
Figure 6. AKAP12 regulates SNAI1 expression through the non-Smad pathways of TGF-β1.
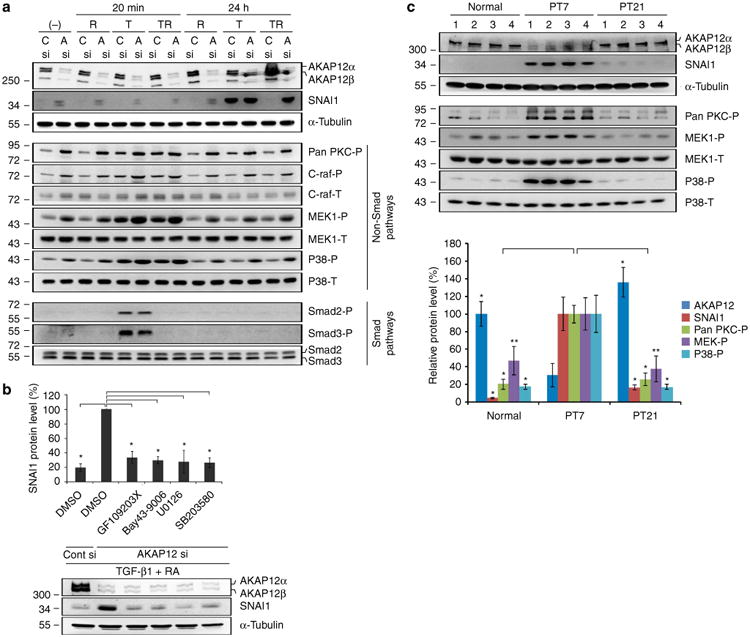
(a) AKAP12-knockdown cells were serum starved for 6 h and then treated with TGF-β1 (10 ng ml−1) and/or RA (10 μM) for 20 min and 24 h. Inhibition of AKAP12 activated the PKC/C-raf/MEK1 and p38 pathways among the examined non-Smad pathways of TGF-β1. Activation of these pathways was closely correlated with SNAI1 expression. This data represent the results from independent three experiments. R, RA; T, TGF-β1; TR, co-treatment of TGF-β1 and RA; C, Cont; A, AKAP12. (b) Specific inhibitors for PKC, C-raf, MEK1 and p38 blocked the upregulation of SNAI1 induced by AKAP12 knockdown. After serum starvation for 24 h, inhibitors were added to AKAP12-knockdown ARPE-19 cells treated with TGF-β1 (10 ng ml−1) and RA (10 μM) for 24 h (mean ± s.d., n = 3, analysis of variance (ANOVA) followed by Tukey-Kramer test: *P<0.0005). GF 109203X (general PKC inhibitor): 5 μM, Bay 43-9006 (Raf-1 inhibitor): 1 μM, U0126 (MEK1 inhibitor): 10 μM, SB203580 (P38 inhibitor): 10 μM. (c) Lesional tissues from four mice per time point were collected and protein levels in the lesional tissue extracts were determined by western blot analysis. AKAP12 expression dynamically changed during the repair process. Phosphorylation of PKC, MEK1, p38 and SNAI1 expression had the opposite pattern to that of AKAP12 levels, similar to the in vitro data. Bands were quantified with Image J. AKAP12, SNAI1 and pan-PKC-P densities were normalized to that of a-tubulin. The densities of MEK-P and p38-P were normalized to that of total MEK and total p38, respectively (mean ± s.d., n = 4 mice per time point, ANOVA followed by Tukey-Kramer test: normal_*P<0.0005, **P = 0.003/PT 21_*P<0.0005, **P = 0.001).