

Abstract
Flow cytometry is a ubiquitous, multiparametric method for characterizing cellular populations. However, this method can grow increasingly complex with the number of proteins that need to be screened simultaneously: spectral emission overlap of fluorophores and the subsequent need for compensation, lengthy sample preparation, and multiple control tests that need to be performed separately must all be considered. These factors lead to increased costs, and consequently, flow cytometry is performed in core facilities with a dedicated technician operating the instrument. Here, we describe a low-cost, label-free microfluidic method that can determine the phenotypic profiles of single cells. Our method employs Node-Pore Sensing to measure the transit times of cells as they interact with a series of different antibodies, each corresponding to a specific cell-surface antigen, that have been functionalized in a single microfluidic channel. We demonstrate the capabilities of our method not only by screening two acute promyelocytic leukemia human cells lines (NB4 and AP-1060) for myeloid antigens, CD13, CD14, CD15, and CD33, simultaneously, but also by distinguishing a mixture of cells of similar size—AP-1060 and NALM-1—based on surface markers CD13 and HLA-DR. Furthermore, we show that our method can screen complex subpopulations in clinical samples: we successfully identified the blast population in primary human bone marrow samples from patients with acute myeloid leukemia and screened these cells for CD13, CD34, and HLA-DR. We show that our label-free method is an affordable, highly sensitive, and user-friendly technology that has the potential to transform cellular screening at the benchside.
Flow cytometry (FCM) is one of the cornerstones of biomedical research and clinical diagnostics. With its ability to screen individual cells for multiple protein epitopes simultaneously and subsequently identify subpopulations of cells, FCM has had a profound impact in a broad range of areas including immunology,1−3 cancer,4,5 and regenerative medicine.6,7 Recent advances in both fluorochrome and laser technologies have dramatically increased the number of proteins that can be screened simultaneously—from 2 to the current state-of-the-art of 208,9—further advancing these fields. Despite this tremendous increased capability, “multi-color” FCM can be difficult to implement given that spectral emission overlap significantly increases with the number of fluorochromes utilized simultaneously, and highly complex analysis is necessary to decouple such overlap.3,9 Additional challenges include the following: the high cost per assay to the user, lengthy sample preparation steps, and multiple control tests that need to be performed separately. Furthermore, because of its overall complexity, the need for frequent calibration, and high cost as an instrument, multicolor FCM is often located in a central facility and operated by a skilled technician.
Most recently, mass cytometry, or CyTOF, which combines FCM with mass spectrometry and can screen more than 70 parameters simultaneously, has been introduced.3,9 Although it is a paradigm-shifting technique, CyTOF does have one distinct disadvantage: cells are vaporized and are therefore not available for collection for secondary analysis or culture. Numerous “lab-on-a-chip” technologies for cell screening have also been introduced. Examples include the true miniaturization of fluorescence-activated cell sorting10−12 and dielectrophoretic or impedance cell characterization.13−17 Although successful in targeted applications, these on-chip technologies have a number of distinct disadvantages, ranging from the need for exogenous labeling with fluorophores or magnetic beads, to the limited parameters that can be screened because the hardware is not yet as sophisticated as that in FCM, to the inability to distinguish cellular subpopulations with similar morphologies or physical properties (e.g., dielectric constants, cell size, etc.).
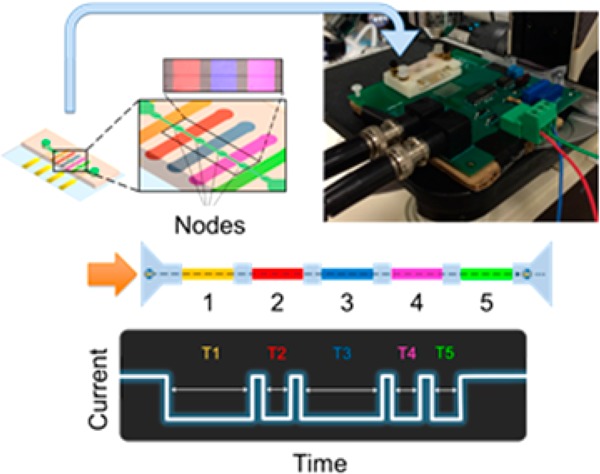
Here, we describe a unique label-free, microfluidic method that employs Node-Pore Sensing (NPS)18 to screen single cells for both size and multiple cell-surface epitopes, simultaneously. NPS is based on measuring the current pulse caused by a cell transiting a microchannel that has been segmented by a series of inserted nodes (Figure 1). Like resistive-pulse sensing (RPS),19−22 i.e., the Coulter-counter principle,23 the magnitude of the current pulse corresponds to cell size; however, unlike RPS, the current pulse in NPS is modulated, reflecting both the number and spacing of the nodes in the channel.18 When the individual segments between the nodes are functionalized with different antibodies corresponding to distinct cell-surface antigens, cells whose antigens can interact specifically with the functionalized antibodies in a particular segment will travel more slowly through that section of the channel than through the isotype-control segment. Surface-marker identification, and ultimately phenotypic profiling, is thus accomplished by comparing transit times within the modulated pulse. Unlabeled cells remain viable and are available for downstream analysis and/or culturing post screening. We demonstrate the versatility of NPS by successfully screening cells from established human cell lines for their specific phenotypic profiles and by distinguishing cell types in a mixed population based on surface-marker profiles. Moreover, we demonstrate the potential clinical value of NPS by immunophenotyping primary human bone marrow samples from acute myeloid leukemia (AML) patients. Overall, we show that NPS goes beyond current screening methods in terms of simplicity and flexibility, cost, and user friendliness.
Figure 1.

Functionalized node-pore device assembly and measurement. (A) The basic node-pore platform consists of a glass substrate with predefined platinum electrodes and gold contact pads. (B) To functionalize the node-pore device with antibodies, a temporary polydimethylsiloxane (PDMS) mold embedded with N individual microchannels, corresponding to N functionalized segments (five are shown here), is positioned onto the substrate transverse to the direction of the ultimate node-pore channel. APTES, sulfo-EGS, Protein G, and antibodies are injected and incubated into the channels to functionalize and pattern the antibodies onto the substrate (C). (D) A slab of PDMS embedded with the node-pore is aligned directly on top of the functionalized substrate such that the channel is perpendicular to the patterned antibodies. (E) The completed node-pore device has functionalized antibodies in the channel between the nodes. (inset) A pseudocolored (ImageJ) fluorescent image of three different patterned antibodies (PE Mouse IgG1, κ Isotype Control (200 μg/mL), Brilliant Violet 421 Rat IgG1, κ isotype control (50 μg/mL), Alexa Fluor 647 Mouse IgG2b, κ Isotype Control (500 μg/mL), all from Biolegend) in a completed node-pore. Scale bar, 500 μm. (F) As a cell transits a node-pore functionalized with five antibodies (top), the modulated current pulse produced (bottom) reflects the interactions between cell surface markers and the functionalized segments. For the schematic shown, the cell expresses two markers that specifically interact with Antibodies 1 and 3, leading to longer transit-times, τAb1 and τAb3, in these segments as compared to the transit-times, τAb2, τAb4, and τAb5, recorded when the cell traverses through the other segments of the node-pore.
Experimental Section
Basic Fabrication of the Node-Pore Platform
Our multimarker screening method is based on performing NPS with a single channel that has been segmented by a series of nodes of larger area (hereafter known as a “node-pore”) (Figure 1). We first utilize standard microfabrication techniques to fabricate on a glass substrate the planar electrodes that enable a four-terminal measurement of the current across the entire node-pore (Figure 1A). Next, we employ soft lithography to create a set of temporary channels whose individual widths are the same size as the segment lengths between the nodes. We align these channels transverse to the direction of the eventual node-pore to thus functionalize each segment between the nodes with a saturating concentration of either a specific antibody corresponding to a particular cell-surface antigen or an isotype-control antibody (Figures 1B,C). We also utilize soft lithography to create the node-pore itself, which we subsequently align on top of, and clamp to, the prepared substrate (Figures 1D,E). For the experiments described here, we utilized either a four-node or five-node device (each device was 18 μm × 18 μm (H × W) with 1150 μm long segments separated by 58 μm wide and 50 μm long nodes). In-plane filters24−26 are included to remove cellular debris and clusters that may clog the device. The flexibility and ease of device design and fabrication allow one to create devices with larger channels in terms of height and width, and more importantly, devices with many more nodes, corresponding to a greater number of surface antigens that could be screened, simultaneously. See Supporting Information (SI) for full details on device preparation.
Data Acquisition and Analysis
For the experiment, ∼1 × 106 cells/mL cells were injected into a prepared node-pore device, and a nonpulsatile pressure (0.07–17.20 kPa) was used to drive the cells through the device. As single cells transited through the device, a four-point terminal measurement of the current across the node-pores was performed using a constant applied DC voltage, as previously published.18,25,26 The measured current was passed through a preamplifier (DL Instrument 1211) before entering a data acquisition board (National Instruments PCI-6035E) for analysis in LabVIEW.
Cell Culture
NB4 cells were cultured at 2 × 106 cells/mL in RPMI 1640 media supplemented with 10% Fetal Bovine Serum (FBS) and 1× penicillin/streptomycin and routinely passaged as described by Lanotte et al.27 AP-1060 cells, obtained from Dr. S. Kogan, University of California-San Francisco, San Francisco, CA, U.S.A., were cultured at 1 × 106 cells/mL in 70% Iscove’s MDM supplemented with 20% FBS, 1× penicillin/streptomycin, and 10% conditioned medium from A5637 cells (ATTC) cultured in RPMI 1640 supplemented with 10% FBS and 1× penicillin/streptomycin. Cells were routinely passaged as described by Sun et al.28 NALM-1 cells (ATCC) were cultured at 1 × 106 cells/mL in RPMI 1640 supplemented with 15% FBS and 1× penicillin/streptomycin and routinely passaged as described by Minowada et al.29 For the screening experiments described, NB4, AP-1060, and NALM-1 cells were sedimented and resuspended at a concentration of ∼1 × 106 cells/mL in their respective (fresh) media prior to being injected into the node-pore device. For mixed sample testing, the samples were resuspended in a 1:1 mixture of each cell-line media.
Primary Human Bone Marrow Samples
Primary bone marrow samples were obtained from patients with AML after obtaining written informed consent to an IRB-approved protocol at The University of Chicago, which allows for the use of AML samples for future scientific research. Bone marrow samples were processed using a standard red-blood cell lysis protocol, frozen viably with 10% DMSO, and stored in liquid nitrogen. For screening, samples were thawed, suspended in prewarmed RPMI 1640 supplemented with 10% FBS and 1× penicillin/streptomycin, and spun at 300g for 3 min. Cells were then resuspended in RPMI 1640 media supplemented with 10% FBS and immediately screened with NPS. All sample screening was performed under a University of California, Berkeley IRB-approved protocol.
Results and Discussion
Operation of the Node-Pore Sensing Platform
As it enters the node-pore, a cell partially blocks the flow of current leading to an initial decrease in current across the node-pore (Figure 1F). The magnitude of the current drop corresponds to the size of the cell (see SI). When the cell enters a node, the current rises, reflecting the fact that the current density of the node, Jnode, is less than that of the channel, Jchannel. The current again decreases when the cell exits the node and enters another segment of the node-pore. Finally, as the cell exits the node-pore, the current once again returns to its baseline value. The continual rise and fall of current as a cell passes through the different nodes and segments of the node-pore is a hallmark of NPS18 and leads to current pulses that have (N + 1) distinct subpulses corresponding to N nodes in the channel. If a cell expresses a surface antigen that can interact specifically with the functionalized antibody in a particular segment of a node-pore with evenly spaced nodes, transient interactions will retard the cell as it transits through that segment, resulting in a longer subpulse duration as compared to that recorded when the cell transits the isotype-control segment (Figure 1F). Antibody arrangement or placement of the isotype-control segment does not influence transit-times, as a cell slows down significantly in the nodes between every measurement region (see Figure S-10 and SI). By functionalizing each segment of a node-pore with a different antibody that corresponds to a different cell-surface marker and by comparing the duration of the subpulses to that of the isotype control, we achieve label-free multimarker screening.
Simultaneous Screening of Single Cells for Four Surface Markers
We screened cells from two different human acute promyelocytic leukemia cell lines, NB4 and AP-1060. NB4 cells were established from a patient who was resistant to all-trans-retinoic acid (ATRA),27 and AP-1060 cells were derived from a patient who was resistant to both ATRA and arsenic trioxide (ATO).28 Although CD13 and CD33 are expressed in the majority of NB4 and AP-1060 cells, CD14 expression is absent.27,28 Moreover, CD15 is expressed moderately in NB4 cells and only weakly in AP-1060 cells.27,28 Figure 2A shows a schematic of the functionalized four-node devices we employed to screen NB4 and AP-1060 cells for these particular markers.
Figure 2.

Surface-marker profiling of leukemia cells using NPS. Acute Promyelocytic Leukemia (APL) human cell lines, NB4 and AP-1060, were screened for CD13, CD14, CD15, and CD33, simultaneously. (A) Schematic of the utilized node-pore device, which was functionalized with four specific antibodies (anti-CD13 Ab, anti-CD14 Ab, anti-CD15 Ab, and anti-CD33 Ab, all at a saturating concentration of 1 mg/mL) and an isotype control antibody (IgG1, 1 mg/mL). The device was 18 μm × 18 μm (H × W) with five 1150 μm long segments separated by four nodes, each 58 μm wide and 50 μm long. (B) Representative current pulses caused by NB4 cells (top, middle) and an AP-1060 cell (bottom) transiting the node-pore device. The modulated pulses are due to each cell traveling through each segment of the node-pore. The width of each subpulse, indicated as τCD13, τCD14, τCD15, τCD33, and τIgG1, corresponds to the transit-time of each cell as it traverses each segment. Scale bar, 0.25 s. (C) Representative normalized transit-times (τnorm = τspecific/τIgG1) of 10 NB4 cells and 10 AP-1060 cells and the resulting distribution for each marker screened. A cell is positive for a particular marker (CD13 = yellow; CD14 = red; CD15 = blue; and CD33 = pink) if τnorm is greater than a threshold cutoff (denoted as a dashed green line and defined as 1+ 2σisotype, where σisotype describes the inherent variability in nonspecific interactions between the cells and the functionalized isotype antibodies (see SI)). Thus, NB4 Cell 1 is CD13+/CD14–/CD15+/CD33+, and AP1060 Cell 3 is CD13+/CD14–/CD15–/CD33+. A summary of cells positive/negative for each marker is shown in the normalized transit-time distribution. A total of 65 NB4 cells and 127 AP-1060 cells were measured. (D) FCM analysis of cells from the same population of NB4 and AP-1060 cells measured with the node-pore device. FCM detailed data can be found in SI, Figures S-2 and S-3. 15 000 NB4 cells and 35 000 AP-1060 cells were screened. A χ2 analysis with a p-value = 0.05 shows that with exception to the AP-1060 CD14 and CD33 results, the two methods are statistically equivalent. See text for details.
Figure 2B shows representative current versus time pulses that were produced when single cells traversed the prepared node-pore. The pulses are clearly modulated, each consisting of five subpulses corresponding to a cell transiting the five different segments of the node-pore. The transit-time of each subpulse is as indicated: τCD13, τCD14, τCD15, τCD33, and τIgG1. To determine whether a cell has interacted specifically with one of the functionalized segments, we normalized the cell’s transit-time in that particular segment, τspecific, with respect to its transit-time through the isotype control, τnorm = τspecific/τisotype. To account for the inherent variability in nonspecific interactions between cells and the functionalized isotype antibodies, we conservatively defined positive expression of an antigen if τnorm > 1 + 2σisotype. Here, σisotype is the standard deviation of the normalized transit-time values we measured when we screened a sample of cells with a node-pore consisting of only multiple isotype-control segments (see SI). Figure 2C shows the phenotypic profiles of 10 individual NB4 and AP-1060 cells. Each dashed box corresponds to a single cell and contains the normalized transit-times corresponding to the markers screened (τCD13, norm = orange, τCD14, norm = red, τCD15, norm = blue, τCD33, norm = pink). The dashed green line corresponds to the cutoff value, 1 + 2σisotype. Thus, NB4 Cell 1 is CD13+/CD14–/CD15+/CD33+, and Cell 10 is CD13+/CD14–/CD15–/CD33+. Likewise, AP-1060 Cell 2 is CD13+/CD14–/CD15–/CD33+, and Cell 7 is CD13–/CD14–/CD15–/CD33+. The normalized transit-time distributions for all cells measured for each marker screened are also shown in Figure 2C. The range of normalized transit-times in each distribution is due to cells having different expression levels of a particular surface marker: as we have shown previously,25,26 cells with higher expression levels will have a greater number of interactions with the functionalized antibodies and in turn longer transit-times as compared to those cells with lower expression levels. With NPS, we found that most NB4 cells express CD13 and CD33 (92% and 83%, respectively), whereas only some express CD15 (45%), and very few express CD14 (5%). Similarly, we found that most AP-1060 cells express CD13 and CD33 (86% and 76%, respectively), far less express CD15 (21%), and very few express CD14 (3%). To validate our results, we performed FCM on samples of cells taken from the same populations screened with the node-pores (Figure 2D). By employing a χ2 test with a p-value = 0.05 (SI), we found that, with the exception of our AP-1060 CD14 and CD33 results, there were no statistically significant differences between the results obtained from NPS and those of FCM. Minor discrepancies may be due to the small sample sizes NPS screened (less than 150 cells versus greater than 15 000 cells for FCM), and/or to the fact that some cells did not have a sufficient number of transient interactions to lead to a detectable transit-time delay in NPS. For CD14, the large χ2 value was not due to a low-detection sensitivity of NPS; rather, it was a result of the fact that only a small number of cells were expected to express this marker. In other words, even minor differences in detection (3% in NPS vs 1% in FCM) would lead to a greatly exaggerated χ2 value (SI). The larger discrepancy with CD33 may be due to weak transient interaction between antibody and antigen in the node-pore versus the permanent binding between the two that occurs during incubation for FCM. As we show below, by incorporating redundancy (i.e., multiple segments functionalized with the same antibody in our device), we can easily overcome this particular limitation in NPS.
Differentiating Mixed Populations and Including Redundancy for Higher Sensitivity in the Node-Pore
We screened a 1:1 mixture of AP-1060 cells and NALM-1 cells for HLA-DR and CD13 using a node-pore that consisted of repeated segments of the same set of antibodies (i.e., anti-HLA-DR antibody and anti-CD13 antibody) (Figure 3B). NALM-1 cells, established from a patient with chronic myeloid leukemia who was in lymphoid blast crisis,29 are similar in size to AP-1060 cells. However, although AP-1060 cells are HLA-DR-/CD13+,28 NALM-1 cells are HLA-DR+/CD13–.30 We chose to employ a redundant-patterned node-pore to increase our screening method’s sensitivity. Using two segments functionalized with the same antibody, we increased the probability of transient interactions that could occur between the cell-surface antigen and that particular antibody.
Figure 3.

Analysis of a 1:1 mixture of AP-1060: NALM-1 cells using a 4-node-pore device (of similar dimension as that in Figure 2) that has repeated regions of antibody functionalization. AP-1060 cells are HLA-DR–/CD13+, while NALM-1 cells are HLA-DR+/CD13–. (A) FCM analysis of the cell mixture confirmed the nearly 1:1 mixture of cells. FCM details can be found in SI, Figure S-4. (B) Schematic of the 4-node-pore device employed. The repeated patterning of the anti-HLA-DR Ab and anti-CD13 Ab combination on either side of the patterned IgG1 was included to increase the sensitivity of screening. All regions were functionalized with 1 mg/mL of antibodies. HLA-DR 1 (region 1) = blue; CD13 1 (region 2) = red, HLA-DR 2 (region 4) = light blue; and CD13 2 (region 5) = pink). (C) Representative normalized transit-time of 10 cells from the mixed sample. As in Figure 2, a cell is determined to be positive for a surface marker if its normalized transit-time is greater than the IgG1 threshold cutoff, which is indicated as a green dashed line (see SI). Although it is HLA-DR–/CD13– in the first half of the device, Cell 1 is HLA-DR+/CD13– in the second half. We consider the cell to be overall HLA-DR+/CD13–. (D) Normalized transit-time distribution of each functionalized segment. A total of 41 cells were measured. By considering those cells like Cell 1, whose normalized transit-time is above the threshold cutoff in at least one of the two similarly functionalized segments as positive for a particular surface marker, the sensitivity of the overall device is greatly increased. A χ2 analysis with a p-value = 0.05 shows that there were no statistically significant differences between the results obtained with NPS and FCM (see SI).
The normalized transit-times of representative cells from the screened mixture are shown in Figure 3C. Using the same analysis as before, where positive expression for a marker corresponds to τnorm > 1 + 2σisotype, we found that in the first half of the device, 39% of the cells were NALM-1, and 44% were AP-1060. In contrast, in the second half of the device, only 32% of the cells were NALM-1, and 41% were AP-1060 (Figure 3D). The different percentages obtained by the two different halves of the device highlight the fact that some cells may not have had sufficient transient interactions to lead to a measurable change in transit-time in a particular functionalized segment. By considering cells as positive for a particular marker if they screened positive in at least one section, we greatly increase the sensitivity and accuracy of our screening method: 41% are thus NALM-1, and 49% are AP-1060. These results are virtually identical to those we obtained by performing FCM on the same population of cells (Figure 3A), and a χ2 test with a p-value = 0.05 confirmed this (SI). Moreover, they showcase our method’s ability to distinguish two different subpopulations accurately in a sample.
Screening Primary Human Bone Marrow Samples from Patients with AML
To demonstrate that NPS could go well beyond screening cell lines and analyze complex cellular subpopulations typically found in patient samples, we screened previously frozen bone-marrow samples from three different AML patients and characterized the blast populations therein. We employed a five-node device to measure expression of CD13, HLA-DR, CD34, and CD45, as shown in Figure 4A. Because the process of freezing and subsequent thawing of primary cells could potentially alter surface-marker expression, we also performed FCM on these samples for comparison.
Figure 4.

FCM and NPS analysis of AML patient bone-marrow samples. (A) The antibody pattern configuration for the five-node-pore used to screen the AML patient bone-marrow samples. Two different isotype-controls, IgG1 and IgG2, were included because of the specific antibodies chosen. (B) NPS cell-size distribution (left) and surface-marker normalized transit-time distributions (right) for each patient sample. Cells greater than 12 μm (dark purple in the cell-size distribution) were considered to be blasts, and the percentage of the blast population is as indicated. (C) FCM analysis of the patient samples. FCM detailed data can be found in SI, Figures S-7–S-9. FCM analyzed ∼4000, 5000, and 9000 cells for Patient 1, 2, and 3, respectively. As with the statistical analysis performed previously (Figures 2 and 3), a χ2 analysis with a p-value = 0.05 shows that, with the exception of Patient 2’s CD34 expression, there are no statistically significant differences between NPS and FCM. See text for a full detailed description.
Blast cells are large, typically 12–20 μm,31 and have low expression of CD45. Although FCM employs side-scatter (SS)/CD45 expression to identify the blast-cell population,32,33 we use cell size and consider only those cells >12 μm (dark purple in Figure 4B) to be blasts. By restricting ourselves to cell size, we could also be including subpopulations such as promyelocytes (13–25 μm),34 myelocytes (12–15 μm),35 and histiocytes (15–25 μm).31 Larger-sized cells, for example, megakaryocytes (35–150 μm),34 are excluded, as they are too large to enter into the node pore utilized. Using our size cutoff of >12 μm, we determined the blast-cell population to be 43%, 44%, and 34% for Patient 1, 2, and 3, respectively. This is in remarkable agreement with the FCM blast percentages of 46%, 36%, and 39% for the respective patients.
Again, defining positive expression for a particular surface marker as τnorm > 1 + 2σisotype (SI), we determined Patient 1, 2, and 3′s normalized blast-cell transit-time distribution for each surface marker, as shown in Figure 4B. The dashed vertical green lines indicate the positive-expression cutoff. Although all three patients showed a high percentage of cells that expressed CD13 and CD45, there was high variability in HLA-DR and CD34 expression. These results were consistent with our FCM data (Figures 4C, S-7–S-9). In fact, by employing a χ2 test with a p-value = 0.05 (SI), we find that, with the exception of Patient 2’s CD34 expression, there were no statistically significant differences between the results obtained with NPS and those of FCM (SI, Table S-4). The large χ2 value obtained for Patient 2’s CD34 expression is again not due to a lack of NPS sensitivity, but rather, due to the extremely low levels of expression expected (2% in FCM) (SI). In general, however, minor differences between NPS and FCM could mainly be attributed to their different gating strategies, with NPS’s size gating resulting in the potential inclusion of cells other than blasts.
Conclusions
NPS is a simple, yet accurate method to screen cells for multiple surface markers simultaneously. Particularly attractive is the fact that NPS is label-free and thus, unlike FCM, no sample preparation is needed. Furthermore, unlabeled cells remain viable after screening and can be collected for downstream analysis or culture (SI). Although the work we have highlighted here focused on screening four surface markers simultaneously using a four or five-node-pore device, many more markers (greater than 10) could be screened by simply adding additional nodes into the device. Although the signal-to-noise ratio drops as the length of the node pore increases, sensitivity is not lost because NPS produces unique electronic signatures that can be easily detected via spectral-domain analysis.18 This is in direct contrast to multicolor FCM, in which careful consideration of fluorophores must be taken into account in order to avoid spectral emission overlap, and in which the use of highly complex analysis is necessary when overlap does occur.3,9 Cells having a very low expression of a particular marker (leading to fewer transient binding events), and cells whose surface marker of interest has a low affinity to the specific antibody in the node-pore would have very modest transit time differences as compared to the isotype control. The time resolution of our data acquisition system is 20 μs.25 This resolution, in combination with employing the redundancy we demonstrated with the node pore, can increase the detection sensitivity of NPS in these specific cases. Currently, our screening rate is ∼15–20 cells/min (corresponding to a single cell occupying the node-pore at any given time). We can significantly increase throughput by operating several node-pores in parallel on a single chip, as we have demonstrated for single pores.36 Likewise, we can employ advanced signal processing, such as matched filtering,37 to analyze multiple cells transiting the node-pore simultaneously. These methods can increase throughput ∼100-fold. While this throughput still would not match that of FCM, NPS’s primary advantages of being label-free and requiring no sample preparation make it highly attractive. NPS could be further developed into an integrated cell screening and sorting platform, such that subpopulations could be sorted for further downstream analysis or culture.
Overall, NPS is a low-cost, simple multiparametric method to characterize cells. Even with consideration of antibody costs, the NPS node-pore platform is far less expensive than FCM. Furthermore, the platform is compact, requiring just two 15 V batteries, an external data-acquisition board, and a current preamplifier. Thus, unlike FCM, NPS could be performed at the benchside or even at the bedside. The node-pore devices are disposable and can be prepared in advance and placed in a refrigerator (or freezer for long-term storage) until needed. NPS’s ability to obtain multiparametric information on cells enables the user to employ a variety of gating strategies to identify a particular subpopulation without having to perform multiple measurements. For example, cell size could be combined with a specific surface marker for a particular gating strategy. Furthermore, the flexibility of soft lithography enables one to use a variety of differently sized channels to accommodate different sized cells. The cell screening workflow in NPS, including the incorporation of all the necessary controls needed, emphasizes the simplicity and user-friendliness of this method. Ultimately, NPS has great potential to be utilized as a label-free, multimarker screening tool for a diverse set of applications in laboratories requiring cell characterization.
Acknowledgments
We thank H. Huang for assistance with the statistical analysis performed in this work, S. Kogan for providing the AP-1060 cell line, and S. Atwater for assistance in interpreting the patient bone marrow sample FCM data. We also thank D. Schaffer, H. Stone, A. Cerchiari, M. Mir, E. Jabart, and T. Sachlos for critical reading of this manuscript. This research was funded by NIH 5R21CA156139-02 (L.L.S. and L.A.G.) and the University of California, Berkeley-Siemens Center of Knowledge Interchange (L.L.S.). K.R.B. is supported by a National Defense Science and Engineering Graduate Fellowship, and L.L.S., by a Bakar Fellowship.
Supporting Information Available
Additional information as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ (J.C.W.) Department of Biomedical Engineering, Tulane University, New Orleans, Louisiana 70118
Author Present Address
⊥ (R.H.) The Carol Franc Buck Breast Care Center, University of California, San Francisco, CA 94143
Author Present Address
∥ (J.H.H.) Department of Materials Science & Engineering, University of Delaware, Newark, Delaware 19716
Author Contributions
K.R.B. and L.L.S. developed the multimarker screening platform. K.R.B., R.H., and L.L.S. designed the experiments. K.R.B., J.C.W., and R.H. fabricated devices and performed experiments. L.A.G. guided the selection of patient bone marrow samples and assisted in the interpretation of the NPS screening results. K.R.B., J.C.W., and L.L.S. performed cell culture. J.H.H. developed the data-analysis tools and performed fluid-dynamic modeling. K.R.B. and L.L.S. analyzed the data and wrote the manuscript.
The authors declare the following competing financial interest: K.R.B. and L.L.S. are founders and shareholders of Nodexus, Inc.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Oliveira J. B.; Notarangelo L. D.; Fleisher T. A. Curr. Opin. Allergy Clin. Immunol. 2008, 8, 499–509. [DOI] [PubMed] [Google Scholar]
- Autissier P.; Soulas C.; Burdo T. H.; Williams K. C. Cytometry, Part A 2010, 77, 410–419. [DOI] [PubMed] [Google Scholar]
- Bendall S. C.; Simonds E. F.; Qiu P.; Amir el A. D.; Krutzik P. O.; Finck R.; Bruggner R. V.; Melamed R.; Trejo A.; Ornatsky O. I.; Balderas R. S.; Plevritis S. K.; Sachs K.; Pe’er D.; Tanner S. D.; Nolan G. P. Science 2011, 332, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A. Y.; True L. D.; LaTray L.; Ellis W. J.; Vessella R. L.; Lange P. H.; Higano C. S.; Hood L.; van den Engh G. Prostate 1999, 40, 192–199. [DOI] [PubMed] [Google Scholar]
- Normolle D. P.; Donnenberg V. S.; Donnenberg A. D. Cytometry, Part A 2013, 83, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagato M.; Heike T.; Kato T.; Yamanaka Y.; Yoshimoto M.; Shimazaki T.; Okano H.; Nakahata T. J. Neurosci. Res. 2005, 80, 456–466. [DOI] [PubMed] [Google Scholar]
- Pasut A.; Oleynik P.; Rudnicki M. A. Methods Mol. Biol. 2012, 798, 53–64. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay P. K.; Gaylord B.; Palmer A.; Jiang N.; Raven M. A.; Lewis G.; Reuter M. A.; Nur-ur Rahman A. K.; Price D. A.; Betts M. R.; Roederer M. Cytometry, Part A 2012, 81, 456–466. [DOI] [PubMed] [Google Scholar]
- Bendall S. C.; Nolan G. P.; Roederer M.; Chattopadhyay P. K. Trends Immunol. 2012, 33, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang H.; Hyun K. A.; Kwon M. H.; Ha K. S.; Joo C.; Jung H. I. Electrophoresis 2013, 34, 3103–3110. [DOI] [PubMed] [Google Scholar]
- Johansson L.; Nikolajeff F.; Johansson S.; Thorslund S. Anal. Chem. 2009, 81, 5188–5196. [DOI] [PubMed] [Google Scholar]
- Wolff A.; Perch-Nielsen I. R.; Larsen U. D.; Friis P.; Goranovic G.; Poulsen C. R.; Kutter J. P.; Telleman P. Lab Chip 2003, 3, 22–27. [DOI] [PubMed] [Google Scholar]
- Su H. W.; Prieto J. L.; Voldman J. Lab Chip 2013, 13, 4109–4117. [DOI] [PubMed] [Google Scholar]
- Ismail A.; Hughes M.; Mulhall H.; Oreffo R.; Labeed F. J. Tissue Eng. Regen. Med. 2012, 9, 162–168. [DOI] [PubMed] [Google Scholar]
- Mernier G.; Duqi E.; Renaud P. Lab Chip 2012, 12, 4344–4349. [DOI] [PubMed] [Google Scholar]
- Vykoukal D. M.; Gascoyne P. R.; Vykoukal J. Integr. Biol. (Camb.) 2009, 1, 477–484. [DOI] [PubMed] [Google Scholar]
- Du E.; Ha S.; Diez-Silva M.; Dao M.; Suresh S.; Chandrakasan A. P. Lab Chip 2013, 13, 3903–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan K. R.; Anwar G.; Chapman M. R.; Nguyen T.; Kesavaraju A.; Sohn L. L. Lab Chip 2013, 13, 1302–1307. [DOI] [PubMed] [Google Scholar]
- Deblois R. W.; Bean C. P. Rev. Sci. Instrum. 1970, 41, 909. [Google Scholar]
- Gregg E. C.; Steidley K. D. Biophys. J. 1965, 5, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitschek H. E. Nature 1958, 182, 234–235. [DOI] [PubMed] [Google Scholar]
- Saleh O. A.; Sohn L. L. Rev. Sci. Instrum. 2001, 72, 4449–4451. [Google Scholar]
- Coulter W. H. U. S. Patent No. 2,656,508, 1953.
- Balakrishnan K.; Sohn L. L. In Methods in Cell Biology; Conn P. M., Ed.; Elsevier: Amsterdam, 2012; pp 369–387. [Google Scholar]
- Carbonaro A.; Mohanty S. K.; Huang H.; Godley L. A.; Sohn L. L. Lab Chip 2008, 8, 1478–1485. [DOI] [PubMed] [Google Scholar]
- Chapman M. R.; Balakrishnan K.; Li J.; Conboy M. J.; Huang H.; Mohanty S. K.; Jabart E.; Hack J.; Conboy I. M.; Sohn L. L. Integrative Biology 2013, 5, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanotte M.; Martin-Thouvenin V.; Najman S.; Balerini P.; Valensi F.; Berger R. Blood 1991, 77, 1080–1086. [PubMed] [Google Scholar]
- Sun Y.; Kim S. H.; Zhou D. C.; Ding W.; Paietta E.; Guidez F.; Zelent A.; Ramesh K. H.; Cannizzaro L.; Warrell R. P.; Gallagher R. E. Leukemia 2004, 18, 1258–1269. [DOI] [PubMed] [Google Scholar]
- Minowada J.; Tsubota T.; Greaves M. F.; Walters T. R. J. Natl. Cancer Inst. 1977, 59, 83–87. [DOI] [PubMed] [Google Scholar]
- Boss M. A.; Delia D.; Robinson J. B.; Greaves M. F. Blood 1980, 56, 910–916. [PubMed] [Google Scholar]
- Loffler H. R.; Haferlach J.; Begemann T. H.; Spring Science & Business Media: Berlin, Germany, 2005. [Google Scholar]
- Brahimi M.; D S.; Touhami H.; Bkadja M. A.. J. Hematol. Thromb. Dis. 2014, 2, DOI: 10.4172/2329-8790.1000e104 [DOI] [Google Scholar]
- Lacombe F.; Durrieu F.; Briais A.; Dumain P.; Belloc F.; Bascans E.; Reiffers J.; Boisseau M. R.; Bernard P. Leukemia 1997, 11, 1878–1886. [DOI] [PubMed] [Google Scholar]
- Handin R. I. L S. E.; Stossel T. P.. Blood: Principles and Practice of Hematology; Lippincott Williams and Wilkins: Philadelphia, PA, 2003; Vol. 1, p 2304. [Google Scholar]
- Henrikson R. C. M., J E.. NMS Histiology; Lippincott Williams & Wilkins: Baltimore, MD, 1997; Vol. 518. [Google Scholar]
- Carbonaro A.; Sohn L. L. Lab Chip 2005, 5, 1155–1160. [DOI] [PubMed] [Google Scholar]
- Lustig M. Private communication.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.