
Figure 7. Schematic model for prompt meningeal reconstruction and its molecular mechanism.
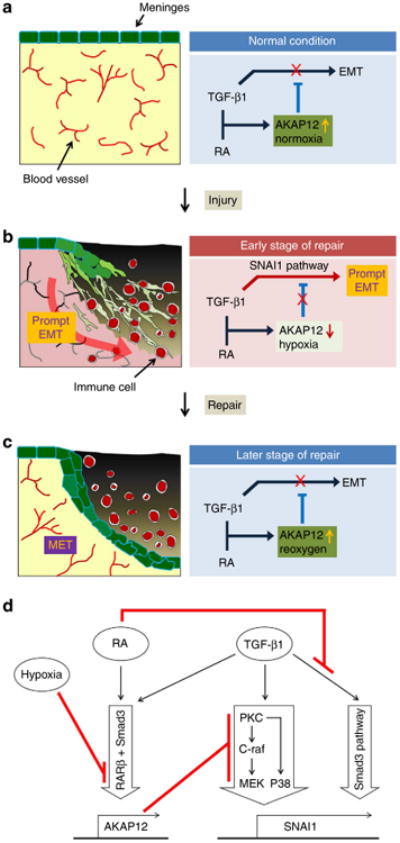
(a) Under normal conditions, meningeal cells maintain a high level of AKAP12 because of the high concentrations of TGF-β1, RA and oxygen. AKAP12 maintains the epithelial properties of the meninges by inhibiting the TGF-β1 pathway. (b) Immediately after injury, hypoxia due to vessel damage de-represses the TGF-β1-SNAI1 pathway by suppressing the expression of AKAP12 resulting in meningeal cells' EMT. Meningeal cells transited to the mesenchymal state by AKAP12 suppression invade into the site of the lesion. (c) At a later stage of repair, neo-microvessels formed by angiogenesis resupply oxygen to the site of the lesion. AKAP12 recovered by reoxygenation represses the TGF-β1-SNAI1 pathway, resulting in re-epithelialization of meningeal cells. (d) The diagram for the molecular mechanism of prompt meningeal reconstruction. Under co-treatment with RA and TGF-β1, the Smad pathway is directly blocked by RA, and non-Smad pathways are indirectly inhibited by high levels of AKAP12, the expression of which is induced by both RA and TGF-β1 under normoxic condition. In the hypoxic condition, AKAP12 expression is suppressed through RARβ repression. Then, the suppression of AKAP12 levels increases the level of SNAI1 by de-repression of the PKC/c-Raf/MEK1 and PKC/P38 pathways of TGF-β1, resulting in EMT.