
Figure 3. Oxygen tension modulates the transition between epithelial and mesenchymal states during meningeal reconstruction by regulating AKAP12 expression.
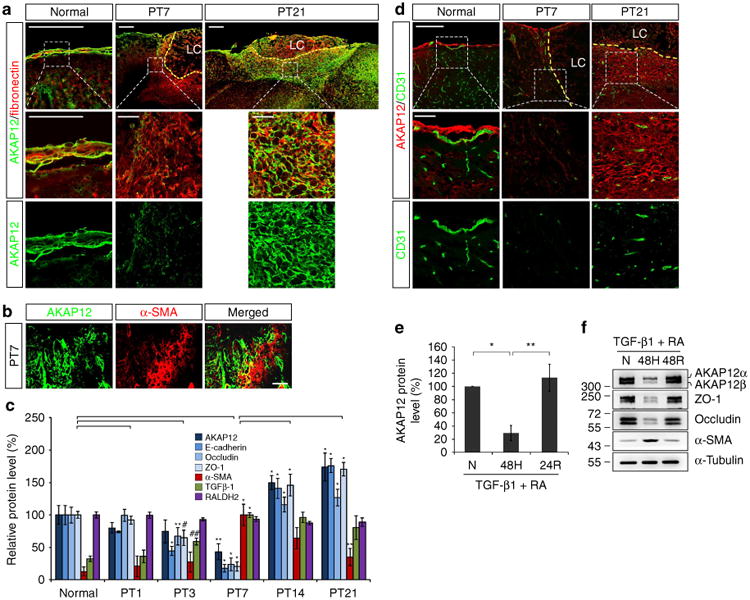
(a) AKAP12 levels were lower in meningeal cells invading into the lesional site than in the meninges. The number of AKAP12 and fibronectin double-positive cells increased over time. Scale bar, 200 μm (upper panels), 50 μm (magnified panels); LC, lesion core. (b) AKAP12 did not co-localize with a-SMA (a marker for motile mesenchymal cells). Scale bar, 40 μm. (c) AKAP12 expression was similar to that of epithelial markers in the repair process. Expression patterns of TGF-β1 and RA were not correlated with the dynamic changes in AKAP12 levels. Protein levels in lesional tissue extracts were determined by western blot analysis (mean ± s.d., n = 3 mice per time point, analysis of variance (ANOVA) followed by Tukey-Kramer test: PT3_*P<0.0005, **P = 0.039, #P = 0.012, ##P = 0.047/PT7_*P<0.0005, **P = 0.006/PT14_*P<0.0005/PT21_*P<0.0005, **P = 0.001). (d) While intact vessels were well organized near the meninges under normal conditions, severe vessel damage was observed near the lesion on day 7 after injury. Neo-microvessels were generated in and near the scar tissue surrounding the lesion core on day 21 after injury. AKAP12 levels were dynamically regulated depending on the state of the vessels. Brain microvessels were stained with an antibody against CD31 (a marker for endothelial cells). Scale bar, 200 μm (upper panels), 50 μm (magnified panels); LC, lesion core. (e) AKAP12 expression was regulated by oxygen tension. After serum starvation for 24 h, ARPE-19 cells were exposed to hypoxic conditions for 48 h and then re-oxygenated for 24 h in the presence of RA (10 μM) and TGF-β1 (10 ng ml−1; mean ± s.d., n = 4, ANOVA followed by Tukey-Kramer test: *P = 0.002, **P = 0.001). (f) Oxygen tension regulates AKAP12 and EMT marker expression in the presence of RA (10 μM) and TGF-β1 (10 ng ml−1). N, normoxia (21% oxygen); H, hypoxia (1% oxygen); R, reoxygenation (21% oxygen). Panels a,b,d,f represent the results from independent experiments repeated at least four times using different animals or conditions.