biofilm fragility.
Twelve-well cluster plates containing 48-h mature biofilms were transferred from the rocker and placed tightly on a titer plate rotating shaker (model 4625; Lab Line). The dishes were rotated at 250 rpm, with a rotation diameter of 3 mm, for 15 s. Images were obtained immediately, using a Nikon Coolpix digital camera.
RESULTS
Sequencing the original mutants.
To be sure that the original null mutants derived from the same natural a/a parent strain (P37005) were valid, we amplified and resequenced the two independently generated CPH1 deletion mutants, a/a cph1Δ/cph1Δ (A) and (B) (4), and the two TEC1 deletion mutants, a/a tec1Δ/tec1Δ (A) and (B) (3). These SAT flipper mutants contain short insertions (28) that are not delineated in the diagrams. The deletions in the duplicates of the alternative mutants are shown (in yellow) in Fig. 1A. The primer sites for amplification of the alternative mutants for sequencing (P1-P2 and P3-P4) are also noted in Fig. 1A. The amplification products of the two CPH1 mutants, a/a cph1Δ/cph1Δ (A) and (B), and the two TEC1 mutants, a/a tec1Δ/tec1Δ (A) and (B), were reduced in size (Fig. 1B), and the sizes were those expected for the remaining sequences (blue regions of Fig. 1A). The amplification products were sequenced and found to contain the expected deletions of the targeted coding regions, as evident in the deduced amino acid sequences, presented in bold in Fig. 1C and D. The complemented mutant strains, a/a tec1Δ/TEC1c (A) and (B), were also validated in the same way (data not shown). We previously showed that the mutant a/a tec1Δ/tec1Δ strains did not produce a TEC1 transcript (3) and that the a/a cph1Δ/cph1Δ strains did not produce a CPH1 transcript (4).
Adhesion to silicone elastomer after 90 min.
In the Douglas model (14–16), the initial inoculum was incubated primarily on catheter material (usually polyvinyl chloride) in yeast nitrogen base medium, with glucose or galactose as the main carbon source, at 37°C in air, with gentle rocking. Here we used silicone elastomer as the substratum and RPMI 1640 medium, both verified by Douglas and coworkers as equally effective as the basic conditions they used for C. albicans biofilm formation in their model (14–16). Cells were incubated on the silicone elastomer for 90 min in RPMI 1640 medium at 37°C in air, and then the elastomer disc was removed from the well, gently rinsed to remove cells that did not initially adhere to the silicone elastomer, and placed in a new well with fresh medium. The preparations were then incubated for 48 h at 37°C in air on a slow rocker to continually create a flow of medium over the disc. Since we originally tested adhesion on plastic rather than on silicone elastomer in our first analysis of the TEC1 deletion mutant (3), we had to verify that there was no advantage between the control and mutants in this first step of biofilm formation (adhesion) in the model employed. We therefore examined adhesion at 90 min on silicone elastomer in RPMI 1640, without rocking, at 37°C in air. Two methods were applied. In the first method, cells were released from the disc by trypsinization and counted. The number of a/a cph1Δ/cph1Δ adherent cells was, on average, slightly higher (8%), and that of the a/a tec1Δ/tec1Δ mutant slightly lower (14% decrease), than that of the control a/a P37005 strain (Fig. 2A), but neither of these differences proved to be significant (Fig. 2B). Enzymatic removal of cells from the elastomer proved not to be 100% efficient, as determined by disc examination after treatment. Therefore, we used a second method, in which adherent cells were fixed onto the silicone elastomer and nuclei were stained and counted directly on the silicone elastomer discs. Three areas of each biofilm (n = 9) were analyzed in each of three independent preparations, for a total of 27 measurements (n = 27). Both independent mutants of the CPH1 deletion mutant and TEC1 deletion mutants (A) and (B) were analyzed. Again, we found that the numbers of adherent cells for the a/a cph1Δ/cph1Δ strains were slightly higher (5% and 2%, respectively) and the numbers for the a/a tec1Δ/tec1Δ strains slightly lower (3% and 4%, respectively). Even though these differences proved to be significant, they were quite minor. Qualitatively, images of the fixed cells on the discs could not be distinguished between control and mutant biofilms (data not shown). These results indicated that there might be small differences in initial adhesion, but they were small enough to conclude that the initial adhering cell populations at the onset of biofilm formation of control and mutant strains were highly similar.
FIG 2.

Initial adherence to silicone elastomer discs after 90 min without rocking (the first step in the biofilm model). The following two methods were used: measurements of cells released from discs in trypsin and measurements of numbers of Syto 9-stained nuclei per microscope field for attached cells on silicone elastomer discs. (A) Concentrations of cells removed from discs by trypsinization for control a/a P37005, a/a cph1Δ/cph1Δ (A), and a/a tec1Δ/tec1Δ (A) preparations. Nine measurements, including three from each of three independent biofilms, were used in the calculations. Values represent the means, and error bars represent standard deviations. (B) P values for differences between trypsinized cell concentrations in panel A. (C) Numbers of nuclei of cells adhering to elastomer discs per microscope field for a/a P37005, a/a cph1Δ/cph1Δ (A), a/a cph1Δ/cph1Δ (B), a/a tec1Δ/tec1Δ (A), and a/a tec1Δ/tec1Δ (B) cells. Twenty-seven measurements, including three areas from each of nine independent biofilms, were used in the calculations. (D) P values for differences between strains in panel C.
Basal yeast-phase cell polylayer.
After adhesion, the next step in biofilm development in the model we employed is the formation of a basal cohesive polylayer composed predominantly of yeast-phase cells, the “basal yeast-phase cell polylayer.” After 4 h of incubation, LSCM of calcofluor white-stained preparations revealed a dense, uniform, cohesive basal polylayer in a/a P37005 control cultures. Both a top view (upper image in each panel) and a side view (90° pitch) (lower image in each panel) of the stacked confocal images are presented for representative 4-h basal yeast-phase cell polylayers of a/a P37005, a/a cph1Δ/cph1Δ, and a/a tec1Δ/tec1Δ preparations, in Fig. 3A, B, and C, respectively. The mean thickness (± standard deviation) of the basal layer of a/a P37005 cells at 4 h was 20 ± 2 μm (n = 9) (Fig. 3A). The average thickness of the basal yeast cell polylayer of a/a cph1Δ/cph1Δ cells after 4 h was 22 ± 4 μm (n = 9) (Fig. 3B), which is statistically indistinguishable from that of a/a control cells (P > 0.05). However, the basal yeast cell polylayer of a/a tec1Δ/tec1Δ cells after 4 h (Fig. 3C) was significantly thinner (14 ± 1 μm; P = 0.0001 for control versus mutant phenotypes). In addition to a difference in thickness, the basal layers of a/a tec1Δ/tec1Δ cells appeared to be patchy, with low-density regions or gaps (Fig. 3C). The average thickness of the 4-h a/a tec1Δ/tec1Δ basal layers, i.e., 14 ± 1 μm (n = 9), is an overestimate, since the sparse areas were not used in thickness calculations. These results demonstrate that although a/a tec1Δ/tec1Δ cells grew primarily in the yeast phase during the period in which the basal yeast cell polylayer was formed, the development and architecture of the polylayer were defective.
FIG 3.

The average thickness of the basal yeast cell polylayer formed by the a/a tec1Δ/tec1Δ mutant after 4 h was smaller than those of the a/a P37005 and a/a cph1Δ/cph1Δ biofilms. Preparations were stained with calcofluor white and scanned by LSCM. The upper image for each strain provides the top surface of the projection of LSCM x-y scans of a representative biofilm (z-interval = 0.25 μm). The lower image for each strain represents a side view of the same projection. (A) a/a P37005 (control). (B) a/a cph1Δ/cph1Δ mutant. (C) a/a tec1Δ/tec1Δ mutant. The mean thickness ± standard deviation for the yeast-phase cell polylayer of each strain is presented underneath the lower image of each panel. Measurements were for three regions of three independent biofilms, resulting in a total (n) of 9 measurements. Bars = 20 μm.
Development of the upper region of biofilms.
After 4 h, a/a P37005 at the top surface of the basal yeast-phase cell polylayer began to extend hyphae. These hyphae grew vertically, and as they continued to elongate, an extracellular matrix formed in the interstitial spaces between hyphae. By 16 h, biofilms had attained half of their final thickness (reaching approximately 50 μm), and by 48 h had achieved their final thickness, as demonstrated originally by Douglas and coworkers (14–16). The extending hyphae formed few lateral yeast-phase cells. And just as Douglas and coworkers demonstrated 20 years ago (14–16), the rate of biofilm growth (in the z axis) continued to decrease dramatically during the last 10 h of development. To compare the final thicknesses of parental and mutant cell biofilms at 48 h, a collection of 500 confocal scans through 125 μm of fixed, calcofluor white-stained preparations was obtained. The scans were stacked and rotated 90° (Fig. 4A to C and D to F, for two independent experiments), and the height (thickness) of each biofilm was measured. Side views of representative biofilms in the two experiments revealed that whereas control a/a cph1Δ/cph1Δ biofilms had similar thicknesses, those of the a/a tec1Δ/tec1Δ mutant were roughly half as thick (Fig. 4A through F). The mean thicknesses (± standard deviations) for parental a/a P37005 biofilms after 48 h at 37°C (in the two independent experiments) were 103 ± 3 μm (n = 9) and 99 ± 4 μm (n = 9) (Table 1). The mean thicknesses of the a/a cph1Δ/cph1Δ mutant biofilms at 37°C in air, for the two independent experiments, were 104 ± 3 μm (n = 9) and 100 ± 2 μm (n = 9) (Table 1). The mean thickness for the a/a control and a/a cph1Δ/cph1Δ biofilms, averaged for the two independent experiments, was 102 μm (Table 1). In marked contrast, the mean thicknesses after 48 h for the a/a tec1Δ/tec1Δ biofilms were 60 ± 6 μm (n = 9) and 54 ± 6 μm (n = 9), with an average of 57 μm. This represented an average decrease of 48% for the a/a tec1Δ/tec1Δ mutants compared to the control strain (Table 1).
FIG 4.
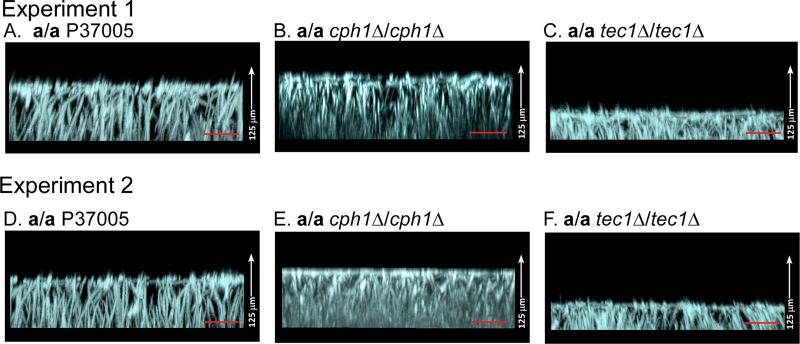
The average thickness of mature 48-h biofilms formed by the a/a tec1Δ/tec1Δ mutant was approximately half that of biofilms formed by the parental strain a/a P37005 and the a/a cph1Δ/cph1Δ mutant. Preparations were stained with calcofluor white and scanned by LSCM. Representative biofilms from two independent experiments (1 and 2) are presented in panels A to C and panels D to F. (A and D) Side views of LSCM image projections of representative biofilms formed by the parental strain (a/a) P37005 in the two independent experiments. (B and E) Side views of representative biofilms formed by strain a/a cph1Δ/cph1Δ (A). (C and F) Side views of representative biofilms formed by strain a/a tec1Δ/tec1Δ (A). The means ± standard deviations for thicknesses (n = 9) from experiments 1 and 2 at 37°C are presented in Table 1. Bars = 20 μm.
TABLE 1.
Effects of minority opaque cells and temperature on thicknesses of 48-h biofilms formed by a/a control, a/a Δcph1/Δcph1, and a/a Δtec1/Δtec1 cells
| Temp (°C) | Strain | Expt | Addition of opaque cells | Biofilm thickness (μm) (mean ± SD) | % increase in thickness | P value for difference with and without opaque cells | P value for difference between 28°C and 37°C |
|---|---|---|---|---|---|---|---|
| 37 | a/a P37005 | 1 | − | 103 ± 3 | <0.0001 | ||
| a/a P37005 | 2 | − | 99 ± 4 | ||||
| a/a P37005 | 1 | + | 118 ± 10 | +15 | 0.003 | <0.0001 | |
| a/a cph1Δ/cph1Δ | 1 | − | 104 ± 3 | <0.0001 | |||
| a/a cph1Δ/cph1Δ | 2 | − | 100 ± 2 | ||||
| a/a cph1Δ/cph1Δ | 1 | + | 121 ± 4 | +16.4 | 0.001 | <0.0001 | |
| a/a tec1Δ/tec1Δ | 1 | − | 60 ± 6 | <0.0001 | |||
| a/a tec1Δ/tec1Δ | 2 | − | 54 ± 6 | ||||
| a/a tec1Δ/tec1Δ | 1 | + | 61 ± 6 | +2 | NSa | <0.0001 | |
| 28 | a/a P37005 | 1 | − | 82 ± 2 | |||
| a/a P37005 | 1 | + | 90 ± 2 | +10 | 0.001 | ||
| a/a cph1Δ/cph1Δ | 1 | − | 95 ± 3 | ||||
| a/a cph1Δ/cph1Δ | 1 | + | 99 ± 5 | +4 | 0.027 | ||
| a/a tec1Δ/tec1Δ | 1 | − | 49 ± 2 | ||||
| a/a tec1Δ/tec1Δ | 1 | + | 52 ± 2 | +4 | 0.01 |
NS, not significant.
Effects of temperature and addition of 10% opaque cells.
In the majority of our original studies of white cell biofilm regulation (3–5, 19, 21, 22, 36, 37), we allowed biofilms to develop at 28°C and, in many experiments, added 10% opaque cells (50% a/a-50% α/α) as a source of pheromone. We showed that control cell a/a biofilms were slightly thicker when opaque cells were added (2–5, 21, 22, 36, 37) and slightly thicker at 37°C than at 28°C (1). In the present study, we performed all experiments at 37°C with homogeneous white cell populations (i.e., with no added minority opaque cells). To test whether the addition of opaque cells or the temperature affected mutant cell biofilm formation, we compared biofilm formation (thicknesses) among the control strain a/a P37005, a/a cph1Δ/cph1Δ (A), and a/a tec1Δ/tec1Δ (B) at either 37°C or 28°C, with or without the addition of 10% opaque cells. The increase in temperature from 28°C to 37°C caused a small but significant increase in thickness for all tested strains but did not affect the major decrease (approximately 50%) in the thickness of a/a tec1Δ/tec1Δ biofilms or the relative normalcy of a/a cph1Δ/cph1Δ biofilms (Table 1). At 37°C, the addition of 10% opaque cells caused increases in a/a P37005 and a/a cph1Δ/cph1Δ biofilm thicknesses of 15 to 16% but caused only a 2% increase in a/a tec1Δ/tec1Δ biofilm thickness (Table 1). At 28°C, the increases for a/a P37005 and a/a cph1Δ/cph1Δ biofilms were smaller but still significant. Also, adding opaque cells had no significant effect on adhesion at 90 min, i.e., the first step in the formation of the basal yeast cell polylayer. Measurements performed by counting the cells washed off the silicone elastomer were statistically indistinguishable between 100% white cells and 90% white cells plus 10% (a/a plus α/α) opaque cells for both control and mutant strains (data not shown). These results indicate that the original mutant studies at 28°C in the presence of 10% opaque cells (a source of pheromone), presented previously (3, 5), hold true for cultures at 37°C in the absence of added minority opaque cells. Also note that we previously showed that homogeneous white cell populations release pheromone of the opposite mating type in a paracrine fashion to stimulate biofilm formation in the absence of opaque cells (37).
Hypha orientation in the upper region.
Whereas the lower layer of a 48-h control cell biofilm was composed primarily of yeast-phase cells, the upper layer, which accounts for approximately 80% of a mature biofilm, was composed of vertically oriented hyphae embedded in an extracellular matrix (Fig. 4A and D). Calcofluor white stained hyphal walls intensely and the matrix weakly. The calcofluor white-stained a/a control preparations, which exhibited vertically oriented hyphae, were imaged by LSCM with reduced IR laser power (<3%) to accentuate the intensely stained hyphae, at the expense of visualizing matrix staining (Fig. 4A and D).
To clearly resolve hyphal orientation in the z axis as well as to assess the uniformity of distribution in the x-y axes, a 20-μm stack of LSCM image scans from the center of a 48-h biofilm was viewed for three biofilms of each strain (first three images in each panel in Fig. 5). In the last image of each panel in Fig. 5, the region of the observed stacks is represented by a red band in the side view of the preparation. The generally punctate profiles in the top views of the stacked midsection LSCM images of the parental a/a P37005 biofilm reflect hypha cross sections and thus a vertical orientation (Fig. 5A). Similar results were obtained for a/a cph1Δ/cph1Δ biofilms (Fig. 5B). This was not true for a/a tec1Δ/tec1Δ biofilms. The top (0°) views of the stacked midsection scans of a/a tec1Δ/tec1Δ biofilms revealed mostly tangential profiles (Fig. 5C), indicating that the hyphae in the center of the biofilm had bent and were oriented obliquely or horizontally, not vertically. By tracking hyphae from scan to scan in this region, it also appeared that the a/a tec1Δ/tec1Δ hyphae were much shorter than the a/a P37005 or a/a cph1Δ/cph1Δ hyphae (data not shown).
FIG 5.

Long hyphae are oriented vertically in a/a P37005 and a/a cph1Δ/cph1Δ biofilms, whereas short hyphae are disorganized in a/a tec1Δ/tec1Δ biofilms. Eighty x-y images (0.25-mm step intervals) representing a distance of 20 μm were extracted from the midregion of the LSCM x-y z-stack collected for each biofilm. Each panel includes top views of the extracted stack for three representative biofilms of a strain and a vertical view, with a red band representing the extracted stack. Preparations were stained with calcofluor white. (A) a/a P37005. (B) a/a cph1Δ/cph1Δ mutant. (C) a/a tec1Δ/tec1Δ mutant. The punctate patterns in panels A and B reflect a vertical orientation, and the short targeted sections in panel C reflect a random orientation. Bars = 20 μm.
When the IR laser power was increased to visualize the matrix, the vertical hyphae were found to be encapsulated by a dense matrix. In the upper regions of a/a P37005 and a/a cph1Δ/cph1Δ biofilms, the matrix staining was more intense than that of the stunted and disorganized a/a tec1Δ/tec1Δ biofilm (data not shown).
Biofilm fragility.
One of the hallmarks of both bacterial and fungal biofilms is their capacity to remain anchored and intact in niches in which they experience disruptive forces, such as fluid flow. The less adhesive a biofilm is to a surface and the less cohesive basal cells are to one another, the more vulnerable a biofilm will be to a mechanical disruption such as fluid flow. To test biofilm fragility, we transferred the 12-well plates containing biofilm discs formed after 48 h by a/a 37005, a/a cph1Δ/cph1Δ, and a/a tec1Δ/tec1Δ cells from the gentle rocker upon which they were developed to a rapid rotator (Fig. 6A). The entire 12-well plate containing the biofilm discs was rotated for 15 s. Biofilms from all three strains were rotated simultaneously, so biofilms from each strain experienced identical sheer forces. The treatment had little effect on biofilms formed by control a/a P37005 cells (Fig. 6B). In two independent experiments, 100% (n = 24) of the control biofilms remained attached to the silicone elastomer discs on which they had formed (Fig. 6B). Small tears were observed in only 2 of 24 a/a control biofilms (Fig. 6B). There was no control biofilm fragmentation (Fig. 6B). The biofilm of the a/a cph1Δ/cph1Δ mutant also remained intact after rotation. However, for both experiments, 2 of the 24 a/a cph1Δ/cph1Δ biofilms, although remaining completely intact, were released from the supporting silicone elastomer (Fig. 6C). There was no fragmentation, even in the case of the released biofilms (Fig. 6C, asterisks). In marked contrast, the biofilms formed by the a/a tec1Δ/tec1Δ mutant were fragile. After rapid rotation, every a/a tec1Δ/tec1Δ biofilm was released from the elastomer on which it had formed, and every one of them fragmented (Fig. 6D). Multiple fragments were distributed throughout the medium in the wells (Fig. 6D). In some cases, the a/a tec1Δ/tec1Δ biofilms exhibited large tears. These results indicate that biofilms of the a/a tec1Δ/tec1Δ mutant, but not those of the a/a control or a/a cph1Δ/cph1Δ strain, are weakly adhesive to the silicone elastomer biofilm and are highly fragile (i.e., less cohesive), fragmenting when exposed to shear forces caused by rapid rotation.
FIG 6.

A simple rotation assay revealed that biofilms of the a/a tec1Δ/tec1Δ mutant, but not the a/a P37005 and a/a cph1Δ/cph1Δ strains, are highly fragile. Each 12-well plate containing 48-h silicone elastomer-supported biofilms was transferred from a slow rocker to a titer plate shaker and rotated for 15 s at 250 rpm. (A) Rocking and rotation assay. (B) All biofilms of parental strain a/a P37005 in two experiments remained attached to silicone elastomers after rotation. One of them showed a single tear, but the remaining 11 were intact. (C) Biofilms of the a/a cph1Δ/cph1Δ mutant remained intact, but two were released from the silicone elastomer during rotation. The asterisks denote release from the silicone elastomer. (D) All biofilms of the a/a tec1Δ/tec1Δ mutant were released from the silicone elastomers and underwent fragmentation after rotation.
White blood cell penetration.
A major difference between MTL-homozygous (a/a or α/α) and MTL-heterozygous (a/α) biofilms is in their penetrability by human phagocytic white blood cells. When DiI-labeled polymorphonuclear leukocytes or cells of the leukemic cell line HL-60 expressing GFP are dispersed on a mature a/α biofilm, they will not penetrate the major portion of the biofilm after 90 min of incubation (1, 5). Rather, they remain on or entangled in the upper edge (5%) of the biofilm, which is composed of the bent apices of vertical hyphae and contains a very diffuse matrix. However, when human polymorphonuclear leukocytes or HL-60 cells are overlaid on a mature a/a or α/α biofilm, they penetrate 70 to 80% through the biofilm after 3 h (1, 5, 7, 20). When HL-60 cells expressing GFP were dispersed on the biofilms formed by a/a P37005 and a/a cph1Δ/cph1Δ cells (data not shown), they penetrated 80% through the biofilms. However, when dispersed on biofilms formed by a/a tec1Δ/tec1Δ biofilms, they penetrated 100% through the biofilms, accumulating on the silicone elastomer substratum (data not shown).
Complementation.
To be sure that the aberrant biofilm phenotypes of a/a tec1Δ/tec1Δ mutants (A) and (B) were not due to a secondary mutation but rather to the deletion of TEC1, we generated the complemented strains a/a tec1Δ/TEC1c A-1, A-2, B-1, and B-2. Complementation of the TEC1 deletion mutants resulted in biofilms that exhibited the same level of adhesion as that of control cells after 90 min of incubation (data not shown). The thickness of control a/a P37005 cells was 107 ± 3 μm (n = 9), and those of the complemented strains a/a tec1/TEC1c A-1, A-2, B-1, and B-2 were 109 ± 3 μm (n = 9), 112 ± 3 μm (n = 9), 100 ± 4 μm (n = 9), and 106 ± 2.2 μm (n = 9), respectively. None were significantly different from that of a/a P37005 biofilms, but all were almost twice as thick as the parental mutants a/a tec1Δ/tec1Δ (A) (see Fig. S1B in the supplemental material) and a/a tec1Δ/tec1Δ (B) (see Fig. S1E). The final architecture of the biofilms formed by the complemented strains was similar to that of the parental strain a/a P37005 (see Fig. S1A). The TEC1-complemented deletion mutants also exhibited no signs of fragility (data not shown).
DISCUSSION
Previously, using the Douglas model of biofilm formation (14, 15), we demonstrated that although MTL-heterozygous a/α cells and MTL-homozygous a/a and α/α white-phase cells form architecturally similar and complex biofilms, several characteristics related to commensalism and pathogenesis differ dramatically, including permeability to low- and high-molecular-weight molecules, penetrability by phagocytic white blood cells, and susceptibility to fluconazole (1, 5, 19). However, while a/α biofilms did not facilitate mating of seeded opaque cells, a/a and α/α biofilms did so (7). We demonstrated by mutational analyses of the components of the pheromone response pathway in a/a cells (ste2Δ/ste2Δ, ste3Δ/ste3Δ, ste4Δ/ste4Δ, ste11Δ/ste11Δ, hst7Δ/hst7Δ, cek1Δ/cek1Δ cek2Δ/cek2Δ, and cph1Δ/cph1Δ mutations) that MTL-homozygous but not MTL-heterozygous biofilm formation is regulated by this MAP kinase pathway but that this pathway does not target Cph1 (4, 5) as it does in the mating response of opaque cells (4, 38). Rather, the MAP kinase pathway targets Tec1 (3). Tec1 is the same transcription factor targeted by the signal transduction pathway in the formation of a/α biofilms (17, 39). Next, by mutational analyses of select components of the Ras1/cAMP pathway (ras1Δ/ras1Δ, cdc35Δ/cdc35Δ, tpk2Δ/tpk2Δ, pde2Δ/pde2Δ, efg1Δ/efg1Δ, Δtec1Δ/tec1Δ, and bcr1Δ/bcr1Δ mutations) and overexpression of downstream components in deletion mutants of upstream components, we demonstrated that this pathway targets the transcription factor cascade Efg1 → Tec1 → Bcr1 (17, 39) in the regulation of a/α but not MTL-homozygous biofilm formation (5). This transcription factor cascade was previously partially identified by Mitchell and coworkers (17, 40), and Efg1 was previously implicated (18, 41), using protocols that resulted in robust a/α biofilm formation (1). In all of our preceding studies involving the formation of biofilms, we used the same procedure for biofilm formation. Cells were first grown in suspension in supplemented Lee's medium to stationary phase and then allowed to adhere to a silicone elastomer disc in RPMI 1640 medium buffered with MOPS for 90 min. After rinsing to remove nonadherent cells, the cells on the discs were allowed to develop into a biofilm in air at 37°C for 48 h, with gentle rocking to create fluid flow, which inhibited conditioning of the microenvironment. The discovery by Lin et al. (25) that the MAP kinase pathway targets Cph1, not Tec1, in regulating MTL-homozygous biofilm formation was not relevant to our results, given that they used a completely different system for biofilm development, including different methods for preparing cells, a different substrate, a different medium, and a shorter time frame for biofilm development. In repeating their method for preparing biofilms to be used in their wild-type and mutant comparisons, we recently showed (1) that none of the major features of sexual biofilms exhibited by a/a and α/α biofilms formed in the model originally pioneered by Douglas and coworkers (14–16, 42, 43) were exhibited by the preparations of Lin et al. (25). The preparations obtained with the models used by Lin et al. (25) were one-fifth as thick, fragile, and devoid of a dense cohesive and adhesive basal polylayer of yeast cells, and they produced no dominant upper region of vertically oriented hyphae embedded in a dense extracellular matrix (1). We therefore continued our analysis of the role of Tec1 in a/a biofilm development by using the Douglas model (15, 16).
Stages of biofilm formation in the model employed.
To understand which aspects of a/a biofilm development Tec1 regulates, we performed a comparison between a/a P37005 and a/a tec1Δ/tec1Δ biofilm formation, in which the stages of (control) biofilm formation were analyzed using confocal microscopy technologies that were not formerly applied. The following stages of a/a TEC1/TEC1 strain P37005 are outlined in Fig. 7. Cells from a stationary-phase white unbudded yeast-phase cell suspension culture of strain P37005, grown in supplemented Lee's growth medium (31), were suspended in RPMI 1640 medium and added to a well containing a silicone elastomer disc (0 h).
FIG 7.

Stages of normal (a/a P37005) biofilm development under the conditions employed in this study. The stages were deduced by LSCM. (A) Planktonic white-phase (yeast-phase) cells grown to stationary phase in supplemented Lee's medium (31) were resuspended in RPMI 1640 medium and inoculated into a well containing a silicone elastomer disc. (B) This preparation was incubated without rotation at 37°C in air for 90 min, during which over 20% of cells adhered to the silicone elastomer (stage 1). (C) At 1.5 h, the elastomer disc with adherent cells only was transferred to a new well containing fresh RPMI 1640 medium and incubated for 4 subsequent hours, during which the cells multiplied in the budding yeast phase. This produced a uniformly distributed basal yeast-phase cell polylayer of approximately 8 to 10 cells thick (∼20 μm thick) (stage 2). (D) Between 6 and 10 h, yeast-phase cells at the upper edge of the basal layer began to form hyphae (“germ tubes”) (stage 3). (E) Between 10 and 20 h, the hyphae grew vertically to lengths of approximately 40 μm. These hyphae were relatively equidistant from one another and encased in a self-produced extracellular matrix (stage 4). (F) Between 20 and 48 h, hyphae continued to elongate vertically and extracellular matrix continued to be deposited, but the rate of biofilm extension decreased and then stopped (stage 5). At the termination of this period, the distal tips of the vertical hyphae became bent. In the model, budding cells are depicted as small circles, hyphae as tubes, and matrix as a blue haze.
(i) Stage 1: adhesion.
The culture was incubated (without movement or rocking) in air at 37°C for 1.5 h, during which cells formed an evenly dispersed layer.
(ii) Stage 2: formation of basal yeast-phase cell polylayer.
At 1.5 h, the original supernatant was removed, nonadherent cells rinsed away, and the disc placed in fresh RPMI 1640 medium. The culture was then slowly rocked. During the first 4 h, cells multiplied primarily in the yeast phase, forming a cohesive, uniformly thick basal yeast cell polylayer of approximately 8 to 10 cells thick (∼20 μm). Note that in stage 2, growth of the “lower region” of the biofilm was almost exclusively in the budding yeast phase, not the hyphal phase.
(iii) Stage 3: hypha formation and matrix distribution (formation of half of the upper layer of the biofilm).
Between 6 and 10 h, the cells at the top of the basal yeast-phase cell polylayer underwent the bud-hypha transition and extended incipient hyphae (referred to as “germ tubes” in earlier literature [44]) that lacked septa at the yeast cell-hypha interface (45, 46). The incipient tubes extended in the z axis (vertically). As the hyphae grew, the matrix was deposited in the interstitial space.
(iv) Stage 4: rapid vertical hypha extension and matrix deposition.
Between approximately 10 and 20 h, the hyphae extended linearly, forming true hyphae with compartments separated by septa, without indentations at the septa and containing large vacuoles. There was little or no lateral branching or yeast cell budding at septal junctions. This 10-h period of hyphal extension and matrix deposition was rapid, accounting for formation of approximately 40% of the volume (height) of the final biofilm.
(v) Stage 5: completion of the mature biofilm.
The final period of development, between 20 and 48 h, was an extension of stage 4, except that the rate of hyphal extension decreased dramatically. At the end of this stage, vertical hyphae were approximately 80 μm long and distributed vertically and roughly equidistant from one another within the supporting matrix, except in the very top 5 μm. In the latter region, the hyphae bent in random directions and there was little matrix deposition.
Several aspects of this developmental scenario are worth considering. First, it is clear that development is separated into two major portions, i.e., the development of the basal yeast-phase cell polylayer and the development of the hypha-dominated upper layer plus encapsulating matrix. Second, it is not clear which or if both of the two cell types (basal yeast-phase cells and hyphae) contribute to the matrix. The distinction of these two phases makes it extremely important that the procedure used for forming biofilms in a study be described carefully and that the stages and cell types be described in space and time. Third, note that there is little hyphal branching or lateral yeast cell formation at the septa that separate hyphal compartments. Fourth, bending of the apical portions of the hyphae suggests but does not prove that the matrix supports a vertical orientation. Fifth, we did not observe yeast cell formation at the hypha apices or a mechanism for yeast-phase cell dispersal (47) after 48 h in the model we employed. Biofilm development for longer incubation periods does result in the formation and release of such cells in the dispersal process. These cells have been shown to be more adhesive and virulent than yeast-phase cells grown planktonically (47). Finally, note that the stages of a/a biofilm development described here for a/a cells are similar to those for a/α biofilm development with the same model.
Abnormalities in tec1Δ/tec1Δ biofilm development.
The a/a tec1Δ/tec1Δ mutant appeared to be capable of adherence, basal yeast cell polylayer formation, hypha formation, and matrix deposition, but each of these capabilities was either slightly or dramatically defective, leading to dramatic changes in architecture. First, the original planktonic a/a tec1Δ/tec1Δ yeast-phase cells were slightly less adherent to silicone elastomer (stage 1) than parental control cells in stage 1 of the developmental program. Adhering cells then formed a thinner, less uniform basal yeast-phase cell polylayer in the next 4 h, in stage 2. The cells in the polylayer, however, were phenotypically similar (primarily yeast-phase cells) to those in the basal layer of wild-type biofilms. The polylayer grew predominantly by budding in the yeast phase. The decrease in thickness was due either to a lower growth rate or to the release of cells during development of the basal yeast-phase cell polylayer. The release of tec1Δ/tec1Δ biofilms due to rapid rotation suggests that the latter is true. Cells of the less uniform tec1Δ/tec1Δ polylayer extended tubes at approximately the same time as that for basal yeast-phase control cells, but the tubes continued to elongate into hyphae in a variety of directions rather than vertically during stages 4 and 5. A matrix did form that encapsulated the incipient hyphae in a/a tec1Δ/tec1Δ biofilms, but it appeared to be diminished (stage 5). The hyphae did not attain the lengths of those in control cultures during stages 4 and 5. Mutant hyphae remained oriented in all directions, either because of a defective matrix or because of the lower matrix density. Alternatively, these architectural defects may have resulted from defects in the actual process of hyphal growth. These defects resulted, in turn, in a diminutive matrix and repressed biofilm thickness. The final a/a tec1Δ/tec1Δ biofilm was approximately half as thick as the average wild-type biofilm at 48 h. Therefore, Tec1 appears to play roles both in the formation of the basal yeast-phase cell polylayer in the lower layer of the biofilm and in the extension of long vertical hyphae and matrix deposition. The increased fragility and release from the silicone elastomer in response to the shear force caused by strong rotation may reflect decreases in cohesion and adhesion, respectively.
Tec1 and matrix formation.
For a/α cells, it has been shown, using the same model, that deleting TEC1 also results in a biofilm that is 40% thinner (5), which is approximately the same as the reduction found here for a/a tec1Δ/tec1Δ cells. In a/α cells, Tec1 regulates the transcription factor Bcr1 in the formation of a normal biofilm (5, 17, 40). Deleting BCR1 resulted in a diminished and architecturally defective biofilm, primarily in the later stages of development (5). However, overexpressing BCR1 in a tec1Δ/tec1Δ mutant did not completely rescue the TEC1 deletion phenotype (5). These observations led to the hypothesis that Tec1 not only regulates BCR1 but also regulates other genes involved in the formation of a normal biofilm. Bcr1, which is regulated by Tec1, appears to regulate the period of hypha elongation and matrix deposition that results in impenetrability by human white blood cells, impermeability to low- and high-molecular-weight molecules, and drug resistance (13). These observations led to the subsequent hypothesis that in MTL-homozygous biofilm (a/a or α/α) formation, Tec1 plays a role similar to that in a/α biofilm formation but activates a transcription factor other than Bcr1 (yet to be identified), which in turn regulates the formation of a matrix that not only supports vertical hypha formation but also facilitates mating and results in increased penetrability, permeability, and drug susceptibility (13). The results presented here support the role proposed for Tec1 in this hypothesis. Experiments are now ongoing to identify the transcription factors regulated by Tec1 that are responsible for the characteristics of biofilms.
Supplementary Material
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00224-14.
REFERENCES
- 1.Daniels KJ, Park YN, Srikantha T, Pujol C, Soll DR. 2013. Impact of environmental conditions on the form and function of Candida albicans biofilms. Eukaryot Cell 12:1389–1402. doi: 10.1128/EC.00127-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daniels KJ, Srikantha T, Lockhart SR, Pujol C, Soll DR. 2006. Opaque cells signal white cells to form biofilms in Candida albicans. EMBO J 25:2240–2252. doi: 10.1038/sj.emboj.7601099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahni N, Yi S, Daniels KJ, Huang G, Srikantha T, Soll DR. 2010. Tec1 mediates the pheromone response of the white phenotype of Candida albicans: insights into the evolution of new signal transduction pathways. PLoS Biol 8:e1000363. doi: 10.1371/journal.pbio.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yi S, Sahni N, Daniels KJ, Pujol C, Srikantha T, Soll DR. 2008. The same receptor, G protein, and mitogen-activated protein kinase pathway activate different downstream regulators in the alternative white and opaque pheromone responses of Candida albicans. Mol Biol Cell 19:957–970. doi: 10.1091/mbc.E07-07-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yi S, Sahni N, Daniels KJ, Lu KL, Srikantha T, Huang G, Garnaas AM, Soll DR. 2011. Alternative mating type configurations (a/alpha versus a/a or alpha/alpha) of Candida albicans result in alternative biofilms regulated by different pathways. PLoS Biol 9:e1001117. doi: 10.1371/journal.pbio.1001117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slutsky B, Staebell M, Anderson J, Risen L, Pfaller M, Soll DR. 1987. “White-opaque transition”: a second high-frequency switching system in Candida albicans. J Bacteriol 169:189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park YN, Daniels KJ, Pujol C, Srikantha T, Soll DR. 2013. Candida albicans forms a specialized “sexual” as well as “pathogenic” biofilm. Eukaryot Cell 12:1120–1131. doi: 10.1128/EC.00112-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hickman MA, Zeng G, Forche A, Hirakawa MP, Abbey D, Harrison BD, Wang YM, Su CH, Bennett RJ, Wang Y, Berman J. 2013. The ‘obligate diploid' Candida albicans forms mating-competent haploids. Nature 494:55–59. doi: 10.1038/nature11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Legrand M, Lephart P, Forche A, Mueller FM, Walsh T, Magee PT, Magee BB. 2004. Homozygosity at the MTL locus in clinical strains of Candida albicans: karyotypic rearrangements and tetraploid formation. Mol Microbiol 52:1451–1462. doi: 10.1111/j.1365-2958.2004.04068.x. [DOI] [PubMed] [Google Scholar]
- 10.Lockhart SR, Zhao R, Daniels KJ, Soll DR. 2003. Alpha-pheromone-induced “shmooing” and gene regulation require white-opaque switching during Candida albicans mating. Eukaryot Cell 2:847–855. doi: 10.1128/EC.2.5.847-855.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Odds FC, Bougnoux ME, Shaw DJ, Bain JM, Davidson AD, Diogo D, Jacobsen MD, Lecomte M, Li SY, Tavanti A, Maiden MC, Gow NA, d'Enfert C. 2007. Molecular phylogenetics of Candida albicans. Eukaryot Cell 6:1041–1052. doi: 10.1128/EC.00041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tavanti A, Davidson AD, Fordyce MJ, Gow NA, Maiden MC, Odds FC. 2005. Population structure and properties of Candida albicans, as determined by multilocus sequence typing. J Clin Microbiol 43:5601–5613. doi: 10.1128/JCM.43.11.5601-5613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soll DR. 2014. The evolution of alternative biofilms in an opportunistic fungal pathogen: an explanation for how new signal transduction pathways may evolve. Infect Genet Evol 22:235–243. doi: 10.1016/j.meegid.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Hawser SP, Douglas LJ. 1994. Biofilm formation by Candida species on the surface of catheter materials in vitro. Infect Immun 62:915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawser SP, Baillie GS, Douglas LJ. 1998. Production of extracellular matrix by Candida albicans biofilms. J Med Microbiol 47:253–256. doi: 10.1099/00222615-47-3-253. [DOI] [PubMed] [Google Scholar]
- 16.Hawser SP, Douglas LJ. 1995. Resistance of Candida albicans biofilms to antifungal agents in vitro. Antimicrob Agents Chemother 39:2128–2131. doi: 10.1128/AAC.39.9.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nobile CJ, Mitchell AP. 2005. Regulation of cell-surface genes and biofilm formation by the C. albicans transcription factor Bcr1p. Curr Biol 15:1150–1155. doi: 10.1016/j.cub.2005.05.047. [DOI] [PubMed] [Google Scholar]
- 18.Ramage G, VandeWalle K, Lopez-Ribot JL, Wickes BL. 2002. The filamentation pathway controlled by the Efg1 regulator protein is required for normal biofilm formation and development in Candida albicans. FEMS Microbiol Lett 214:95–100. doi: 10.1111/j.1574-6968.2002.tb11330.x. [DOI] [PubMed] [Google Scholar]
- 19.Srikantha T, Daniels KJ, Pujol C, Kim E, Soll DR. 2013. Identification of genes upregulated by the transcription factor Bcr1 that are involved in impermeability, impenetrability and drug-resistance of Candida albicans a/alpha biofilms. Eukaryot Cell 12:875–888. doi: 10.1128/EC.00071-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srikantha T, Daniels KJ, Pujol C, Sahni N, Yi S, Soll DR. 2012. Nonsex genes in the mating type locus of Candida albicans play roles in a/alpha biofilm formation, including impermeability and fluconazole resistance. PLoS Pathog 8:e1002476. doi: 10.1371/journal.ppat.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yi S, Sahni N, Pujol C, Daniels KJ, Srikantha T, Ma N, Soll DR. 2009. A Candida albicans-specific region of the alpha-pheromone receptor plays a selective role in the white cell pheromone response. Mol Microbiol 71:925–947. doi: 10.1111/j.1365-2958.2008.06575.x. [DOI] [PubMed] [Google Scholar]
- 22.Yi S, Sahni N, Daniels KJ, Lu KL, Huang G, Garnaas AM, Pujol C, Srikantha T, Soll DR. 2011. Utilization of the mating scaffold protein in the evolution of a new signal transduction pathway for biofilm development. mBio 2:e00237-10. doi: 10.1128/mBio.00237-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soll DR. 2011. Evolution of a new signal transduction pathway in Candida albicans. Trends Microbiol 19:8–13. doi: 10.1016/j.tim.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Soll DR. 2014. The role of phenotypic switching in the basic biology and pathogenesis of Candida albicans. J Oral Microbiol 6:22993. doi: 10.3402/jom.v6.22993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin CH, Kabrawala S, Fox EP, Nobile CJ, Johnson AD, Bennett RJ. 2013. Genetic control of conventional and pheromone-stimulated biofilm formation in Candida albicans. PLoS Pathog 9:e1003305. doi: 10.1371/journal.ppat.1003305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramage G, Vandewalle K, Wickes BL, Lopez-Ribot JL. 2001. Characteristics of biofilm formation by Candida albicans. Rev Iberoam Micol 18:163–170. [PubMed] [Google Scholar]
- 27.Lockhart SR, Pujol C, Daniels KJ, Miller MG, Johnson AD, Pfaller MA, Soll DR. 2002. In Candida albicans, white-opaque switchers are homozygous for mating type. Genetics 162:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reuss O, Vik A, Kolter R, Morschhauser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127. doi: 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 29.Park YN, Morschhauser J. 2005. Tetracycline-inducible gene expression and gene deletion in Candida albicans. Eukaryot Cell 4:1328–1342. doi: 10.1128/EC.4.8.1328-1342.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee KL, Buckley HR, Campbell CC. 1975. An amino acid liquid synthetic medium for the development of mycelial and yeast forms of Candida albicans. Sabouraudia 13:148–153. doi: 10.1080/00362177585190271. [DOI] [PubMed] [Google Scholar]
- 31.Bedell GW, Soll DR. 1979. Effects of low concentrations of zinc on the growth and dimorphism of Candida albicans: evidence for zinc-resistant and -sensitive pathways for mycelium formation. Infect Immun 26:348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson JM, Soll DR. 1987. Unique phenotype of opaque cells in the white-opaque transition of Candida albicans. J Bacteriol 169:5579–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao R, Daniels KJ, Lockhart SR, Yeater KM, Hoyer LL, Soll DR. 2005. Unique aspects of gene expression during Candida albicans mating and possible G(1) dependency. Eukaryot Cell 4:1175–1190. doi: 10.1128/EC.4.7.1175-1190.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hauert AB, Martinelli S, Marone C, Niggli V. 2002. Differentiated HL-60 cells are a valid model system for the analysis of human neutrophil migration and chemotaxis. Int J Biochem Cell Biol 34:838–854. doi: 10.1016/S1357-2725(02)00010-9. [DOI] [PubMed] [Google Scholar]
- 35.Millius A, Weiner OD. 2010. Manipulation of neutrophil-like HL-60 cells for the study of directed cell migration. Methods Mol Biol (Clifton, NJ) 591:147–158. doi: 10.1007/978-1-60761-404-3_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sahni N, Yi S, Daniels KJ, Srikantha T, Pujol C, Soll DR. 2009. Genes selectively up-regulated by pheromone in white cells are involved in biofilm formation in Candida albicans. PLoS Pathog 5:e1000601. doi: 10.1371/journal.ppat.1000601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yi S, Sahni N, Daniels KJ, Lu KL, Huang G, Srikantha T, Soll DR. 2011. Self-induction of a/a or alpha/alpha biofilms in Candida albicans is a pheromone-based paracrine system requiring switching. Eukaryot Cell 10:753–760. doi: 10.1128/EC.05055-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramirez-Zavala B, Weyler M, Gildor T, Schmauch C, Kornitzer D, Arkowitz R, Morschhauser J. 2013. Activation of the Cph1-dependent MAP kinase signaling pathway induces white-opaque switching in Candida albicans. PLoS Pathog 9:e1003696. doi: 10.1371/journal.ppat.1003696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finkel JS, Xu W, Huang D, Hill EM, Desai JV, Woolford CA, Nett JE, Taff H, Norice CT, Andes DR, Lanni F, Mitchell AP. 2012. Portrait of Candida albicans adherence regulators. PLoS Pathog 8:e1002525. doi: 10.1371/journal.ppat.1002525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nobile CJ, Andes DR, Nett JE, Smith FJ, Yue F, Phan QT, Edwards JE, Filler SG, Mitchell AP. 2006. Critical role of Bcr1-dependent adhesins in C. albicans biofilm formation in vitro and in vivo. PLoS Pathog 2:e63. doi: 10.1371/journal.ppat.0020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis RE, Lo HJ, Raad II, Kontoyiannis DP. 2002. Lack of catheter infection by the efg1/efg1 cph1/cph1 double-null mutant, a Candida albicans strain that is defective in filamentous growth. Antimicrob Agents Chemother 46:1153–1155. doi: 10.1128/AAC.46.4.1153-1155.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baillie GS, Douglas LJ. 2000. Matrix polymers of Candida biofilms and their possible role in biofilm resistance to antifungal agents. J Antimicrob Chemother 46:397–403. doi: 10.1093/jac/46.3.397. [DOI] [PubMed] [Google Scholar]
- 43.Douglas LJ. 2003. Candida biofilms and their role in infection. Trends Microbiol 11:30–36. doi: 10.1016/S0966-842X(02)00002-1. [DOI] [PubMed] [Google Scholar]
- 44.Odds FC. 1988. Candida and candidosis: a review and bibliography, 2nd ed. Bailliere Tindall, London, United Kingdom. [Google Scholar]
- 45.Merson-Davies LA, Odds FC. 1989. A morphology index for characterization of cell shape in Candida albicans. J Gen Microbiol 135:3143–3152. [DOI] [PubMed] [Google Scholar]
- 46.Soll DR. 1986. The regulation of cellular differentiation in the dimorphic yeast Candida albicans. Bioessays 5:5–11. doi: 10.1002/bies.950050103. [DOI] [PubMed] [Google Scholar]
- 47.Uppuluri P, Pierce CG, Thomas DP, Bubeck SS, Saville SP, Lopez-Ribot JL. 2010. The transcriptional regulator Nrg1p controls Candida albicans biofilm formation and dispersion. Eukaryot Cell 9:1531–1537. doi: 10.1128/EC.00111-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.