

Recent genome-wide association studies have identified single nucleotide polymorphisms (SNPs) at 7 loci influencing multiple myeloma (MM) risk. We generated expression quantitative trait loci (eQTL) data on malignant plasma cells on 848 patients to fine map the risk loci associations and gain insight into their functional basis. At 7p15.3 the strongest association with MM risk is provided by rs4487645, which is associated with allele-specific cis-regulation of the MYC-interacting gene CDCA7L with P-value=4.1×10−25. rs4487645 resides within intron 80 of DNAH11 and only 47 kb upstream of the transcription start site of CDCA7L. rs4487645 maps within a binding site for interferon regulatory factor 4 (IRF4) in a strong enhancer element upstream of CDCA7L. IRF4 is a critical transcriptional regulator in B-cell development. Since rs4487645 represents the strongest signal for MM at 7p15.3 the data are compatible with increased expression of CDCA7L being the functional basis of the 7p15.3 association exerting its effects through an extended pathway involving IRF4 and MYC. None of the SNPs were eQTLs specific to cytogenetic subtypes or trans-regulating eQTLs.
Genome-wide association studies (GWAS) identified SNPs at chromosomes 2p23.3, 3p22.1, 3q26.2, 6p21.33, 7p15.3, 17p11.2, and 22q13.1 influencing MM risk.1,2 The genetic and functional basis of these associations have, however, yet to be deciphered. Most GWAS-detected SNPs influencing cancer risk map to non-coding regions of the genome implying that their functional effect is mediated through differential expression rather than change in protein sequence.3 Therefore, study of expression quantitative trait loci (eQTL) provides a means of delineating the basis of GWAS signals. A recently published sequencing study of lymphoblastoid cell lines showed that 28% of 14,000 genes had one or more eQTLs, implicating inherited variation as an important determinant of gene expression.4 Moreover, 16% of 6500 variants in the disease- and trait-related GWAS catalog (www.genome.gov/gwasstudies/) were identified as eQTLs.4
In this study, we analyzed eQTL data generated on malignant plasma cells from 848 MM patients to investigate whether MM risk SNPs influence gene expression in MM cells.
Collection of samples and clinico-pathological information from patients was undertaken with informed consent and relevant ethical review board approval in accordance with the tenets of the Declaration of Helsinki. Plasma cells were CD138-purified from bone marrow aspirates as previously published from two case series which had been the subject of a previous GWAS and an additional series of 277 German patients.1,2 The 665 German MM patients (389 male, mean age 59±9 years) used for eQTL discovery were patients of the Heidelberg University Clinic and the German-speaking Myeloma Multicenter Group. The 183 UK MM patients (107 male, mean age 64±10 years) had been enrolled in the UK Medical Research Council Myeloma IX trial. Both German and UK patients had been genotyped using Illumina Human OmniExpress-12 v.1.0 arrays. General genotyping quality control assessment was carried out as previously described,1,2 and all SNPs and samples presented in this study passed the required thresholds. Fluorescence in situ hybridization and gene expression profiling of CD138-purified plasma cells using Affymetrix U133 2.0 plus arrays were performed as described.2,5,6 Quality control was carried out using NUSE and RLE metrics to identify poorly performing arrays, which were excluded from the analysis.6 Expression data have been deposited in ArrayExpress (E-MTAB-2299) and Gene Expression Omnibus (GSE21349). Analyses were undertaken using R (v.2.8). As chip definition file (CDF) we used the Affymetrix U133 2.0 plus array custom (CDF) (v.17) mapping to Entrez genes (http://brainarray.mhri.med.umich.edu/Brainarray/Database/CustomC DF/). Microarray probes binding to polymorphic sites were identified using PiP Finder (http://bit.ly/pipfinder); probes (n=16,964/313772) were excluded from the CDF using CustomCDF (101 probesets with <3 probes were discarded). Expression data were normalized using GC-RMA. We excluded genes with log2 expression <3.5 in at least 95% of samples. After QC, and confining our analysis to autosomal genes, expression data of 9116 genes was available. The filtered set was analyzed using probabilistic estimation of expression residuals (PEER) to infer broad variance components in the data.7 For the German analysis, batch effect was included as a co-variate. PEER was used to adjust expression data for known and hidden intervening variables, such as cytogenetic subgroups. As previously advocated, we set the maximum number of unobserved factors (hidden confounders) to 100 or 25% of the case number;7 more specifically, 100 for the German series and 46 for the UK series. PEER computed residuals of expression were used for eQTL analyses. We studied the relationship between MM risk SNPs and expression of genes located within 1 Mb (cis-eQTL analysis), and all other genes (trans analysis) using MatrixEQTL under a linear model.8 In the case of cis-eQTL analyses on the 7 SNPs and by average 28 genes/SNP we used the Bonferroni adjusted P-value of 0.0003 (0.05/(28×7)= 0.00026). For the detection of trans effects we analyzed the German set using a P-value threshold of 0.05 and tested all identified SNP-gene combinations in the UK data set, considering a joint threshold of P<10−6. Effect sizes (beta) and standard errors (SE) of eQTLs were calculated using R. Prediction of untyped genotypes was based on data from the 1000 Genomes Project (Phase I integrated variant set release v.3) using IMPUTE2 after pre-phasing using SHAPE-IT. Imputed data were analyzed using SNPTEST (v.2.5) to account for uncertainties in SNP prediction. Meta analysis was performed using Metal as described.1
After quality control, expression data on plasma cells were available from 665 patients of the German and 183 patients of the UK GWAS series. cis-eQTL analysis provided data on 196 genes (Online Supplementary Table S1). Table 1 summarizes the cis-eQTL analysis on the 7 SNPs associated with MM risk. The strongest cis-eQTL was provided by the relationship between rs4487645 genotype and CDCA7L expression (Table 1). The association was consistent in both series, with the risk allele being associated with a higher CDCA7L expression (respective P-values 3.3×10–21 and 2.0×10−5 (Table 1 and Online Supplementary Figure S1). Two other MM risk SNPs showed evidence of eQTL but associations were not significant after adjustment for multiple comparisons (P<0.0003) (Online Supplementary Appendix): rs1052501 and TRAK1, rs2285803 and HLA-B and HLA-C (Table 1). Similarly no significant trans associations were shown after adjustment for multiple testing.
Table 1.
Impact of multiple myeloma risk SNPs (left columns) and other SNPs (right columns) on expression of proximal genes in malignant plasma cells of 665 German and 183 UK patients.
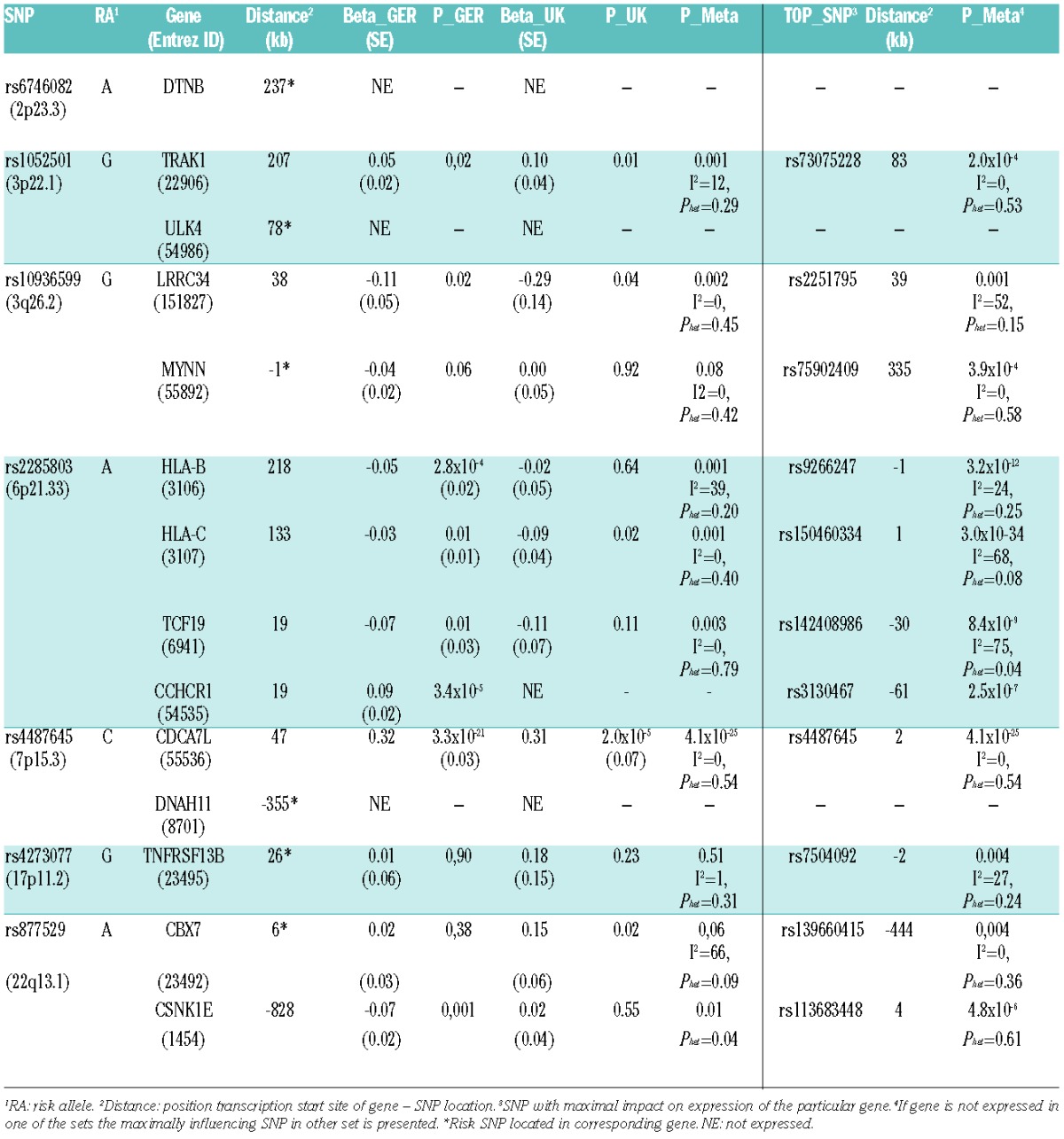
An analysis including imputed and genotyped SNPs located 1Mb each direction from the transcription start site of CDCA7L showed that rs4487645 had the strongest cis effect on the expression of this gene (Table 1, right columns). According to results of a conditional analysis rs4487645 genotype was sufficient to capture the association between variation at 7p15.3 and CDCA7L expression (Figure 1A and B). While rs4487645 resides within intron 80 of DNAH11 it is only 47 kb upstream of the transcription start site of CDCA7L. Since DNAH11 is not expressed in malignant plasma cells, and rs4487645 represents the strongest signal for MM at 7p15.3, the data are collectively consistent with this eQTL being the functional basis of the association. CDCA7L and c-Myc physically interact by the conserved leucine zipper domain of CDCA7L and the c-Myc NH2-terminal domain.9 CDCA7L potentiates c-Myc transforming activity, and can complement a transformation-defective c-Myc mutant.10 Our data showed that CDCA7L and MYC expression did not correlate in MM cells. rs4487645 maps within a binding site for interferon regulatory factor 4 (IRF4, alias myeloma oncogene 1) in a strong enhancer element upstream of CDCA7L (Regulome DB and HaploReg data for B-lymphoblastoid cell lines). In the present analysis, rs4487645 did not influence IRF4 expression. IRF4 is a critical transcriptional regulator in B-cell development,11–13 it is expressed in most stages of B-cell developmental and it has critical functions in plasma cell development.14 Specifically, IRF4 orchestrates pre-B-cell development by limiting pre-B-cell expansion and by promoting pre-B-cell differentiation.15 IRF4 is an essential gene in MM directing a broad expression program in MM cells; knockdown of IRF4 induces a rapid non-apoptotic cell death in MM cell lines.11,12 MYC is a direct target gene of IRF4 through regulation of MYC mRNA levels.
Figure 1.

Relationship between rs4487645 (7p15.3) genotype and CDCA7L expression in CD138+ selected plasma cells from 665 German and 183 UK myeloma patients. Expressions sets were analyzed using PEER6,7 to infer broad variance components in the data and computed residuals of expression were used for eQTL analyses.
Figure 2.

eQTL meta-analysis of the association between SNP genotype and CDCA7L expression at 7p15.3 (A) and the same data conditioned on rs4487645 (B). Directly genotyped SNPs are denoted by circles and imputed SNPs by squares. Associations −log10 P values (y-axis) of the SNPs are shown according to their chromosomal position (x-axis). The MM risk SNP rs4487645 is represented by a purple circle in (A) and (B). The color code of each symbol reflects the extent of LD with the risk SNP. Genetic recombination rates (cM/Mb), estimated using HapMap CEU samples, are shown in light blue lines. Physical positions are based on NCBI build 36 of the human genome. Also shown are the relative positions of genes and transcripts mapping to the region of association. On the bottom are shown all transcripts and the chromatin-state segmentation track (ChromHMM) for lymphoblastoid cells based on HapMap ENCODE Project data.
To examine if the risk alleles had different local effects on gene expression in cytogenetic subgroups of MM, we performed eQTL analyses in hyperdiploid, t(4;14) and t(11;14) MM. Expression data were pre-processed separately for each subgroup using PEER. We could not detect subgroup specific eQTLs. The association between CDCA7L expression and rs4487645 genotype showed no heterogeneity between these subgroups (data not shown).
Major strengths of the present study were that expression data were generated in malignant plasma cells obtained from the same patients on whom GWAS analyses were carried out. Other strengths were the sample size and the availability of two independent sample sets. However, our data may not necessarily fully recapitulate gene expression pertinent in early stages of plasma cell transformation. Additional limitations are that the analysis is restricted to genes for which probes were featured on the Affymetrix array. While most genes within close cis regions were captured by the array it is notable that TERC, which is likely a priori to be the basis of the chromosome 3p association, was not featured.2
In conclusion, our data are consistent with the rs4487645-CDCA7L eQTL being responsible for the chromosome 7p11.2 association with MM risk, probably exerting its effects through an extended pathway involving IRF4, and MYC.
Acknowledgments
We thank the staff of the CTRU University of Leeds and the NCRI haematology Clinical Studies Group. Finally we are grateful to all patients for their participation.
Footnotes
Funding: funding was provided by the Dietmar-Hopp-Stiftung, Germany, the Deutsche Krebshilfe (110131), Germany, the EU 7th framework program OVER-MyR, the German Ministry of Education and Science (CAMPSIMM (01ES1103) and CLIOM-MICS (01ZX1309)), and the Deutsche Forschungsgemeinschaft (SI 236/8-1, SI236/9-1, ER 155/6-1, and SFB/Transregio TRR79). In the UK Myeloma UK and Leukaemia Lymphoma Research provided the principal funding for this study. Additional funding was provided by Biological Research Centre of the National Institute for Health Research at the Royal Marsden Hospital NHS Trust.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Broderick P, Chubb D, Johnson DC, et al. Common variation at 3p22.1 and 7p15.3 influences multiple myeloma risk. Nat Genet. 2012;44(1):58–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chubb D, Weinhold N, Broderick P, et al. Common variation at 3q26.2, 6p21.33, 17p11.2 and 22q13.1 influences multiple myeloma risk. Nat Genet. 2013; 45(10):1221–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fletcher O, Houlston RS. Architecture of inherited susceptibility to common cancer. Nat Rev Cancer. 2010;10(5):353–361. [DOI] [PubMed] [Google Scholar]
- 4.Lappalainen T, Sammeth M, Friedländer MR, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker BA, Leone PE, Jenner MW, et al. Integration of global SNP-based mapping and expression arrays reveals key regions, mechanisms, and genes important in the pathogenesis of multiple myeloma. Blood. 2006;108(5):1733–1743. [DOI] [PubMed] [Google Scholar]
- 6.Meissner T, Seckinger A, Rème T, et al. Gene expression profiling in multiple myeloma--reporting of entities, risk, and targets in clinical routine. Clin Cancer Res. 2011; 17(23):7240–7247. [DOI] [PubMed] [Google Scholar]
- 7.Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc. 2012; 7(3):500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012; 28(10):1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ou XM, Chen K, Shih JC. Monoamine oxidase A and repressor R1 are involved in apoptotic signaling pathway. Proc Natl Acad Sci USA. 2006;103(29):10923–10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang A, Ho CS, Ponzielli R, et al. Identification of a novel c-Myc protein interactor, JPO2, with transforming activity in medulloblastoma cells. Cancer Res. 2005;65(13):5607–5619. [DOI] [PubMed] [Google Scholar]
- 11.Shaffer AL, Emre NCT, Lamy L, et al. IRF4 addiction in multiple myeloma. Nature. 2008; 454(7201):226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaffer AL, Emre NCT, Romesser PB, Staudt LM. IRF4: Immunity. Malignancy! Therapy? Clin Cancer Res. 2009;15(9):2954–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jourdan M, Caraux A, De Vos J, et al. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood. 2009;114(25):5173–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Silva NS, Simonetti G, Heise N, Klein U. The diverse roles of IRF4 in late germinal center B-cell differentiation. Immunol Rev. 2012; 247(1):73–92. [DOI] [PubMed] [Google Scholar]
- 15.Pathak S, Ma S, Trinh L, et al. IRF4 is a suppressor of c-Myc induced B cell leukemia. PLoS One. 2011;6(7):e22628. [DOI] [PMC free article] [PubMed] [Google Scholar]