

Abstract
Menkes disease (MIM 309400) is an X-linked, neurodegenerative disorder resulting from deficient activity of copper-dependent enzymes and caused by alterations in the APT7A gene. In its classic form, it manifests in boys with hypotonia, seizures, skin and joint laxity, hair twisting (pili torti), cerebrovascular tortuosity, and bladder diverticulae. Menkes disease phenotypes have been reported in females with X; autosome translocations- disrupting ATP7A gene function- or ATP7A gene alterations. Those females manifest variable clinical findings, some of which, such as pili torti, seizure presence and/or age of onset, cerebrovascular tortuosity, degree of intellectual disability, bladder divericulae are largely under-reported and under-studied. Here, we report three females with Menkes disease and variant phenotypes, sharing characteristic features; one with classic Menkes disease and two with Menkes disease variants. We conclude that Menkes disease in females manifests with a variable spectrum of clinical findings but a few are uniformly present such as neurodevelopmental disability, hypotonia, and connective tissue findings. Others, such as seizures, cerebral atrophy, and cerebrovascular tortuosity may be present but are under-reported and under-studied. We propose that the diagnosis of Menkes disease or variants in females with suspicious clinical findings is an important one to consider as early treatment with parenteral copper may be considered. The effect of this treatment on the disease course in females with MD is unknown and remains to be seen.
Keywords: Menkes disease, Menkes syndrome, ATP7A, kinky hair syndrome, pili torti, copper deficiency
Introduction
Menkes disease (MIM 309400) is an X-linked neurodegenerative disorder resulting from subnormal Cu transport and resultant deficient activity of Cu-dependent enzymes. This transport is facilitated via an ATPase encoded by the ATP7A gene on chromosome Xq21.1. This is a rare disorder with an estimated incidence of 1 in 100,000 births [Kaler SG, 2010].
Classic features include hypotonia, developmental delay, seizures, skin and joint laxity, “twisted” hair (pili torti) with or without hypopigmentation, and connective tissue findings, such as cerebrovascular tortuosity and bladder diverticulae. A milder form is occasionally seen in males with mild intellectual disability, muscle weakness, tremor, ataxia, connective tissue signs, pili torti, and later-onset seizures [Kaler SG, 2010].
MD variant phenotypes have been reported in some females, mostly associated with X- autosome translocations involving the ATP7A locus. Heterozygous females are thought to be asymptomatic; however, half of obligate carrier females show regions of pili torti [Moore and Howell, 1985]. No females with classic MD have been reported to our knowledge.
Early treatment with parenteral copper (Cu) has been tried in some boys with classic MD and improved the neurodevelopmental outcome in a subset [Kaler et al, 2008; Kaler et al, 2010]. Cu treatment has never been studied in females with MD or variants.
Here, we describe the clinical manifestations and laboratory findings in a female with classic MD and two others with MD variants; we also summarize the published reports of females with MD.
Clinical Report
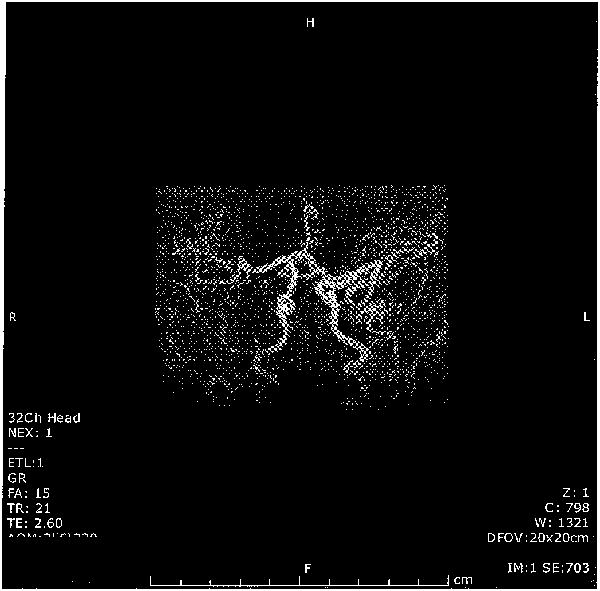
Patient 1 is a 22 month old girl who presented at 7 months old with hypotonia. Physical exam showed myopathic face with sagging cheeks, “kinky” hair with silvery streaks, skin and joint laxity, and truncal hypotonia. On microscopic hair examination, 20% of sampled hair shafts showed pili torti. She had low serum Cu at 19 mcg/dL (normal 24-152) and low serum ceruloplasmin at 9 mg/dL (normal 15-43) at 7 months of age. Radiographs showed “wormian” bones and general osteopenia. She has marked cerebrovascular tortuosity (Figure I) and delayed myelination. She has no bladder diverticulae. ATP7A gene analysis showed a heterozygous, frameshift mutation in exon 17 (c.3445delC), predicted to be pathogenic. SNP-array CGH analysis showed a maternally-inherited, heterozygous 500Kb deletion at Xq28 (not including the ATP7A gene), associated with X-linked intellectual disability. Parenteral Cu therapy was initiated at 8 months old and, at 22 months old, she has severe global developmental delay and hypotonia but no seizures. She is gastrostomy tube dependent.
Figure I. MR Angiogram showing marked cerebrovascular tortuosity in patient 1 with classic Menkes disease.
Patient 2 is a 4 year old girl, first evaluated at 2 weeks old for “silvery hair tips.” Initial exam showed excess nuchal skin, normal neurologic signs, and normal head circumference. Family history notes epilepsy in her mother who also had “coarse” hair with silvery tips in infancy. Patient B developed seizures at 5 weeks of age and gastrostomy tube feedings were initiated at 5 months of age due to feeding problems. She had serum low normal Cu at 70 mcg/dL and low normal serum ceruloplasmin at 21 mg/dL. SNP-array CGH was normal. ATP7A gene analysis showed a heterozygous deletion of exons 8-12, predicted to be pathogenic as well as 2 polymorphisms (c.2299G>C, IVS13-290A). She has marked cerebrovascular tortuosity, and brain atrophy. At 4 years old, she has severe hypotonia, marked intellectual impairment, epilepsy managed with two anticonvulsants, dystonia, and microcephaly. She is exclusively gastrostomy tube fed.
Patient 3 is a 7 year old girl, initially evaluated at 15 months of age for poor growth and developmental delay. Examination showed myopathic facies, coarse hair, skin and joint laxity, hypotonia, truncal ataxia, and dystonia. Radiographs showed wormian bones and general osteopenia. Cerebellar atrophy and marked cerebrovascular tortuosity were present. At 7 years old, mitral valve prolapse with mild mitral regurgitation and severe aortic root dilation were noted. She has two large, widely-communicating diverticulae. Serum Cu was low at 11 mg% (normal: 85-150) and ceruloplasmin was low at 13 mg% (normal: 20-43) at initial presentation. ATP7A gene analysis showed the same two heterozygous polymorphisms (c.2299G>C, IVS13-29C>A) as in patient B but no pathogenic mutations or deletions. Parenteral Cu was initiated at 2.5 yrs of age. At 7 years old, she has severe global developmental delay, hypotonia, epilepsy, a movement disorder, and progressive aortic root dilation.
Discussion
This is the first report of classic Menkes disease in a female. We additionally report two females with MD variants and summarize all known reports of affected females with MD and variants in Tables I and II.
Table I.
Clinical findings in all reported affected females with MD and variant phenotypes.
| Report | Hypotonia | ID | Epilepsy | Brain atrophy | CV tortuosity | “Kinky” hair | Cu therapy | Last report |
|---|---|---|---|---|---|---|---|---|
| Barton et al, 1983 | x | x | x | x | 2.5 y/ death | |||
| Favier et al, 1983 | x | x | x | x | 34 y | |||
| Kapur et al, 1987 | x | x | x | x | unknown | |||
| Gerdes et al, 1990 | x | x | x | x | 5 y | |||
| Beck et al, 1994 | x | x | x | unknown | ||||
| Sugio et al, 1998 | x | x | x | x | unknown | |||
| Abusaad et al, 1999 | x | x | x | x | 18 m/ death | |||
| Sirleto et al, 2009 | x | x | x | 26 m | ||||
| Moller et al, 2012 | x | x | x | x | 9 y | |||
| x | x | 22 y | ||||||
| x | x | x | 10 y | |||||
| x | x | 2 y | ||||||
| x | x | x | 41 y | |||||
| x | x | 14 y | ||||||
| x | x | x | 29 y | |||||
| Present report | x | unknown | x | x | x | x | 22 m | |
| x | unknown | x | x | x | 4 y | |||
| x | x | x | x | x | x | 7 y |
ID: Intellectual Disability
CV: Cerebrovascular
Cu: Copper
Y: years
M: months
Table II.
Biochemical and genetic findings in all reported affected females with MD and variant phenotypes.
| Report | Low serum Cu, ceruloplasmin | Cytogenetic abnormality | ATP7A alteration |
|---|---|---|---|
| Barton et al, 1983 | unknown | Mosaic TS | unknown |
| Favier et al, 1983 | unknown | ||
| Kapur et al, 1987 | x | 46,X, t(X;2) (q13;q32.2) | unknown |
| Gerdes et al, 1990 | x | unknown | |
| Beck et al, 1994 | x | 46,X, t(X;1) (q13;q12) | unknown |
| Sugio et al, 1998 | x | 46,X, t(X;21) (q13.3;p11.1) | unknown |
| Abusaad et al, 1999 | x | 46,X, t(X;13) (q13.3;q14.3) | unknown |
| Sirleto et al, 2009 | x | 46,X, t(X;16) (q13.3;p11.2) | unknown |
| Moller et al, 1998 | x | exon 6 deletion | |
| exon 6-9 deletion | |||
| intronic missense | |||
| unknown | c.2179G>A | ||
| unknown | c.2383C>A | ||
| x | exon 1 deletion | ||
| c.532G>T | |||
| Present report | x | Xq28 deletion | c.3445delC |
| exon 8-12 deletion | |||
| x | negative |
TS: Turner syndrome
Ten of 18 reported affected females exhibit low serum Cu and ceruloplasmin levels. As evidenced by the severe manifestations in females with normal serum Cu and ceruloplasmin but a pathogenic change in the ATP7A gene (see patient 2), these biochemical parameters are of limited diagnostic utility although they constitute a useful biomarker for laboratory monitoring while on Cu therapy (if initiated) provided that they were low at diagnosis. It is, thus, recommended that females with suspected MD or variant phenotypes undergo molecular genetic testing even if biochemical parameters are normal, if a high degree of clinical suspicion remains.
Hair abnormalities such as “kinky hair” (pili torti) or scattered hypopigmentation are characteristic findings in females and were reported in 46% of heterozygous females with MD [Moore et al, 1985]; these hair findings are uniformly found in males with classic MD. Patient 1 exhibited characteristic pili torti and scattered hair hypopigmentation while patient 2's first presenting sign was scattered hair hypopigmentation. This clinical finding is characteristic of the disease and hair microscopy becomes an important part of the clinical evaluation.
Seizures and abnormal neuroimaging findings (cerebrovascular tortuosity, cerebral atrophy) are noted in 11 of 18 of all reported females. In a study by Kaler et al [2010], only 12.5% of 24 patients with classic MD diagnosed and treated prior to 6 weeks of age manifested clinical seizures, whereas 46% of them had at least one abnormal EEG tracing. The authors proposed that early identification and treatment in classic MD “improves brain electrical activity and decreases seizure occurrence in classic MD”, although “it could not prevent severe intellectual disability.” In fact, parenteral Cu was initiated in patient 1 in early infancy and she has not developed seizures to date, which may in part be due to early diagnosis and treatment. In patient 2, however, the severe degree of developmental delay and seizures at diagnosis together with the normal serum Cu and ceruloplasmin levels were judged as making it unlikely that Cu therapy would be of clinical benefit, and, thus, it was not initiated.
Current management of MD in females focuses on symptomatic relief and targeted interventions in areas of physical and cognitive disabilities. Treatment with parenteral Cu is not standard of care in either males or females with classic or variant MD but it may be offered on a case-by-case basis recognizing the variable clinical benefit shown in studies of males with classic MD [Kaler, 1998]. Some males, when treated prior to neurologic symptom onset, showed improved clinical outcomes whereas others showed only symptomatic relief from irritability or sleep disturbance with no effect on overall neurocognitive outcome [Kaler, 1998]. Targeted pharmacologic therapies, including parenteral Cu, have not been systematically studied in females with classic or variant MD, likely due to many factors, such as the overall rarity of the disease, great variability in clinical manifestations in females making the diagnosis difficult, and potentially low index of suspicion and/or unfamiliarity with MD in general pediatric practice.
In summary, this report illustrates the variability in the presence and/or severity of clinical manifestations in females with MD and variant phenotypes requiring a high index of clinical suspicion during evaluations. Serum Cu and ceruloplasmin levels in those females can be variable with some showing low levels and others showing normal levels even in the presence of pathogenic ATP7A gene alterations confirming the MD diagnosis. Molecular genetic testing in these cases is fundamental. Importantly, females with MD and variants, if diagnosed and treated early, may benefit from Cu therapy, but the effect of this therapy on the overall disease course in those females remains to be seen.
Acknowledgments
The authors wish to acknowledge the contribution of Philip James, MD, MPH in the diagnostic evaluation and clinical management of patient 3.
Footnotes
The authors have no conflicts of interest to declare.
References
- Abusaad I, Mohammed SN, Ogilvie CM, Ritchie J, Pohl KRE, Docherty Z. Clinical Expression of Menkes Disease in a Girl with X;13 Translocation. Am J Med Gen. 1999;87:354–359. [PubMed] [Google Scholar]
- Beck J, Enders H, Schliephacke M, Buchwald-Saal M, Turner Z. X;1 Translocation in a Female Menkes patient: characterization by fluorescence in situ hybridization. Clin Genet. 1994;46:295–298. doi: 10.1111/j.1399-0004.1994.tb04163.x. [DOI] [PubMed] [Google Scholar]
- Gerdes AM, Tonnesen T, Horn N, Grisar T, Marg W, Muller A, Reinsch R, Barton NW, Guiraud P, Joannard A, Richard MJ, Guttler F. Clinical Expression of Menkes disease in females. Clin Genet. 1990;38:452–459. doi: 10.1111/j.1399-0004.1990.tb03612.x. [DOI] [PubMed] [Google Scholar]
- Kaler SG. Diagnosis and therapy of Menkes syndrome, a genetic form of copper deficiency. Am J Clin Nutr. 1998;67(suppl):1029S–1034S. doi: 10.1093/ajcn/67.5.1029S. [DOI] [PubMed] [Google Scholar]
- Kaler SG, Holmes CS, Goldstein DS, Tang J, Godwin SC, Donsante A, Liew CJ, Sato S, Patronas N. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358(6):605–14. doi: 10.1056/NEJMoa070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler SG, Liew CJ, Donsante A, Hicks JD, Sato S, Greenfield JC. Molecular correlates of epilepsy in early diagnosed and treated Menkes disease. J Inherit Metab Dis. 2010;33(5):583–9. doi: 10.1007/s10545-010-9118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaler SG. ATP7A-Related Copper Transport Disorders. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews™. Seattle (WA): University of Washington, Seattle; 2010. pp. 1993–2013. Internet. [PubMed] [Google Scholar]
- Kapur S, Higgins JV, Delp K, Rogers B. Menkes syndrome in a girl with X-Autosome Translocation. Am J Med Gen. 1987;26:503–510. doi: 10.1002/ajmg.1320260230. [DOI] [PubMed] [Google Scholar]
- Moller LB, Lenartowicz M, Zabot MT, Josiane A, Burglen L, Bennett C, Riconda D, Fisher R, Janssens S, Mohammed S, Ausems M, Turner Z, Horn N, Jensen T. Clinical Expression of Menkes Disease in Females with Normal Karyotype. Orph J Rare Dis. 2012;7:6. doi: 10.1186/1750-1172-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CM, Howell RR. Ectodermal manifestations in Menkes disease. Clin Genet. 1985;28(6):532–40. doi: 10.1111/j.1399-0004.1985.tb00422.x. [DOI] [PubMed] [Google Scholar]
- Sirleto P, Surace C, Santos H, Bertini E, Tomaiuolo AC, Lombardo A, Boenzi S, Bevivino E, Dionisi-Vici C, Angioni A. Lyonization Effects of the t(X;16) Translocation on the Phenotypic Expression in a Rare Female with Menkes Disease. Ped Res. 2009;65(3):347–351. doi: 10.1203/PDR.0b013e3181973b4e. [DOI] [PubMed] [Google Scholar]
- Sugio Y, Sugio Y, Kuwano A, Miyoshi O, Yamada K, Niikawa N, Tsukahara M. Translocation t(X;21)(q13.3;p11.1) in a Girl with Menkes Disease. Am J Med Gen. 1998;79:191–194. doi: 10.1002/(sici)1096-8628(19980923)79:3<191::aid-ajmg7>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Favier A, Boujet C, Joannard Possibility of a Menkes-like disorder of copper metabolism in a girl. J Inher Metab Dis. 1983;6(Suppl 2):89. [Google Scholar]
- Barton NW, Dambrosia JM, Barranger JA. Menkes kinky-hair syndrome: Report of a case in a female infant. Neurology. 1983;33(Suppl 2):154. [Google Scholar]