

Abstract
Neuroblastoma (NB) is the third most common malignancy of childhood, and outcomes for children with advanced disease remain poor; amplification of the MYCN gene portends a particularly poor prognosis. Mxi1 antagonizes N-Myc by competing for binding to Max and E-boxes. Unlike N-Myc, Mxi1 mediates transcriptional repression and suppresses cell proliferation. Mxi1 and Mxi1-0 (an alternatively transcribed Mxi1 isoform) share identical Max and DNA binding domains but differ in amino-terminal sequences. Because of the conservation of these critical binding domains, we hypothesized that Mxi1-0 antagonizes N-Myc activity similar to Mxi1. SHEP NB cells and SHEP cells stably transfected with MYCN (SHEP/MYCN) were transiently transfected with vectors containing full-length Mxi1, full-length Mxi1-0, or the common Mxi domain encoded by exons 2 to 6 (ex2-6). After incubation in low serum, parental SHEP/MYCN cell numbers were reduced compared with SHEP cells. Activated caspase-3 staining and DNA fragmentation ELISA confirmed that SHEP/MYCN cells undergo apoptosis in low serum, while SHEP/MYCN cells transfected with Mxi1 or Mxi1-0 do not. However, SHEP/MYCN cells transfected with Mxi1 or Mxi1-0 and grown in normal serum showed proliferation rates similar to SHEP cells. Mxi ex2-6 did not affect cell number in low or normal serum, suggesting that amino terminal domains of Mxi1 and Mxi1-0 are critical for antagonism. In the absence of N-Myc, Mxi1 and Mxi1-0 induce apoptosis independently through the caspase-8–dependent extrinsic pathway, while N-Myc activates the caspase-9–dependent intrinsic pathway. Together, these data indicate that Mxi1 and Mxi1-0 antagonize N-Myc but also independently impact NB cell survival.
Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor in children and the third most common pediatric cancer overall [1]. It makes up 8% to 10% of all childhood cancers but accounts for a disproportionate 15% of cancer-related deaths in children [1]. Current treatment for advanced stage NB includes high-dose chemotherapy, radiation, and autologous hematopoietic stem cell transplantation. However, despite intensive therapy, outcomes for patients with advanced stage NB remain poor. Frequently, NB initially responds to treatment but then returns, resistant to additional therapeutic attempts. To improve outcomes in children with NB, new therapies must be developed. Understanding the mechanisms by which NB circumvents current therapies could aid in the development of new therapeutic approaches.
The MYCN oncogene is amplified in one third of NB tumors, and amplified MYCN is strongly associated with rapid disease progression and poor prognosis [2], [3], [4], [5]. Although MYCN clearly serves as a marker for advanced NB [6], the precise mechanisms by which MYCN and deregulated N-Myc protein contribute to the pathogenesis of NB remain poorly understood. N-Myc belongs to the basic helix-loop-helix leucine zipper (b-HLH-LZ) superfamily and to the MYC family of proto-oncogenes that act as transcriptional activators of growth-related target genes [7], [8]. Myc proteins dimerize with the ubiquitously expressed Max protein, bind to CACGTG (E-box) sequences [9], [10], [11], and regulate cell proliferation and differentiation [7], [12], [13], [14], [15], [16], [17], cell cycle control [18], [19], [20], and apoptosis [21], [22], [23], [24]. In addition, MYC genes play a pivotal role in the pathogenesis of neoplasia [25]. Genetic alterations resulting in Myc overexpression have been identified in a host of human malignancies, including Burkitt lymphoma, glioblastoma, and medulloblastoma [26], [27].
MYCN was originally cloned from NB cell lines by identifying amplified DNA sequences with partial homology to the MYC proto-oncogene [28], [29]. As with other Myc family members, N-Myc binds to and dimerizes with Max and binds to E-box sequences [30]. Amplification of MYCN has been shown to correlate with elevated expression of N-Myc protein [31]. Ectopic expression of N-Myc in cells leads to neoplastic transformation [32], [33]. Additionally, directed expression of N-Myc under control of the tyrosine hydroxylase promoter leads to the development of NB tumors in transgenic mice [34]. Conversely, MYCN antisense expression in NB cell lines results in reduced cell proliferation, suggesting that antagonism of N-Myc activity might reduce the aggressiveness of MYCN-amplified NB [35].
The MAD gene family encodes proteins that have significant homology to Myc and act as natural Myc antagonists [36], [37], [38], [39]. The MXI1 gene encodes the Mxi1 protein that binds Max and E-box sequences like Myc but instead functions as a transcriptional repressor [40], [41], [42], [43], [44], [45]. Several studies have demonstrated the ability of Mxi1 to counteract Myc-dependent transcription and transformation [27], [46], [47]. The specific mechanisms by which Mxi1 produces its repressive effects and regulates Myc activity remain to be determined.
While screening for upregulated genes in an NB cell line, our laboratory discovered Mxi1-0, a novel Mxi1 isoform, translated from an alternative transcript derived from a promoter upstream of a previously unidentified exon (exon 0) [48]. Because Mxi1 and Mxi1-0 share homology in the Max and E-box binding domains, we hypothesized that like Mxi1, Mxi1-0 functions as a Myc antagonist.
Here, we examine the impact of N-Myc expression on NB cell proliferation in both low and normal serum conditions. In addition, we demonstrate that both Mxi1 and Mxi1-0 are able to counteract the effects of N-Myc in low and normal serum concentrations. Finally, we show that Mxi1/Mxi1-0 activate distinct apoptotic pathways from N-Myc, indicating that these transcription factors possess distinct functions independent of their Myc inhibitory properties. Together, these observations suggest that activation of Mxi1 and Mxi1-0 may represent an attractive therapeutic target that both inhibits N-Myc function and concurrently stimulates NB cell apoptosis.
Materials and Methods
Plasmid Construction
MXI1-0 and MXI1 cDNAs were amplified from a human heart cDNA library (BD Biosciences-Clontech, Palo Alto, CA) using BamH1 sequence containing forward primers (5′-CGGGATCCCATGGGCAAACGCGGGCGG-3′ and 5′-CGCGGATCCTCTAGACCATGGAGCGGGTGAAGATGATC-3′, respectively) and a reverse primer (5′-CGCGGATCCTTAAGCGTAGTCTGGGACGTCGTATGGGTACAAGCTTGAAGTGAATGAAAGTTTGAC-3′) that includes coding sequence for an influenza hemagglutinin (HA) peptide epitope (YPYDVPDYA). This HA tag permits detection of either Mxi1-0-HA or Mxi1-HA using anti-HA antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). The MXI1-0-HA and MXI1-HA cDNAs were subcloned into the pcDNA3.1 eukaryotic expression vector (Invitrogen, Carlsbad, CA). DNA sequencing was performed using the fluorescent dideoxy terminator method of cycle sequencing on a PE/ABd 373a automated DNA sequencer following ABd protocols at the University of Michigan DNA Sequencing Core to confirm appropriate sequence and orientation of each plasmid vector.
Cell Culture
SHEP and SHEP cells stably transfected with MYCN (SHEP/MYCN) human NB cells (gift of Valerie Castle) were maintained in RPMI-1640 supplemented with 10% FBS and 50 μg/ml penicillin/streptomycin (Invitrogen-Gibco BRL, Rockville, MD) at 37°C in a humidified atmosphere with 5% CO2. Cells were plated onto 100 mm × 20 mm tissue culture dishes (Corning, Corning, NY) and passaged with 0.25% trypsin-EDTA (Invitrogen-Gibco). Cells were plated at 1 × 105 cells per well in six-well plates (Corning) for transfections.
Transfection
SHEP or SHEP/MYCN cells were transiently transfected with pcDNA3.1 empty vector or pcDNA3.1 containing MXI1-0-HA, MXI1-HA, or MXIex2-6-HA using TransIT-LT1 (Mirus, Madison, WI). Briefly, 3 μl TransIT-LT1/1 μg plasmid DNA was added to 0.2 ml of Opti-MEM (Invitrogen-Gibco BRL) for 20 minutes. This mixture was then added dropwise to cells in Opti-MEM media. After 24 hours, culture medium containing either 0.5% or 10% FBS was added back to the cells.
Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis and Western Blot Analysis
Lysates from transfected SHEP and SHEP/MYCN cells were collected in protein extraction buffer (160 mM NaCl, 65 mM Tris-HCl, 2 mM EDTA, 1% sodium dodecyl sulfate, pH 8.0) containing complete protease inhibitor cocktail (Roche, Indianapolis, IN). The lysates were boiled after the addition of Laemmli sample buffer and run on a 10% Tris-glycine gel (Bio-Rad, Hercules, CA). Proteins were then transferred to Immobilon-P membranes by wet transfer (Millipore, Bedford, MA). The membranes were blocked with 5% milk and incubated with primary rabbit polyclonal antibodies to HA (1:1000) or N-Myc (1:500; Santa Cruz Biotechnology). After washing, the membranes were incubated with an HRP-linked goat anti-rabbit secondary antibody (1:10,000; Jackson ImmunoResearch, West Grove, PA). Band detection was achieved by the addition of ECL Western Blotting Reagent and exposure to Hyperfilm ECL (Amersham Biosciences, Piscataway, NJ).
Immunocytochemistry
SHEP and SHEP/MYCN cells in 0.5% FBS were fixed with methanol/acetone (1:1) 48 hours after transfection. Cells were then blocked with 3% BSA and incubated with a primary antibody to activated caspase-3 (1:250; Promega, Madison, WI) or HA (1:1,000; Santa Cruz Biotechnology). After washing, the cells were incubated with an Alexa Fluor 488 or 594-linked anti-rabbit secondary antibody (1:100; Invitrogen-Molecular Probes, Eugene, OR). Cells were visualized with a CoolSnap CCD camera (Media Cybernetics, Carlsbad, CA) on a Nikon Eclipse E600 fluorescent microscope (Melville, NY).
3-[4,5-Dimethylthiazol-2-yl]-Diphenyltetrazolium Bromide Assay
The reagent 3-[4,5-dimethylthiazol-2-yl]-diphenyltetrazolium bromide (MTT; Sigma Chemical, St Louis, MO) was added at a final concentration of 0.5 mg/ml (1:10 from a 5 mg/ml stock) to SHEP and SHEP/MYCN cells in 0.5% or 10% FBS 96 hours after transfection. After 1 hour at 37°C, the MTT/media was removed from the cells and DMSO was added for 10 minutes. The DMSO samples were transferred to a 96-well plate (Corning). Colorimetric determination was performed at 490 nm using a SpectraMax Plus microplate reader (Molecular Devices, Sunnyvale, CA).
Bromodeoxyuridine Cell Proliferation ELISA
Proliferation was assessed by bromodeoxyuridine (BrdU) incorporation using a kit as per the manufacturer’s instructions (Roche). Briefly, BrdU was added to transfected SHEP and SHEP/MYCN cells in 0.5% or 10% FBS and proliferation was allowed to occur for 96 hours. The cells were then fixed followed by incubation with a peroxidase-conjugated antibody to BrdU. After washing, tetramethyl-benzidine was added to the cells and color was allowed to develop for 20 minutes. Samples were transferred to 96-well plates (Corning). Colorimetric determination was performed at 370 nm using a SpectraMax Plus microplate reader (Molecular Devices).
DNA Fragmentation Apoptosis ELISA
Apoptosis was determined by the presence of cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) using a kit as per the manufacturer’s instructions (Roche). Briefly, lysis buffer was added to SHEP and SHEP/MYCN 48 hours after transfection and 24 hours after the addition of 10 μM caspase inhibitor control (FMK) or inhibitors to caspase-3, caspase-8, or caspase-9 (R&D Systems, Minneapolis, MN). The lysates were centrifuged for 10 minutes to separate the cytoplasmic fraction from intact nuclei and the supernatant was transferred to a 96-well streptavidin-coated plate. An anti–histone-biotin and anti–DNA-peroxidase mixture was added to the plate and incubated for 2 hours. The plate was then washed, and 2,2′-azinobis[3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt was added for 15 minutes. Colorimetric determination was performed at 405 nm using a microplate reader (Molecular Devices).
Statistical Analyses
The two-tailed Student’s t test was used to compare two groups. Significance was considered to be P < .05. Data are reported as the means ± SD unless otherwise stated. All assays were done in triplicate and repeated at least three times.
Results
N-Myc Promotes NB Cell Proliferation in Normal Serum but Inhibits Proliferation in Low Serum
We first examined whether expression of N-Myc in SHEP NB cells alters cell proliferation. SHEP cells provide a good model for these studies, as they do not express endogenous N-Myc. Although they do express limited amounts of c-Myc, it is at levels much lower than those seen in other malignant cell lines [49]. SHEP and SHEP/MYCN were plated and incubated in either normal serum (10% FBS) or low serum (0.5% FBS) for 4 days to represent optimal growing conditions or stress conditions, respectively. A cell viability assay was used to detect differences in cell number. In normal serum, SHEP/MYCN cells showed higher cell numbers than SHEP cells (Figure 1A). In contrast, in low serum conditions, there were fewer SHEP/MYCN cells than SHEP cells (Figure 1A). To establish that the observed changes in cell number were due to changes in proliferation rather than isolated increases in cell death rate, the experiment was repeated with a BrdU proliferation ELISA (Figure 1B). Similar trends were observed, with SHEP/MYCN cells showing more BrdU incorporation when compared with SHEP cells in normal serum with the converse observed in low serum. These results indicate that N-Myc expression in SHEP cells leads to increased cell proliferation in normal serum conditions but decreased cell numbers under stress conditions.
Figure 1.
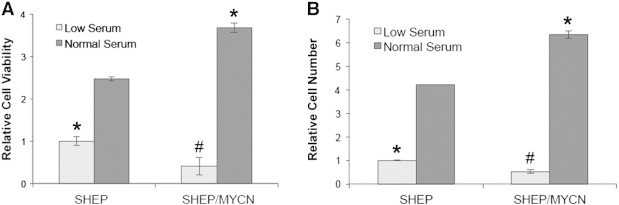
SHEP/MYCN cells exhibit higher cell numbers in normal serum and lower cell numbers in low serum than SHEP cells. SHEP and SHEP/MYCN cells were plated and cultured in RPMI-1640 containing either low serum (0.5% FBS) or normal serum (10% FBS). After 96 hours, an MTT assay (A) or BrdU proliferation ELISA (B) was run, and the samples were read at 490 nm or 370 nm, respectively. *P < .05 when compared to SHEP cells in normal serum. #P < .05 when compared to SHEP cells in low serum.
Mxi1 and Mxi1-0 Antagonize the Effects of N-Myc on Cell Proliferation and Apoptosis
Our laboratory previously described MXI1-0, an alternatively transcribed isoform of MXI1. MXI1-0 retains exons 2 to 6 of MXI1 but splices out exon 1 in favor of an alternative exon (exon 0) approximately 18 kb upstream of exon 1 [48]. However, the precise function of Mxi1-0 in NB is unknown. Since Mxi1 is an N-Myc antagonist, we used this system to determine the effects of Mxi1 and Mxi1-0 on the N-Myc–induced changes previously observed in SHEP and SHEP/MYCN cells. To further investigate the functional role of Mxi1-0, we cloned MXI1 and MXI1-0 into an expression vector. We also cloned exons 2 to 6, which are common between the two transcription factors, without either exon 1 or exon 0 (Mxiex2-6) to determine the importance of either first exon in the function of these two proteins (Figure 2A). Each of these constructs, as well as an empty pcDNA3.1 control vector, was separately transfected into SHEP and SHEP/MYCN cells. To confirm expression and comparable levels of these constructs after transfection, lysates were collected after 48 hours and separated by polyacrylamide gel electrophoresis. All constructs were expressed at relatively similar levels, and SHEP/MYCN cells displayed N-Myc expression as expected (Figure 2B). Expression of these constructs was also confirmed by immunocytochemistry against Mxi1 and Mxi1-0 48 hours after transfection in SHEP and SHEP/MYCN cells (Figure 3). Interestingly, Mxi1-0 appears to reside predominantly in the cytoplasm, while Mxi1 and Mxiex2-6 localize primarily in the nucleus.
Figure 2.
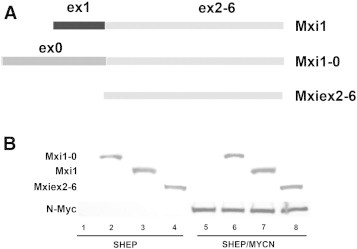
Derivation of MXI1-0 and MXI1 transcripts. (A) MXI1 mRNA is transcribed from a promoter immediately upstream of exon 1, while MXI1-0 transcription initiates upstream of exon 0. The three overexpressed proteins were Mxi1, Mxi1-0, and Mxiex2-6. (B) Western blot of SHEP and SHEP/MYCN cells transfected with either empty pcDNA3.1 vector or pcDNA3.1 containing MXI0-1-HA, MXI1-HA, or MXIex2-6-HA. After 48 hours, lysates were collected, run on sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and probed with antibodies to N-Myc or HA.
Figure 3.
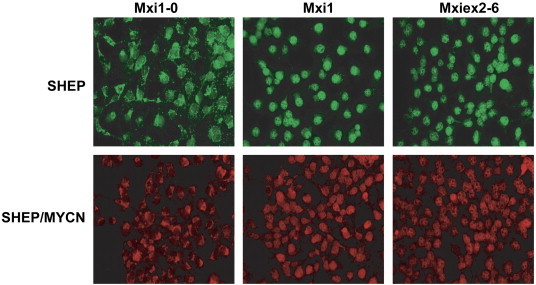
Mxi1-0, Mxi1, and Mxiex2-6 expression in SHEP and SHEP/MYCN cells. SHEP and SHEP/MYCN cells were transfected with pcDNA3.1 containing MXI1-0-HA, MXI1-HA, or MXIex2-6-HA. After 48 hours, the cells were fixed with methanol/acetone and incubated with a primary antibody to HA followed by a secondary antibody tagged with Alexa Fluor 488 (SHEP—green) or 594 (SHEP/MYCN—red). Cells were visualized with a CoolSnap CCD camera on a Nikon Eclipse E600 fluorescent microscope.
After establishing consistent expression through transient transfection, SHEP and SHEP/MYCN cells were transfected with empty vector, MXI1, MXI1-0, or MXIex2-6 (Mxi common region). Twenty-four hours after transfection, cells were placed in low or normal serum for 4 days. In low serum, SHEP cells transfected with Mxi1 or Mxi1-0 showed fewer cells than empty vector or Mxiex2-6 transfected cells (Figure 4A). The same trend was observed for SHEP cells in normal serum (Figure 4B). When SHEP/MYCN cells were incubated in normal serum, the same pattern was observed (Figure 4D). In contrast, in low serum conditions, expression of Mxi1 or Mxi1-0 resulted in increased numbers of SHEP/MYCN cells compared with empty vector or Mxiex2-6 (Figure 4C).
Figure 4.
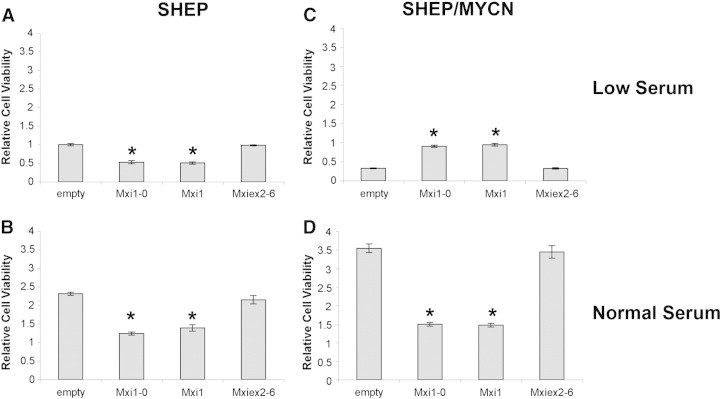
Mxi1-0 and Mxi1 antagonize N-Myc–dependent proliferation. SHEP (A and B) and SHEP/MYCN (C and D) cells were transfected with either empty pcDNA3.1 vector or pcDNA3.1 containing MXI1-0-HA, MXI1-HA, or MXIex2-6-HA. Cells were plated and cultured in RPMI-1640 containing either low serum (0.5% FBS—A and C) or normal serum (10% FBS—B and D). After 96 hours, an MTT assay was performed. *P < .05 when compared to empty vector.
Since the MTT cell viability assay only measures cell number at a specific time point and does not distinguish between cell proliferation and cell death, experiments were performed using a BrdU proliferation ELISA to investigate whether changes observed were due to cell proliferation. The trends observed above were again seen with SHEP cells in low and normal serum (Figure 5, A and B, respectively). Similarly, SHEP/MYCN cells in normal serum (Figure 5D) displayed less BrdU incorporation with Mxi1 or Mxi1-0 transfected cells than with empty vector or Mxiex2-6 transfected cells. Furthermore, SHEP/MYCN cells in low serum showed increased BrdU incorporation when transfected with Mxi1 or Mxi1-0 when compared with transfection with empty vector or Mxiex2-6; this is also consistent with the results seen using the MTT cell viability assay (Figure 5C). Together, these experiments suggest that Mxi1 and Mxi1-0 expression leads to decreased SHEP cell numbers regardless of serum concentration. In SHEP/MYCN cells, however, Mxi1 and Mxi1-0 both abrogated the N-Myc–induced increase in cell number in normal serum and prevented the decrease in cell number in low serum, suggesting that both act as N-Myc antagonists. Intriguingly, these observations raise the possibility that Mxi1 and Mxi1-0 may also have effects distinct from solely antagonizing N-Myc.
Figure 5.
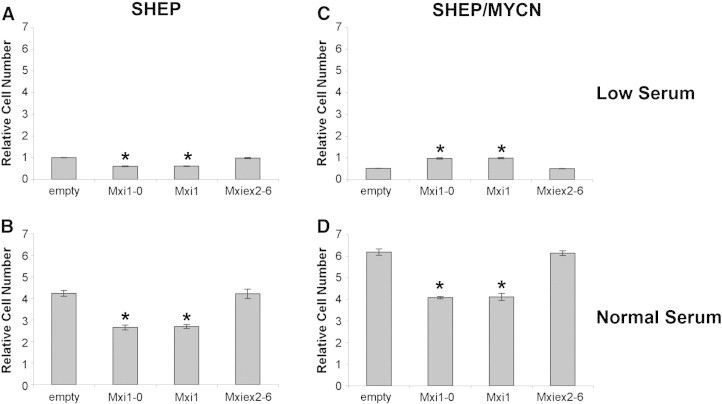
Mxi1-0 and Mxi1 block N-Myc–dependent proliferation. SHEP (A and B) and SHEP/MYCN (C and D) cells were transfected with either empty pcDNA3.1 vector or pcDNA3.1 containing MXI1-0-HA, MXI1-HA, or MXIex2-6-HA. Cells were plated and cultured in RPMI-1640 containing either low serum (0.5% FBS—A and C) or normal serum (10% FBS—B and D). After 96 hours, a BrdU proliferation ELISA was performed. *P < .05 when compared to empty vector.
N-Myc Expression Triggers Apoptosis in the Presence of Low Serum
We next explored the mechanism by which N-Myc expression is associated with reduced SHEP/MYCN cell numbers in low serum. On the basis of previous observations, we hypothesized that N-Myc expression leads to increased rates of apoptosis in the presence of low serum [50], [51]. We again exposed SHEP and SHEP/MYCN cells to either low or normal serum. After 48 hours, rate of apoptosis was quantified using a DNA fragmentation ELISA (Figure 6). Not surprisingly, the concentration of serum had no effect on the amount of apoptosis in SHEP cells in the absence of N-Myc. In contrast, incubating SHEP/MYCN cells in low serum led to a significant increase in apoptosis. This suggests that N-Myc triggers apoptosis in the presence of low serum.
Figure 6.
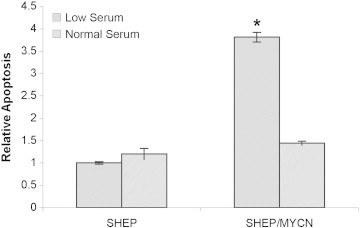
SHEP/MYCN cells undergo apoptosis in low serum. SHEP and SHEP/MYCN cells were plated and cultured in RPMI-1640 containing either low serum (0.5% FBS) or normal (10% FBS) serum. After 48 hours, a DNA fragmentation apoptosis ELISA was performed. *P < .05 when compared to SHEP at 0.5% FBS.
To confirm that apoptosis was occurring, we examined SHEP and SHEP/MYCN cells by immunohistochemical detection of activated caspase-3 after incubation in low serum for 48 hours. Abundant expression of activated caspase-3 in SHEP/MYCN cells transfected with empty vector confirmed that expression of N-Myc leads to apoptosis in low serum (Figure 7A). There was no substantial apoptosis seen in SHEP cells, consistent with what we observed using MTT and BrdU assays. We next examined whether N-Myc–mediated apoptosis could be abrogated by expression of Mxi1 and Mxi1-0. SHEP and SHEP/MYCN cells were transiently transfected with Mxi1, Mxi1-0, or Mxiex2-6 vectors before growth in low serum medium. As can be seen in Figure 7A, apoptosis triggered by N-Myc was blocked by expression of Mxi1 and Mxi1-0, indicating antagonism of N-Myc by Mxi1 and Mxi1-0. However, the Mxi common region, Mxiex2-6, did not block apoptosis, suggesting that exon 0 and exon 1 are critical for N-Myc antagonism. Interestingly, SHEP cells transfected with Mxi1 or Mxi1-0 also showed activated caspase-3 expression suggesting that they, too, were undergoing apoptosis, while SHEP cells transfected with empty vector showed no signs of apoptosis (Figure 7B). This observation further supports the hypothesis that Mxi1 and Mxi1-0 may exert effects outside of their N-Myc inhibitory capacity.
Figure 7.
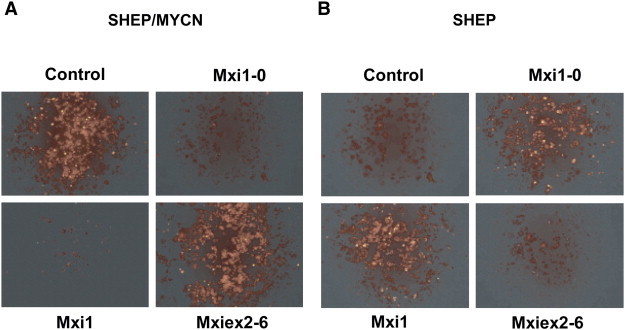
SHEP and SHEP/MYCN cells have different patterns of apoptosis in low serum in the presence of Mxi1 and Mxi1-0. SHEP/MYCN (A) and SHEP (B) cells were transfected with either pcDNA3.1 empty vector or pcDNA3.1 containing MXI1-0-HA, MXI1-HA, or MXIex2-6-HA. Cells were plated and cultured in RPMI-1640 containing low serum (0.5% FBS) for 48 hours. The cells were then fixed before incubation with a primary antibody to activated caspase-3 for immunohistochemistry.
Mxi1 and Mxi1-0 Activate Apoptosis through a Pathway Distinct from N-Myc
To elucidate the mechanism of apoptosis in response to N-Myc and Mxi1/Mxi1-0 expression, we repeated the MTT cell viability assay with transfected SHEP and SHEP/MYCN but added caspase-3, caspase-8, or caspase-9 inhibitors or a caspase inhibitor control in low serum media. SHEP cells grown in low serum showed a pattern similar to that seen in normal serum with caspase-3 and caspase-8, but not caspase-9, preventing the difference in cell proliferation between cells transfected with Mxi1 or Mxi1-0 and cells transfected with empty vector or Mxiex2-6 (Figure 8A). Consistent with what was seen in previous experiments, SHEP/MYCN cells in low serum showed higher cell viability when transfected with Mxi1 or Mxi1-0 than empty vector or Mxiex2-6 transfected cells. The baseline decrease in cell viability mediated by N-Myc was blocked by inhibitors to caspase-3 and caspase-9 but not caspase-8 (Figure 8B). These results indicate that N-Myc mediates apoptosis in low serum through the intrinsic apoptosis pathway by signaling through caspase-3 and caspase-9. In contrast, the apoptotic pathway activated by Mxi1 and Mxi1-0 in the presence of N-Myc (SHEP) involves the extrinsic pathway (caspase-8).
Figure 8.
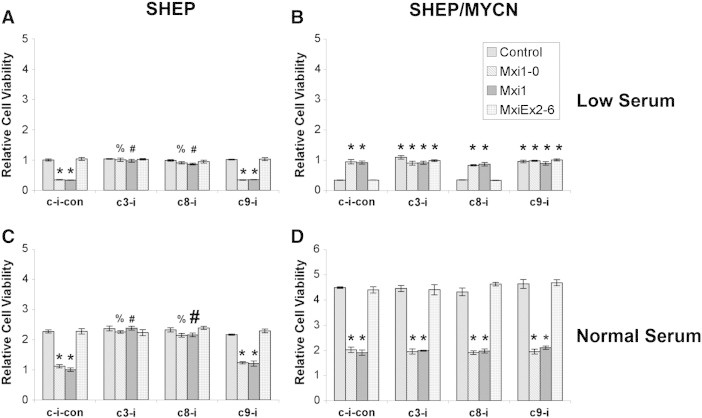
N-Myc affects apoptosis through caspase-3/caspase-9. SHEP (A and C) and SHEP/MYCN (B and D) cells were transfected with either pcDNA3.1 empty vector or pcDNA3.1 containing MXI1-0-HA, MXI1-HA, or MXIex2-6-HA. Cells were plated and cultured in RPMI-1640 containing either low serum (0.5% FBS—A and B) or normal serum (10% FBS—C and D) with or without inhibitors to caspase-3 (c3-i), caspase-8 (c8-i), or caspase-9 (c9-i) or a caspase inhibitor control (c-i-con). After 48 hours, an MTT assay was performed. *P < .05 when compared to empty vector with caspase inhibitor control. %P < .05 when compared to Mxi1-0 with caspase inhibitor control. #P < .05 when compared to Mxi1 with caspase inhibitor control.
To assess the contributions of apoptosis compared with cell growth inhibition under standard conditions, the above experiments were repeated in normal serum. For SHEP cells grown in normal serum transfected with Mxi1 or Mxi1-0, MTT levels were less than those in SHEP cells transfected with empty vector or Mxiex2-6 (Figure 8C). The addition of an inhibitor to caspase-3 abrogated this difference, with cells transfected with Mxi1 or Mxi1-0 showing the same amount of MTT uptake as empty vector or Mxiex2-6 transfected cells (Figure 8C). An inhibitor to caspase-8 also blocked this decrease, but the caspase-9 inhibitor did not. In contrast, when SHEP/MYCN cells were grown in normal serum, none of the caspase inhibitors blocked the decrease in cell viability seen with Mxi1 or Mxi1-0 transfection when compared to empty vector or Mxiex2-6 transfection (Figure 8D). These data confirm the observation that both Mxi1 and Mxi1-0 expression are able to block N-Myc–induced proliferation in SHEP/MYCN cells in normal serum rather than causing apoptosis. However, these results also indicate that Mxi1 and Mxi1-0 induce apoptosis in the absence of N-Myc, suggesting that Mxi1 and Mxi1-0 have functions independent of inhibiting N-Myc activity.
To confirm these differences, we used DNA fragmentation ELISA to specifically examine apoptosis. SHEP cells grown in low serum transfected with Mxi1 or Mxi1-0 showed increased apoptosis compared with empty vector or Mxiex2-6 transfected cells (Figure 9A). As expected, the caspase-3 and caspase-8 inhibitors blocked Mxi1 and Mxi1-0–induced apoptosis, with levels similar to those seen with empty vector and Mxiex2-6. In contrast, SHEP/MYCN cells growing in low serum underwent apoptosis by activating caspase-9 rather than caspase-8 as seen in SHEP cells. Additionally, Mxi1 or Mxi1-0 transfection blocked the apoptosis seen in empty vector or Mxiex2-6 transfected SHEP/MYCN cells grown in low serum (Figure 9B). Furthermore, the caspase-9 inhibitor did not block apoptosis in SHEP cells grown in low serum, and this same pattern was observed for SHEP cells grown in normal serum (Figure 9C). SHEP/MYCN cells grown in normal serum showed the same level of apoptosis regardless of transfection with empty vector, Mxi1, Mxi1-0, or Mxiex2-6, confirming that Mxi1 and Mxi1-0 were not contributing to apoptosis in these cells (Figure 9D). Given the activation of distinct apoptosis pathways, these data confirm the hypothesis that Mxi1 and Mxi1-0 are able to affect proliferation independently of their antagonism of N-Myc.
Figure 9.
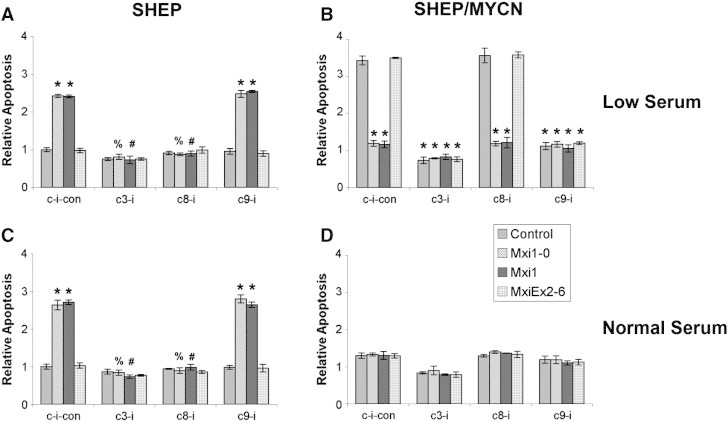
N-Myc and Mxi1-0/Mxi1 induce apoptosis through different caspase pathways. SHEP (A and C) and SHEP/MYCN (B and D) cells were transfected with either pcDNA3.1 empty vector or pcDNA3.1 containing MXI1-0-HA, MXI1-HA, or MXIex2-6-HA. Cells were plated and cultured in RPMI-1640 containing either low serum (0.5% FBS—A and B) or normal serum (10% FBS—C and D) with or without inhibitors to caspase-3 (c3-i), caspase-8 (c8-i), or caspase-9 (c9-i) or a caspase inhibitor control (c-i-con). After 48 hours, a DNA fragmentation apoptosis ELISA was performed. *P < .05 when compared to empty vector with caspase inhibitor control. %P < .05 when compared to Mxi1-0 with caspase inhibitor control. #P < .05 when compared to Mxi1 with caspase inhibitor control.
Discussion
NB is the most common extracranial solid tumor of childhood, and despite intensive treatments, outcomes for children with advanced disease remain poor [1]. Current therapies are intensive but still cure only a fraction of patients with advanced stage NB; to achieve better outcomes in this disease, new treatment modalities are needed. Amplification of the MYCN oncogene is well known to be a predictor of poor outcome [5], [6]. The Myc family of proteins plays a central role in the regulation of cell growth and proliferation. Additionally, they have been implicated in the etiology of several malignancies [25]. However, the mechanisms by which N-Myc leads to aggressive and treatment-resistant disease are unclear. A better understanding of the impact of excess N-Myc in the NB cell should lead to the development of targeted new therapies.
The MAD family proteins, including Mxi1, are intrinsic inhibitors of N-Myc function. Mxi1 plays an important role in controlling cell growth and proliferation, and its loss of function has been implicated in the formation of various malignancies, such as B-cell lymphoma and glioblastoma [26], [27]. Furthermore, expression of Mxi1 in prostate cancer cells leads to decreased growth and proliferation [47]. In glioblastoma, Mxi1 overexpression also leads to growth arrest and induction of apoptosis [27]. Furthermore, Mxi1 may be involved in modulating tumor cell physiology. In NB, Mxi1 expression is induced after exposure to hypoxic conditions and has been shown to be involved in a hypoxia pathway involving hypoxia-inducible factor 1-alpha that can inhibit c-Myc–mediated apoptosis [52], [53]. Furthermore, Mxi1 has been suggested to play a role in von Hippel-Lindau–deficient tumorigenesis [54]. In the present study, overexpression of Mxi1 in SHEP cells leads to apoptosis, which is not surprising given its inhibitory effects on cell growth and proliferation. Furthermore, Mxi1 antagonizes the effects of N-Myc when expressed in SHEP/MYCN cells. In low serum, Mxi1 expression preserves cell viability, while in normal serum, decreased cell proliferation is observed. These results support an antagonistic function for Mxi1 in NB.
Mxi1-0 is an alternatively spliced variant of Mxi1. It contains an alternative first exon (exon 0) while sharing exons 2 to 6 with Mxi1 [48]. In most systems, Mxi1-0 has not been shown to inhibit Myc proteins but may actually act as an antagonist of Mxi1 by exerting a dominant negative effect. For example, in glioblastoma, Mxi1-0 is unable to inhibit c-Myc–dependent transcription [48]. A similar dominant negative effect has also been demonstrated in esophageal adenocarcinoma [55]. Furthermore, Mxi1-0 appears to localize in the cytoplasm instead of the nucleus [48], suggesting that Mxi1-0 may act by binding critical co-factors, such as Max and Sin3, and inhibit the function of Mxi1. We observed the same pattern of localization in NB cells (Figure 3). In addition, Mxi1-0 expression, along with Mxi1, is increased under hypoxic conditions [53]. However, no changes were observed in N-Myc–dependent gene transcription, indicating that despite higher levels of Mxi1, it does not effectively inhibit N-Myc transcriptional activity [53]. Intriguingly, we have recently demonstrated that Mxi1-0 expression is upregulated in the presence of N-Myc, with a concurrent decrease in Mxi1 expression [56]. This could be a mechanism by which N-Myc decreases the activity of one of its inhibitors (Mxi1). However, the results from the present study suggest that like Mxi1, Mxi1-0 has N-Myc inhibitory properties as well, at least when expressed at high levels. Given its ability to bind Max and Sin3 like Mxi1, it is tempting to speculate that Mxi1-0 is capable of modulating transcriptional activity in the proper context.
In the present study, we have investigated the role of Mxi1 and its homolog, Mxi1-0, in the modulation of N-Myc function. Our findings examine the inter-relationships among Mxi1, Mxi1-0, and N-Myc and demonstrate a complex set of regulatory interactions among these factors (Figure 10). In normal serum, N-Myc promotes cell growth and proliferation, while in the presence of low serum, N-Myc triggers apoptosis, a process requiring activation of caspase-9. A similar pro-apoptotic role has been demonstrated for c-Myc after exposure of cells to hypoxic conditions [57]. Under normal physiologic conditions, Myc proteins induce cell growth and proliferation; however, under conditions of stress (hypoxia, low growth factors), they can switch to a pro-apoptotic role. Dysregulation of these functions may provide a permissive environment for malignancies to thrive despite the non-ideal physiologic conditions often found in tumors.
Figure 10.
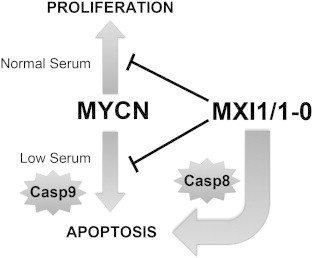
Model of NB cell apoptosis induction under normal and low serum conditions. N-Myc induces proliferation in normal serum and apoptosis through caspase-9 (Casp9) in low serum. Mxi1-0 and Mxi1 block these N-Myc–induced effects. In the absence of amplified MYCN, Mxi1-0 and Mxi1 induce apoptosis through caspase-8 (Casp8), regardless of serum concentration.
Intriguingly, our studies demonstrate that Mxi1 and Mxi1-0 induce apoptosis independently of N-Myc and that this activity is dependent on their distinct amino terminal regions. The domain common to Mxi1 and Mxi1-0 (exons 2-6) is unable to affect apoptosis, although resulting alterations in protein structure due to the elimination of amino terminal amino acids of the protein may interfere with proper folding of the protein. Unlike the N-Myc–mediated pathway, Mxi1 and Mxi1-0 activate caspase-8 to trigger apoptosis. Caspase-8 is part of the extrinsic pathway of apoptosis, which is typically activated by binding of death receptors of the tumor necrosis factor receptor family (for review, see [58]). Binding of the death receptor activates the Fas-associated death domain, leading to the creation of the death-inducing signaling complex [58]. The death-inducing signaling complex is able to recruit and activate caspase-8 and caspase-10, leading to activation of the apoptosis cascade. Caspase-9, however, is activated through the internal pathway that is triggered by mitochondrial cytochrome c release [58]. Our data indicate that Mxi1 and Mxi1-0 are able to induce apoptosis by a mechanism independent of their N-Myc inhibitory functions. The mechanism by which Mxi1 and Mxi1-0 exert their pro-apoptotic function is not clear but suggests a potential larger role for regulation of cell growth and proliferation in NB outside of modulating the transcriptional activity of N-Myc.
In summary, Mxi1 and Mxi1-0 function in NB to inhibit the transcriptional activity of N-Myc. Additionally, they possess independent anti-proliferative activity in the absence of N-Myc. Given their role in preventing NB cell proliferation, Mxi1 and Mxi1-0 or their downstream effectors may be potential targets to prevent NB growth and to sensitize it to therapeutic agents. Further understanding of the mechanisms of action of these two factors may shed light on other potential targets for therapy.
Acknowledgements
We appreciate the constructive input of Barry Wolf, Emily Fox, and Datong Zheng. We are grateful to Valerie Castle for sharing reagents. M.B.A. was supported by a Hyundai Hope on Wheels Hope grant, an Alex’s Lemonade Stand Research Award, National Institutes of Health grant K12 5K12HD043494, and support from the Howell family. D.S.W. was supported by National Cancer Institute grant 1R01CA92171 and the Strokes Against Cancer Foundation.
References
- 1.Maris J.M., Hogarty M.D., Bagatell R., Cohn S.L. Neuroblastoma. Lancet. 2007;369(9579):2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.Brisse H.J., McCarville M.B., Granata C., Krug K.B., Wootton-Gorges S.L., Kanegawa K., Giammarile F., Schmidt M., Shulkin B.L., Matthay K.K. Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Project. Radiology. 2011;261(1):243–257. doi: 10.1148/radiol.11101352. [DOI] [PubMed] [Google Scholar]
- 3.Brodeur G.M. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3(3):203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 4.Maris J.M., Matthay K.K. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17(7):2264. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 5.Seeger R.C., Brodeur G.M., Sather H., Dalton A., Siegel S.E., Wong K.Y., Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985;313(18):1111–1116. doi: 10.1056/NEJM198510313131802. [DOI] [PubMed] [Google Scholar]
- 6.Brodeur G.M., Seeger R.C., Schwab M., Varmus H.E., Bishop J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224(4653):1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 7.Evan G.I., Littlewood T.D. The role of c-myc in cell growth. Curr Opin Genet Dev. 1993;3(1):44–49. doi: 10.1016/s0959-437x(05)80339-9. [DOI] [PubMed] [Google Scholar]
- 8.Jones N. Introduction: Transcription factors, differentiation and cancer. Semin Cancer Biol. 1990;1:1–3. [Google Scholar]
- 9.Blackwood E.M., Kretzner L., Eisenman R.N. Myc and Max function as a nucleoprotein complex. Curr Opin Genet Dev. 1992;2(2):227–235. doi: 10.1016/s0959-437x(05)80278-3. [DOI] [PubMed] [Google Scholar]
- 10.Kato G.J., Lee W.M., Chen L.L., Dang C.V. Max: functional domains and interaction with c-Myc. Genes Dev. 1992;6(1):81–92. doi: 10.1101/gad.6.1.81. [DOI] [PubMed] [Google Scholar]
- 11.Kato G.J., Wechsler d.S., Dang C.V. DNA binding by the Myc Oncoproteins. In: Benz C.C., Liu E.T., editors. Oncogenes and Tumor Suppressor Genes in Human Malignancies. Kluwer Academic Publishers; Boston: 1992. pp. 313–325. [Google Scholar]
- 12.Claassen G.F., Hann S.R. Myc-mediated transformation: the repression connection. Oncogene. 1999;18(19):2925–2933. doi: 10.1038/sj.onc.1202747. [DOI] [PubMed] [Google Scholar]
- 13.Cole M.D. The myc oncogene: its role in transformation and differentiation. Annu Rev Genet. 1986;20:361–384. doi: 10.1146/annurev.ge.20.120186.002045. [DOI] [PubMed] [Google Scholar]
- 14.Dang C.V., Lee L.A. Medical Intelligence Unit. R.G. Landes Company; Austin, Texas, USA: 1995. c-MYC Function in NeoplasiaAustin; p. 196. [Google Scholar]
- 15.DePinho R.A., Schreiber-Agus N., Alt F.W. myc Family oncogenes in the development of normal and neoplastic cells. Adv Cancer Res. 1991;57:1–46. doi: 10.1016/s0065-230x(08)60994-x. [DOI] [PubMed] [Google Scholar]
- 16.Henriksson M., Lüscher B. Proteins of the Myc Network: Essential Regulators of Cell Growth and Differentiation. Adv Cancer Res. 1996;68:109–182. doi: 10.1016/s0065-230x(08)60353-x. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt E.V. The role of c-myc in cellular growth control. Oncogene. 1999;18(19):2988–2996. doi: 10.1038/sj.onc.1202751. [DOI] [PubMed] [Google Scholar]
- 18.Bodrug S.E., Warner B.J., Bath M.L., Lindeman G.J., Harris A.W., Adams J.M. Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J. 1994;13(9):2124–2130. doi: 10.1002/j.1460-2075.1994.tb06488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Obaya A.J., Mateyak M.K., Sedivy J.M. Mysterious liaisons: the relationship between c-Myc and the cell cycle. Oncogene. 1999;18(19):2934–2941. doi: 10.1038/sj.onc.1202749. [DOI] [PubMed] [Google Scholar]
- 20.Philipp A., Schneider A., Vasrik I., Finke K., Xiong Y., Beach D., Alitalo K., Eilers M. Repression of cyclin D1: a novel function of MYC. Mol Cell Biol. 1994;14(6):4032–4043. doi: 10.1128/mcb.14.6.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amati B., Brooks M.W., Levy N., Littlewood T.D., Evan G.I., Land H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell. 1993;72(2):233–245. doi: 10.1016/0092-8674(93)90663-b. [DOI] [PubMed] [Google Scholar]
- 22.Evan G.I., Wyllie A.H., Gilbert C.S., Littlewood T.D., Land H., Brooks M., Waters C.M., Penn L.Z., Hancock D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69(1):119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 23.Harrington E.A., Fanidi A., Evan G.I. Oncogenes and cell death. Curr Opin Genet Dev. 1994;4(1):120–129. doi: 10.1016/0959-437x(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 24.Prendergast G.C. Mechanisms of apoptosis by c-Myc. Oncogene. 1999;18(19):2967–2987. doi: 10.1038/sj.onc.1202727. [DOI] [PubMed] [Google Scholar]
- 25.Nesbit C.E., Tersak J.M., Prochownik E.V. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18(19):3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 26.Guo X.L., Pan L., Zhang X.J., Suo X.H., Niu Z.Y., Zhang J.Y., Wang F., Dong Z.R., Da W., Ohno R. Expression and mutation analysis of genes that encode the Myc antagonists Mad1, Mxi1 and Rox in acute leukaemia. Leuk Lymphoma. 2007;48(6):1200–1207. doi: 10.1080/10428190701342018. [DOI] [PubMed] [Google Scholar]
- 27.Wechsler D.S., Shelly C.A., Petroff C.A., Dang C.V. MXI1, a putative tumor suppressor gene, suppresses growth of human glioblastoma cells. Cancer Res. 1997;57(21):4905–4912. [PubMed] [Google Scholar]
- 28.Kohl N.E., Kanda N., Schreck R.R., Bruns G., Latt S.A., Gilbert F., Alt F.W. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell. 1983;35(2 Pt 1):359–367. doi: 10.1016/0092-8674(83)90169-1. [DOI] [PubMed] [Google Scholar]
- 29.Schwab M., Alitalo K., Klempnauer K.H., Varmus H.E., Bishop J.M., Gilbert F., Brodeur G., Goldstein M., Trent J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 1983;305(5931):245–248. doi: 10.1038/305245a0. [DOI] [PubMed] [Google Scholar]
- 30.Wenzel A., Cziepluch C., Hamann U., Schurmann J., Schwab M. The N-Myc oncoprotein is associated in vivo with the phosphoprotein Max(p20/22) in human neuroblastoma cells. EMBO J. 1991;10(12):3703–3712. doi: 10.1002/j.1460-2075.1991.tb04938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bordow S.B., Norris M.D., Haber P.S., Marshall G.M., Haber M. Prognostic significance of MYCN oncogene expression in childhood neuroblastoma. J Clin Oncol. 1998;16(10):3286–3294. doi: 10.1200/JCO.1998.16.10.3286. [DOI] [PubMed] [Google Scholar]
- 32.Schwab M., Varmus H.E., Bishop J.M. Human N-myc gene contributes to neoplastic transformation of mammalian cells in culture. Nature. 1985;316(6024):160–162. doi: 10.1038/316160a0. [DOI] [PubMed] [Google Scholar]
- 33.Small M.B., Hay N., Schwab M., Bishop J.M. Neoplastic transformation by the human gene N-myc. Mol Cell Biol. 1987;7(5):1638–1645. doi: 10.1128/mcb.7.5.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiss W.A., Aldape K., Mohapatra G., Feuerstein B.G., Bishop J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16(11):2985–2995. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt M.L., Salwen H.R., Manohar C.F., Ikegaki N., Cohn S.L. The biological effects of antisense N-myc expression in human neuroblastoma. Cell Growth Differ. 1994;5(2):171–178. [PubMed] [Google Scholar]
- 36.Chin L., Liegeois N., DePinho R.A., Schreiber-Agus N. Functional interactions among members of the Myc superfamily and potential relevance to cutaneous growth and development. J Investig Dermatol Symp Proc. 1996;1(2):128–135. [PubMed] [Google Scholar]
- 37.Hurlin P.J., Ayer D.E., Grandori C., Eisenman R.N. The Max transcription factor network: involvement of Mad in differentiation and an approach to identification of target genes. [Review] [42 refs] Cold Spring Harb Symp Quant Biol. 1994;59:109–116. doi: 10.1101/sqb.1994.059.01.014. [DOI] [PubMed] [Google Scholar]
- 38.Luscher B. Function and regulation of the transcription factors of the Myc/Max/Mad network. Gene. 2001;277(1–2):1–14. doi: 10.1016/s0378-1119(01)00697-7. [DOI] [PubMed] [Google Scholar]
- 39.Zhou Z.Q., Hurlin P.J. The interplay between Mad and Myc in proliferation and differentiation. Trends Cell Biol. 2001;11(11):S10–S14. doi: 10.1016/s0962-8924(01)02121-3. [DOI] [PubMed] [Google Scholar]
- 40.Alland L., Muhle R., Hou H., Jr., Potes J., Chin L., Schreiber-Agus N., DePinho R.A. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387(6628):49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- 41.Ayer D.E., Lawrence Q.A., Eisenman R.N. Mad-Max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell. 1995;80(5):767–776. doi: 10.1016/0092-8674(95)90355-0. [DOI] [PubMed] [Google Scholar]
- 42.Harper S.E., Qiu Y., Sharp P.A. Sin3 corepressor function in Myc-induced transcription and transformation. Proc Natl Acad Sci U S A. 1996;93(16):8536–8540. doi: 10.1073/pnas.93.16.8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao G., Alland L., Guida P., Schreiber-Agus N., Chen K., Chin L., Rochelle J.M., Seldin M.F., Skoultchi A.I., DePinho R.A. Mouse Sin3A interacts with and can functionally substitute for the amino-terminal repression of the Myc antagonist Mxi1. Oncogene. 1996;12(5):1165–1172. [PubMed] [Google Scholar]
- 44.Schreiber-Agus N., Chin L., Chen K., Torres R., Rao G., Guida P., Skoultchi A.I., DePinho R.A. An amino-terminal domain of Mxi1 mediates anti-Myc oncogenic activity and interacts with a homolog of the yeast transcriptional repressor SIN3. Cell. 1995;80(5):777–786. doi: 10.1016/0092-8674(95)90356-9. [DOI] [PubMed] [Google Scholar]
- 45.Zervos A.S., Gyuris J., Brent R. Mxi1, a protein that specifically interacts with Max to bind Myc-Max recognition sites. Cell. 1993;72(2):223–232. doi: 10.1016/0092-8674(93)90662-a. [DOI] [PubMed] [Google Scholar]
- 46.Manni I., Tunici P., Cirenei N., Albarosa R., Colombo B.M., Roz L., Sacchi A., Piaggio G., Finocchiaro G. Mxi1 inhibits the proliferation of U87 glioma cells through down-regulation of cyclin B1 gene expression. Br J Cancer. 2002;86(3):477–484. doi: 10.1038/sj.bjc.6600065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taj M.M., Tawil R.J., Engstrom L.D., Zeng Z., Hwang C., Sanda M.G., Wechsler D.S. Mxi1, a Myc antagonist, suppresses proliferation of DU145 human prostate cells. Prostate. 2001;47(3):194–204. doi: 10.1002/pros.1063. [DOI] [PubMed] [Google Scholar]
- 48.Engstrom L.D., Youkilis A.S., Gorelick J.L., Zheng D., Ackley V., Petroff C.A., Benson L.Q., Coon M.R., Zhu X., Hanash S.M. Mxi1-0, an alternatively transcribed Mxi1 isoform, is overexpressed in glioblastomas. Neoplasia. 2004;6(5):660–673. doi: 10.1593/neo.04244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qing G., Li B., Vu A., Skuli N., Walton Z.E., Liu X., Mayes P.A., Wise D.R., Thompson C.B., Maris J.M. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell. 2012;22(5):631–644. doi: 10.1016/j.ccr.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fulda S., Lutz W., Schwab M., Debatin K.M. MycN sensitizes neuroblastoma cells for drug-induced apoptosis. Oncogene. 1999;18(7):1479–1486. doi: 10.1038/sj.onc.1202435. [DOI] [PubMed] [Google Scholar]
- 51.Packham G., Porter C.W., Cleveland J.L. c-Myc induces apoptosis and cell cycle progression by separable, yet overlapping, pathways. Oncogene. 1996;13(3):461–469. [PubMed] [Google Scholar]
- 52.Corn P.G., Ricci M.S., Scata K.A., Arsham A.M., Simon M.C., Dicker D.T., El-Deiry W.S. Mxi1 is induced by hypoxia in a HIF-1-dependent manner and protects cells from c-Myc-induced apoptosis. Cancer Biol Ther. 2005;4(11):1285–1294. doi: 10.4161/cbt.4.11.2299. [DOI] [PubMed] [Google Scholar]
- 53.Lofstedt T., Fredlund E., Noguera R., Navarro S., Holmquist-Mengelbier L., Beckman S., Pahlman S., Axelson H. HIF-1alpha induces MXI1 by alternate promoter usage in human neuroblastoma cells. Exp Cell Res. 2009;315(11):1924–1936. doi: 10.1016/j.yexcr.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 54.Wong W.J., Simon M.C. The role of MXI1 in VHL deficient tumorigenesis. Cancer Biol Ther. 2008;7(10):1628–1629. doi: 10.4161/cbt.7.10.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boult J.K., Taniere P., Hallissey M.T., Campbell M.J., Tselepis C. Oesophageal adenocarcinoma is associated with a deregulation in the MYC/MAX/MAD network. Br J Cancer. 2008;98:1985–1992. doi: 10.1038/sj.bjc.6604398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Armstrong M., Mody R., Ellis D., Hill A., Erichsen D., Wechsler D. N-Myc Differentially Regulates Expresion of MXI1 Isoforms in Neuroblastoma. Neoplasia. 2013;15:1363–1370. doi: 10.1593/neo.11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brunelle J.K., Santore M.T., Budinger G.R., Tang Y., Barrett T.A., Zong W.X., Kandel E., Keith B., Simon M.C., Thompson C.B. c-Myc sensitization to oxygen deprivation-induced cell death is dependent on Bax/Bak, but is independent of p53 and hypoxia-inducible factor-1. J Biol Chem. 2004;279(6):4305–4312. doi: 10.1074/jbc.M312241200. [DOI] [PubMed] [Google Scholar]
- 58.Howley B., Fearnhead H.O. Caspases as therapeutic targets. J Cell Mol Med. 2008;12(5A):1502–1516. doi: 10.1111/j.1582-4934.2008.00292.x. [Review] [DOI] [PMC free article] [PubMed] [Google Scholar]