

Abstract
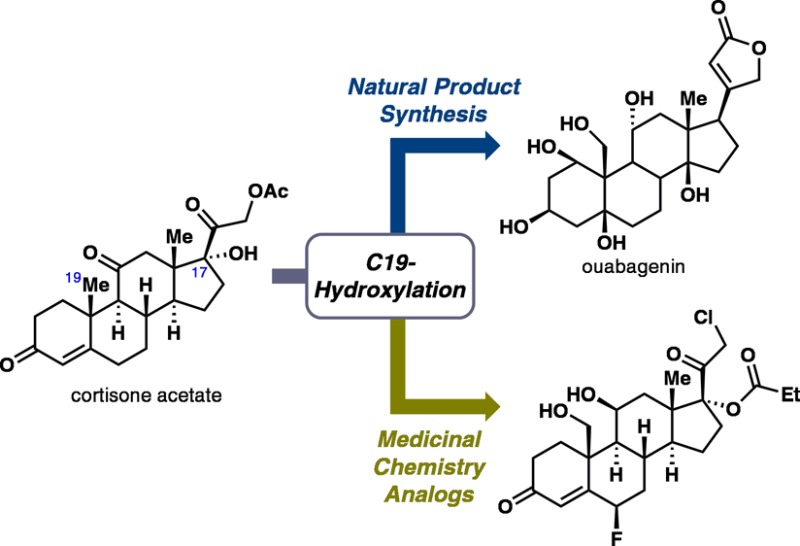
The natural product ouabagenin is a complex cardiotonic steroid with a highly oxygenated skeleton. This full account describes the development of a concise synthesis of ouabagenin, including the evolution of synthetic strategy to access hydroxylation at the C19 position of a steroid skeleton. In addition, approaches to install the requisite butenolide moiety at the C17 position are discussed. Lastly, methodology developed in this synthesis has been applied in the generation of novel analogues of corticosteroid drugs bearing a hydroxyl group at the C19 position.
Introduction
The term “cardiac glycosides”—often used interchangeably with “cardiotonic steroids”—refers to a class of steroidal natural products exhibiting positive inotropic activity.1 These molecules possess the capacity to increase the cardiac output through their inhibitory interaction with the extracellular surface of the membrane-bound sodium pump (Na+/K+–ATPase) through stabilization in the E2-P transition state, resulting in the increase of intracellular sodium concentration and the buildup of intracellular calcium concentration in the sarcoplasmic reticulum. This process ultimately results in a more powerful contraction of the mycocyte.
Cardiac glycosides possess several characteristic features2 (Figure 1A): (i) glycosylation, if any, is found at the C3 position of the steroidal framework; (ii) in contrast to the common steroidal skeleton, both their A/B and C/D rings are of cis configuration; (iii) a β-configured tertiary alcohol is present at C14 (and often at C5); and finally, (iv) an unsaturated lactone ring is found at the C17 position. The C17 lactone domain further defines the subclass of cardiac glycosides: those with an unsaturated butyrolactone moiety, typically afforded by plant sources, are called cardenolides and those with an unsaturated 2-pyrone moiety, typically afforded by animal sources, are called bufadienolides.3
Figure 1.
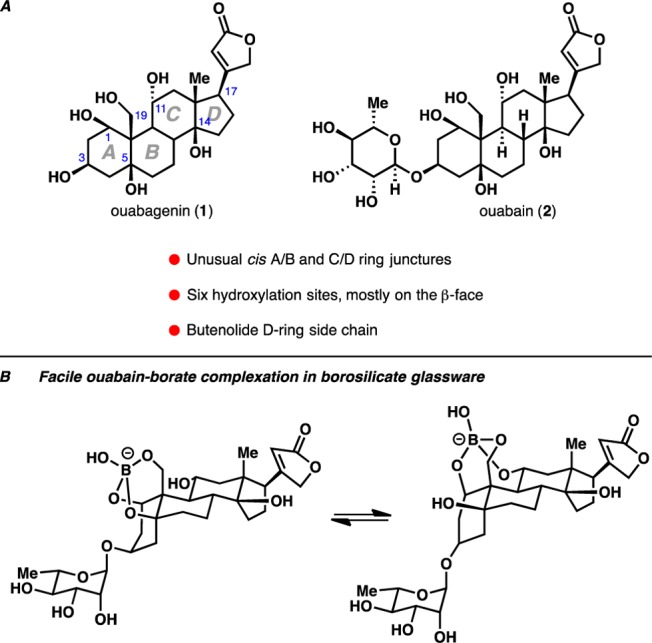
(A) Structures of cardiotonic steroid ouabagenin (1) and its parent glycoside ouabain (2). (B) Interaction of ouabain (2) with borosilicate glassware.
In 1888, a highly oxidized member of the cardenolide family, ouabain (2), was isolated by Arnaud4 from the roots and barks of the African ouabaio tree. Its aglycone, ouabagenin (1), was later isolated in 1942 by Mannich and Siewert,5 who also proposed the correct structure for the aglycone and the parent glycoside. These molecules have attracted considerable attention due to the discovery of naturally occurring ouabain (or a ouabain-like compound) in mammals. In fact, buildup of endogenous ouabain has recently been proposed as one genetic molecular mechanism for hypertension in animal models.6 From a chemical synthesis point of view, the predominant β-orientation of the hydroxyl groups of ouabagenin and ouabain presents an additional layer of complexity as ouabain (2) has been reported to undergo facile complexation with borosilicate glassware (Figure 1B).7
Our laboratory became enamored with cardiotonic steroids for both chemical and biological reasons. Chemically, there was no scalable solution (semisynthetic or fully synthetic) to the synthesis of highly oxygenated steroid systems such as ouabain (2), thus presenting an opportunity for innovation. From a biological vantage point, we were interested in the medicinal chemistry of highly oxygenated steroids that would result from such an effort. This full account traces the evolution of our synthetic strategy that ultimately led to a scalable solution to the puzzle posed by the ouabain problem and enabled initial medicinal chemistry explorations of uniquely oxidized steroid derivatives.
Results and Discussion
Historical Context and Key Precedents
Ouabain and related natural products are classic targets for synthesis. Semisynthesis campaigns for the preparation of the cardenolides and the related bufadienolides date back to the early 1970s with the landmark synthesis of batrachotoxinin8 by Werhli and co-workers. Since then, numerous synthetic efforts have resulted in the semisynthesis of strophanthidol and its parent glycoside (strophanthidin) by Yoshii,9 the semisynthesis of digitoxigenin by Wiesner10 and Kabat,11 and bufalin by Wiesner12 and Yoshii.13 In addition, the first total synthesis of digitoxigenin14 was reported by Stork and co-workers in 1996, and the total synthesis of rhodexin A15 was achieved by Jung and co-workers in 2011. More recently, an elegant total synthesis of 19-hydroxysarmentogenin starting from carvone was disclosed by Inoue and co-workers.16
Synthetic studies toward the most oxidized members of the cardenolide family, ouabagenin (1) and ouabain (2), have also been conducted, culminating in a landmark total synthesis of these compounds in 2008 by Deslongchamps and co-workers featuring a polyanionic cascade as the key skeletal construction step (Scheme 1).17 In addition, other elegant approaches have been developed, including a novel Heck annulation by Overman and co-workers,18 and a Diels–Alder approach by Jung and co-workers.19
Scheme 1. Previous Endeavors toward the Synthesis of Ouabagenin (1).

Given the success of pharmaceutical research on semisynthetic steroids, we anticipated that a semisynthetic approach would be the most viable means to achieve a scalable synthesis of ouabagenin (1). With pragmatic considerations in mind, our retrosynthetic analysis used a “look-ahead” approach20 commencing with readily available cortisone acetate (9; Figure 2). This starting material was chosen because of (i) its bulk availability at a reasonable cost ($1.2/g); (ii) the existence of “pre-built” oxidations at C3 and C11, which would facilitate access to the requisite hydroxyl groups at these two carbons; and (iii) the facile cleavage of the C17–C20 bond to give a ketone moiety at C17,21 which would provide us with a functional handle for the butenolide appendage.
Figure 2.
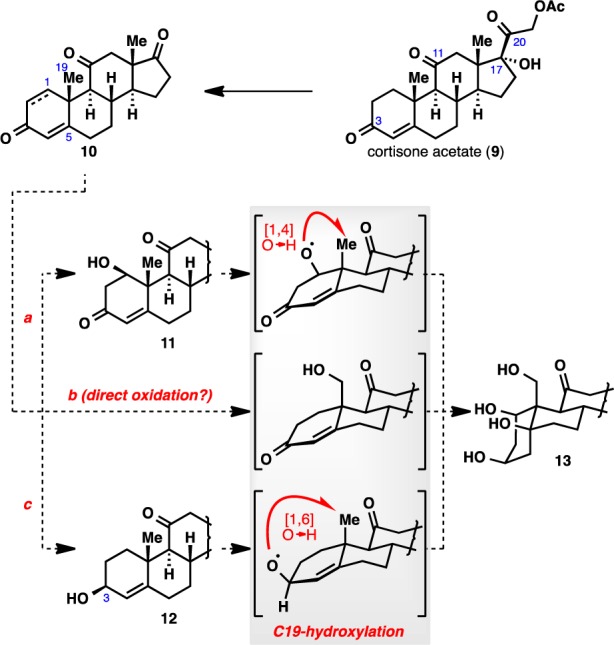
Preliminary strategic considerations for elaboration of the A ring.
The C19 Oxidation Problem
An overwhelming amount of literature evidence has suggested that the A ring of a steroid skeleton exhibits higher reactivity than the rest of the tetracyclic framework.22 Thus, we posited that the A ring be fully elaborated before the rest of the molecule in the approach toward ouabagenin (1). Several scenarios exist for the elaboration of the A ring using the putative steroid intermediate 10 derived from cortisone acetate (9) following C17–C20 cleavage (Figure 2). Given the multitude of hydroxyl groups to be introduced, a “pre-built” oxidation state within the A ring would be desirable. In pathway a, it is envisioned that either the C1 or C5 hydroxyl group could be introduced via selective hydration of the dienone moiety. However, introduction of the hydroxyl group at C19 would still be problematic due to the geometrically unfavorable 1,4-relationship to the requisite hydroxyl group at either C1 or C5.23 The same problem would also be encountered with the use of the hydroxyl group at C3 to direct oxidation onto the C19 position, as they exist in a 1,6-relationship (pathway c). Literature evidence suggested that C19 functionalization by way of alkoxy radical generation at C3 proceeded only in poor yield.24 Thus, it is apparent that the oxidation at the C19 carbon is unlikely to be accessed via the use of other functionalities on the A ring as directing groups, and would have to be attained via other means (pathway b), preferably early in the synthesis.
Installation of functionality at C19 is a classic unsolved problem in steroid chemistry dating back to the days of Woodward.25 A representative panel of strategies that have been developed to solve this problem is shown in Scheme 2. While the bromohydrin functionalization route (Scheme 2A) is a well-established method to access the C19 oxidation in steroid semisynthesis literature, this strategy was not pursued due to the lengthiness of operations required. Our studies thus commenced with the reinvestigation of an intriguing report of metalloporphyrin-catalyzed hydroxylation of the steroidal C19 position26 as this would represent the most direct access to the desired C19-hydroxyl compound (Scheme 2B). The described reaction utilized iron-porphyrin 18 in the presence of N-methylimidazole and cumyl hydroperoxide to furnish hydroxylated product 19 in moderate yield among other oxidation products. In our hands, this result could not be reproduced, as 17 proved to be unreactive toward oxidation under the reported conditions. Disappointingly, the use of a more reactive perfluorinated iron-porphyrin catalyst27 resulted only in epoxidation of the C4–C5 olefin of 17.
Scheme 2. A Reinvestigation of Previous Approaches Regarding the C19 Functionalization of a Steroid Scaffold.

Attention was then turned to a report from Rindone and co-workers (Scheme 2C) of a direct dichloromethylation of estrone under aerobic reaction conditions with cobalt(II) salen as catalyst.28 This report was viewed as an alternative solution for the installation of the requisite oxidation state at the C19 position. In our hands, formation of two products was observed with NMR spectra identical to the ones reported by the authors. X-ray analysis, however, established that the reported dichloromethyl adduct 22 was instead an oxidized dimer of estrone (23). In the original publication, the structure of product 22 was established based on the presence of a singlet at 5.39 ppm by 1H NMR spectroscopy. However, the actual structure of the product 23 suggests that this singlet belongs to the o-quinone proton. In addition, a multitude of conditions were attempted to effect a para-functionalization of estrone but most conditions only resulted in the formation of ortho-functionalized products. The preference for ortho-functionalization could be rationalized by the high thermodynamic penalty associated with breaking the aromaticity of the A ring in the para-functionalization pathway.
These failures led us to retool our synthesis plan and consider the possibility of introducing the hydroxyl group at C19 with the use of an appropriate functionality embedded at the C11 carbon. Literature evidence suggested that hypoiodite photochemistry would lead to nonselective functionalizations of both C19 and C18 carbons.29 On the other hand, a Norrish type II photochemistry could pave the way for selective functionalization of the C19 methyl group (Scheme 2D). Jeger and co-workers were able to introduce a hydroxyl moiety at C19 by first effecting a Norrish type II photochemistry to generate cyclobutanol 25 in modest yield, followed by an oxidative fragmentation with Pb(OAc)4.30 Encouraged by this finding, we arrived at the final retrosynthesis for ouabagenin as depicted in Figure 3. The general premise of this strategy was to relay the C17 and C19 ketone oxidation states to neighboring and distal carbon atoms. This so-called “redox-relay” approach would potentially simplify the installation of four hydroxyl groups (C1, C5, C14 and C19) and minimize functional group incompatibilities along the way. Butenolide installation at a late stage would result in target 27 and allow divergent exploration of D-ring substituents. Redox-relay from the C17 ketone would lead to 28. Subsequent hydroxyl-directed bis-oxygenation (C1 and C5) simplifies the target to C19-oxygenated steroid 29, which is essentially one oxidation away from readily available cortisone acetate (9).
Figure 3.
Final retrosynthetic analysis of ouabagenin.
Norrish Type II Photochemistry and Cyclobutanol Fragmentation
Oxidative cleavage of the C17–C20 bond of cortisone acetate (9) generated adrenosterone (30) in a straightforward manner (Scheme 3). Preliminary photochemical investigations suggested that the presence of ketone moieties at C3 and C17 led to the formation of a complex mixture of products, presumably via competitive photoexcitation. Thus, we elected to convert these two ketone moieties to the corresponding ketals. Although 31 could be converted to the desired cyclobutanol 32, the reaction gave only a modest yield of desired product (43%) and suffered from formation of undesired side products, most notably compound 33, which arises from Norrish type I cleavage of the C9–C11 bond.
Scheme 3. Norrish Type II Photochemistry on Steroid 31.
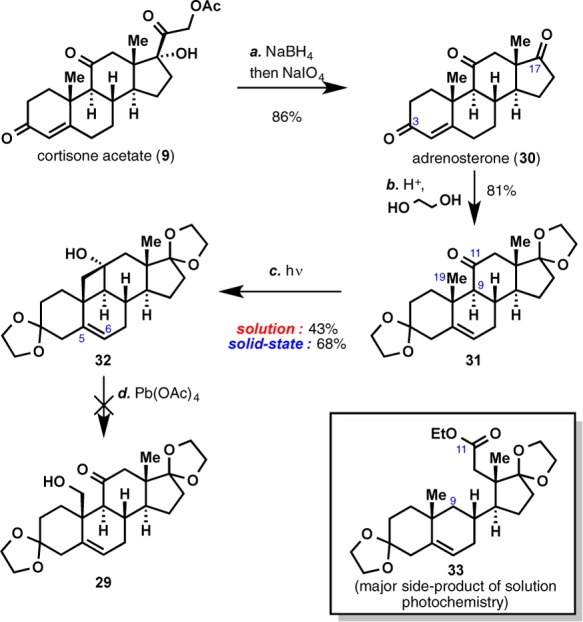
Reagents and conditions: (a) NaBH4 (0.6 equiv), 0 °C, 135 min; then NaIO4 (5 equiv), 23 °C, 12 h, 86%; (b) p-TsOH (0.1 equiv), ethylene glycol (30 equiv), PhCH3, 135 °C, 6 h, 81%; (c) hν, Vycor filter, 48–120 h; (d) Pb(OAc)4, C6H6, 80 °C, 1 h.
In 2004, a total synthesis of herbertenolide31 was accomplished by Garcia-Garibay and co-workers and featured for the first time the application of a solid-state Norrish type I photoreaction in total synthesis. Remarkably, the authors reported a highly chemoselective formation of cyclopentane 35, which stood in stark contrast to the nonselective outcome obtained with conventional solution photochemistry (Scheme 4A). In addition, Scheffer and co-workers also reported a highly differential outcome of a Norrish type II photoreaction32 when conducted in solution and in solid-state: significant improvement in the formation of cyclobutanols 37 and 38 was observed when solid-state photochemistry was employed in place of conventional solution photochemistry (Scheme 4B). Encouraged by these precedents, we elected to conduct our photochemistry in the solid state. Gratifyingly, the use of solid-state photochemistry led to an appreciable increase in the yield of cyclobutanol formation (43% to 68%) while minimizing the formation of undesired α-cleavage product 33 (see Scheme 3).
Scheme 4. Examples of Solid-State Photochemistry Precedents.

Unfortunately, the oxidative fragmentation protocol described by Jeger proved unsuccessful on 32 as a complex mixture of products was observed, none of which corresponded to the desired C19-functionalized product 29. Presumably, the C5–C6 olefin of 32 is incompatible with the strong oxidant/harsh conditions employed, leading to the formation of various undesired products.
Following precedents by Snider, Phillips, and several other research groups,33 a range of inorganic oxidants was also screened to effect the oxidative fragmentation of the C11–C19 bond, but none led to formation of the desired product (Scheme 5A). It was then reasoned that a hypervalent iodine species could undergo a ligand exchange reaction with cyclobutanol 32 and the resulting adduct could then fragment oxidatively to give either a hydroxyl or an acetate group at C19. Unfortunately, no reaction was observed with both IBX and DMP even at elevated temperatures, and undesired fragmentation of the C9–C11 bond to hemiketal 40 and 41 was observed with PhI(OAc)2 and hypoiodite photolysis conditions (I2/PhI(OAc)2), respectively.34
Scheme 5. Endeavors toward Oxidative Fragmentation of Cyclobutanol 32.
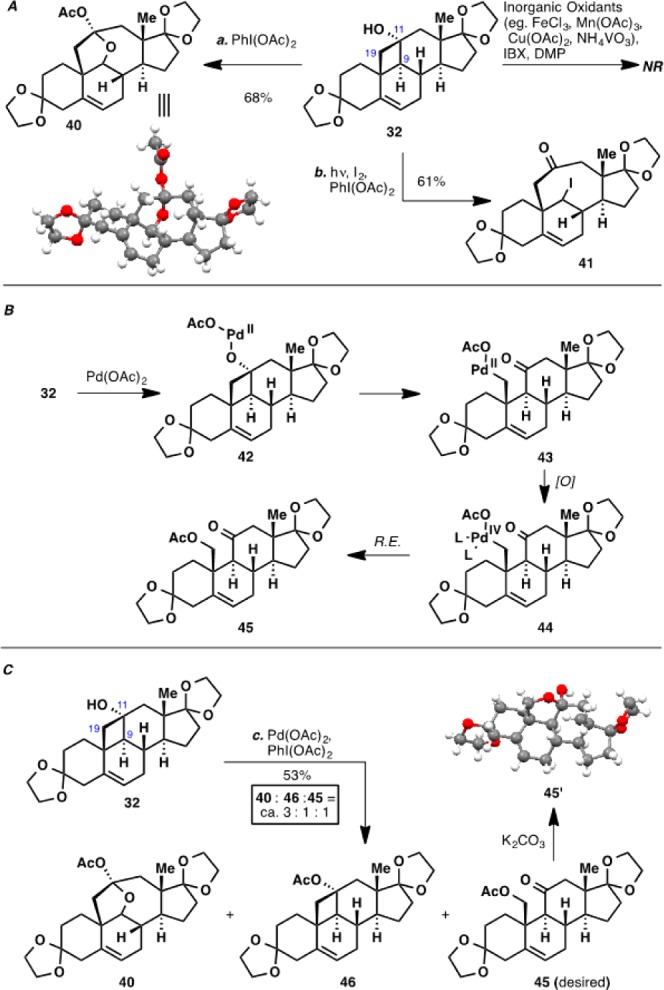
Reagents and conditions: (a) PhI(OAc)2 (1.5 equiv), Ac2O, 80 °C, 3 h, 68%; (b) hν, I2 (1.5 equiv), PhI(OAc)2 (2 equiv), CH2Cl2, 23 °C, 20 min, 61%; (c) Pd(OAc)2 (0.2 equiv), PhI(OAc)2 (1.5 equiv), Ac2O, 80 °C, 3 h.
Building on precedents set by Uemura and co-workers,35 we next envisaged a palladium-mediated fragmentation reaction (Scheme 5B). It was shown that cyclobutanols can be engaged in a β-carbon elimination from the initial palladium(II) alcoholate adduct. Thus, it was hoped that the resulting metalloketone intermediate could be intercepted by an oxidant to generate a PdIV species, which would in turn undergo reductive elimination to furnish acetate 45.
To this end, 32 was heated with Pd(OAc)2 and PhI(OAc)2, with Ac2O as solvent at 80 °C. The desired C19 functionalized compound 45 was indeed formed, albeit in minor amounts, validating our hypothesis (Scheme 5C). The major product from this reaction turned out to be hemiketal 40, indicating significant background reaction of the cyclobutanol with the oxidant itself. Unfortunately, despite extensive experimentations, we were not able to improve the yield of formation of acetate 45.
Eventually, the desired C19 functionalization was effected by using either Barluenga’s reagent36 or N-iodosuccinimide37 under photolytic conditions, with the latter being preferred simply due to the lower cost of the reagent. A proposed mechanism for this transformation is put forth in Figure 4. In addition, since both conditions b (see Scheme 5) and d/e (see Figure 4) should generate an identical hypoiodite intermediate, it is entirely possible that additives from the latter conditions exert subtle geometrical changes in the reaction transition state such that the O–I bond is anti-periplanar to the C11–C19 bond, leading to a chemoselective fragmentation to iodide 47.
Figure 4.
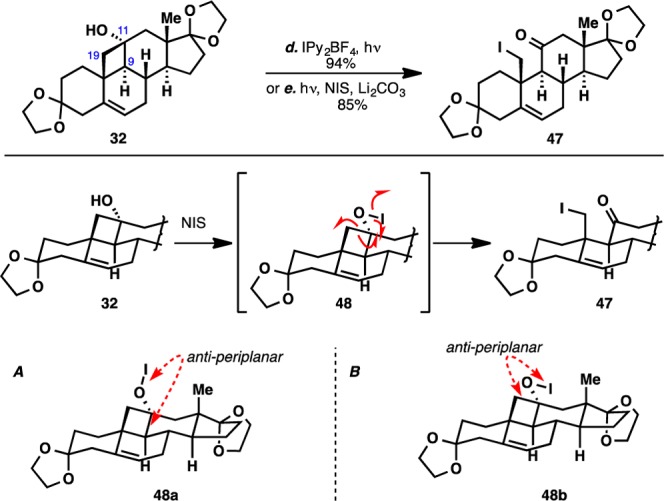
NIS-assisted oxidative fragmentation and proposed mechanism. A and B show two conformations of the O–I bond, leading to the cleavage of different C–C bonds.
With iodide 47 now in hand, selective deketalization at C3 was achieved by using TiCl4, and the iodide at C19 was hydrolyzed with AgOAc to furnish enone alcohol 49 (Scheme 6). In contrast to a similar epoxidation with a simpler C19 methyl substrate,38 epoxidation of the C4–C5 olefin proceeded in a completely diastereoselective fashion. Dehydrogenation of the C1–C2 bond was then achieved with SeO2 to give 50, as the use of other reagents such as IBX39 or HIO3 led to formation of over-oxidized products. Lastly, another diastereoselective epoxidation furnished diepoxide 51, a landmark intermediate en route to full elaboration of the A ring of ouabagenin (1).
Scheme 6. Elaboration of Cyclobutanol 32 to Diepoxide 51.
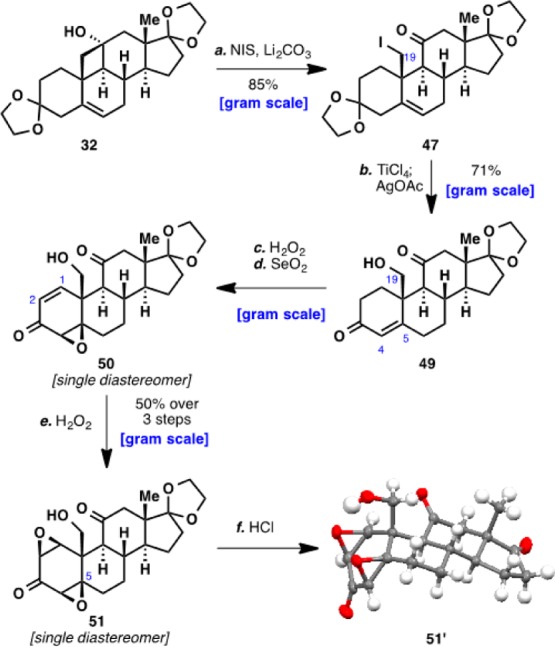
Reagents and conditions: (a) hν, N-iodosuccinimide (3 equiv), Li2CO3 (3.5 equiv), MeOH, PhCH3, 23 °C, 20 min, 85%; (b) TiCl4 (1 M in CH2Cl2, 1 equiv), CH2Cl2, −10 °C, 15 min; AgOAc (1.5 equiv), THF, H2O, 50 °C, 2 h, 71%; (c) H2O2 (35 wt% in H2O, 6 equiv), 3 M NaOH (1 equiv), MeOH, 0 °C, 75 min; (d) SeO2 (1.1 equiv), PhCl, 90 °C, 10 h; (e) H2O2 (35 wt% in H2O, 6 equiv), 3 M NaOH (1 equiv), MeOH, 0 °C, 75 min, 50% (over three steps); (f) conc. HCl (1 equiv), CH2Cl2, 23 °C, 1 h, 87%.
Diepoxide Fragmentation and Synthesis of Protected “Ouabageninone”
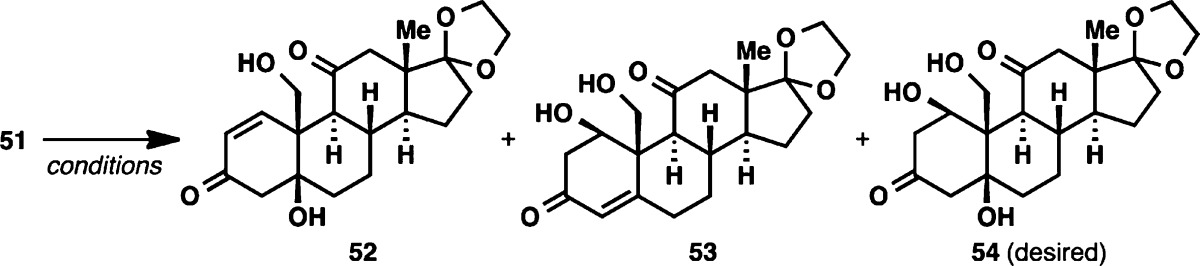
As was observed by Jung and co-workers in their model study,19 reductive opening of diepoxide 51 proved to be daunting. A wide range of conditions40 (Table 1) were tried, but most conditions resulted only in the formation of regioisomeric mixtures of A-ring enones. Hydrogenation conditions (entry 10), despite multiple precedents by Porco,41 led to reduction of the C3 ketone while leaving the two epoxides intact.
Table 1. Screening of Conditions for Diepoxide Fragmentation.
| entry | reagent | solvent | product(s) |
|---|---|---|---|
| 1 | PhSeNa | 52 + 53 | |
| 2 | NaTeH | 52 + 53 | |
| 3 | NaI/NaOAc/HOAc | no reaction | |
| 4 | Zn/MeOH | 52 + 53 | |
| 5 | SmI2 | 52 + 53 | |
| 6 | Li/naphthalene | 52 + 53 | |
| 7 | Mg/MeOH | no reaction | |
| 8 | N2H4 | no reaction | |
| 9 | Cr(OAc)2 | decomposition | |
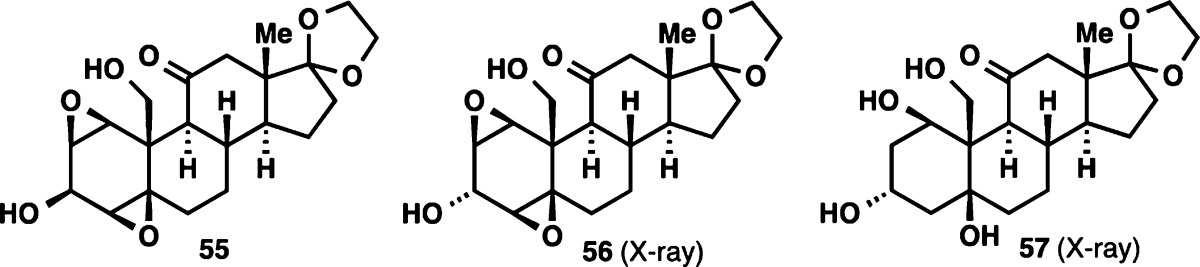
| 10 | Pt/C, H2 | ethyl acetate | 55 + 56 |
| 11 | Al–Hg | THF:EtOH:sat. NaHCO3 (2:1:0.15) | 54 (15%) + 57 |
| 12 | Al–Hg | THF:EtOH (2:1) | no reaction |
| 13 | Al–Hg | EtOH:sat. NaHCO3 (1:0.15) | 54 (<5%) + 57 |
| 14 | Al–Hg | sat. NaHCO3 | 54 (51%) + 57 |
| 15 | Al–Hg (portion-wise addition) | sat. NaHCO3 | 54 (64%; 56%a) |
| 16 | Al–Hg | sat. K2CO3 | no reaction |
| 17 | Al–Hg | sat. LiCl | no reaction |
| 18 | Al–Hg | sat. NaCl | 54 (<10%) + 57 |
| 19 | Al–Hg | sat. NH4Cl | 54 (19%) + 57 |
| 20 | Al–Hg | pH 7.4 buffer | 54 (41%) + 57 |
Gram-scale reaction.
Gratifyingly, triol 54 was accessible via treatment of 51 with in situ-generated aluminum amalgam. Poor conversion to 54 was observed when this reaction was run in the more conventional biphasic solvent mixture, leading to an extensive solvent screening for the reaction (entries 11–20), which eventually revealed saturated NaHCO3 as the optimal medium (entries 14 and 15). It is worth noting that with these “on-water” conditions,42 we observed formation of over-reduced product 57, suggesting that this aqueous medium provided a higher reduction potential for the reagent than conventional organic solvents.
Cognizant of the ligating ability of the A ring of ouabain (2),7 we elected to introduce an appropriate protecting group on the A ring. While attempts to tie the three hydroxyl groups in 54 as an orthoformate were unsuccessful, clean acetonide formation could be effected in acidic acetone to form 58. Reduction of the C3 ketone of 58 with LiBEt3H (Scheme 7) proceeded in a completely diastereoselective fashion and also protected the remaining two free hydroxyl moieties as the boronate ester to give 59.43 The ketone moiety of the C ring was next reduced under thermodynamic conditions (Li/NH3) to furnish an α-configured alcohol at C11. Following deketalization, the C17 ketone moiety of 60 was subjected to Saegusa oxidation to provide the corresponding conjugated enone. Isomerization of the olefin at C15–C16 out of conjugation could be effected with a SiO2/iPr2EtN mixture to give 61,44 although this necessitated the use of octafluorotoluene as solvent in order to minimize undesired epimerization of the C14 stereocenter (the C14 β-hydrogen epimer of the conjugated enone cannot be isomerized to give 61). To introduce the last hydroxyl group at C14, the C14–C15 olefin was subjected to Mukaiyama hydration.45 Once again, solvent optimization leading to the use of dioxane was necessary in order to achieve a satisfactory diastereomeric ratio of hydration products. With the completion of this step, we arrived at the protected form of the ketonic core of our target molecule (62, termed “ouabageninone”), and to date, more than 500 mg of this compound has been prepared. In addition, as a testament to the versatility of this route for further diversification, we demonstrated that 61 could undergo radical fluorination46 to produce 63 (Scheme 8). In light of recent advances in radical functionalization of alkenes,47 compound 61 could be viewed as a viable platform for further generation of new steroidal skeletons, and ultimately, novel analogues of the cardenolides and bufadienolides.
Scheme 7. Elaboration of Triol 54 To Protected Ouabageninone (62).
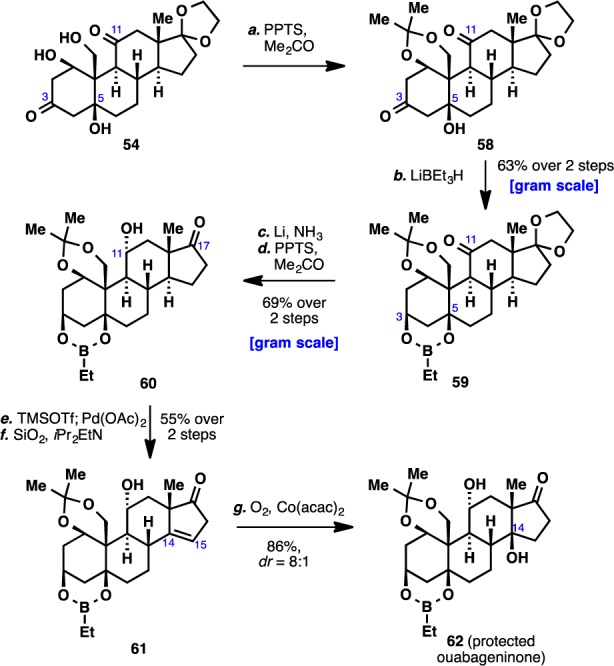
Reagents and conditions: (a) PPTS (0.2 equiv), CaSO4 (2.5 equiv), Me2CO, 23 °C, 20 h; (b) LiBEt3H (1 M in THF, 1.1 equiv), −78 °C, 30 min, 92%; (c) Li (60 equiv), NH3, THF, −78 °C, 30 min; (d) PPTS (1.5 equiv), Me2CO, 70 °C, 16 h, 69% over 2 steps; (e) TMSOTf (3 equiv), Et3N (4 equiv), CH2Cl2, 0 to 23 °C, 30 min; Pd(OAc)2 (1.2 equiv), MeCN, 23 °C, 3 h, then FeCl3 (1 equiv), 0 °C, 10 min; (f) SiO2, iPr2EtN (55 equiv), C6F5CF3, 23 °C, 45 min, 55% over 2 steps; (g) Co(acac)2 (0.2 equiv), PhSiH3 (3 equiv), O2 (1 atm), dioxane, 23 °C, 3 h, 86%.
Scheme 8. Radical Fluorination of Intermediate 61.
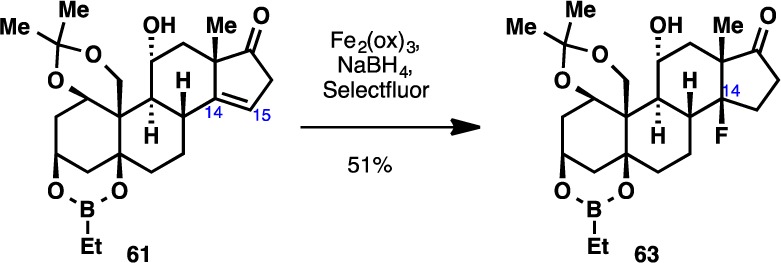
Reagents and conditions: Fe2(oxalate)3 (4 equiv), NaBH4 (6.4 equiv), Selectfluor (4 equiv), 3:4:2 MeCN:THF:H2O, 0 °C, 20 min, 51%.
Model Study for Butenolide Installation
With a scalable route to ouabageninone secured, attention was then turned to introduction of the butenolide moiety with the correct stereochemical orientation. To this end, a model study employing estrone as starting material was conducted (Scheme 9). Using a similar route to Scheme 7, the tertiary hydroxyl group at C14 was introduced to afford 64 (see the Supporting Information for its preparation). It was initially envisioned that the butenolide moiety would be appended via a palladium-catalyzed cross-coupling, followed by a chemo- and diastereoselective reduction of the C16–C17 olefin. Thus, ketone 64 was first converted to the corresponding vinyl iodide using Barton’s protocol.48 A wide range of conditions were tried for the butenolide attachment, but initial results were discouraging as the desired product 68 could only be obtained in very poor yield (Table 2). Eventually, it was found that the use of Fürstner’s modified Stille coupling protocol49 led to the formation of dienoate 68 in a synthetically useful yield.
Scheme 9. Attempted Installation of a Butenolide Moiety on an Estrone Model System.
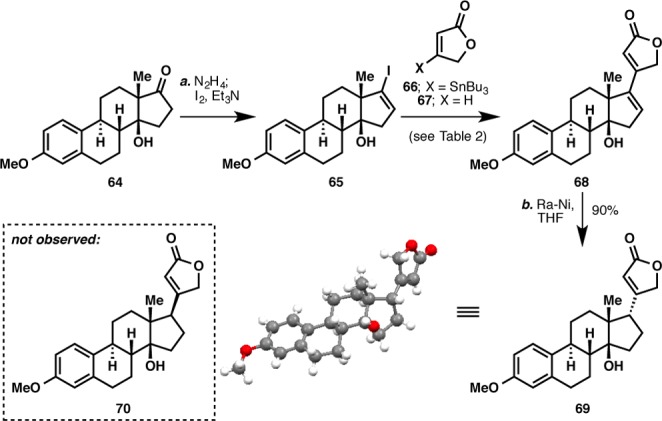
Reagents and conditions: (a) N2H4 (10 equiv), Et3N (10 equiv), EtOH, 50 °C, 5 h; I2 (3 equiv), Et3N (4 equiv), THF, 23 °C, 10 min; (b) Ra-Ni (10 wt equiv), THF, 23 °C, 14 h, 90%.
Table 2. Optimization of the Pd Coupling Reaction of 65 with 66 (X = SnBu3) or 67 (X = H)a.
| entry | reaction conditions | X | result |
|---|---|---|---|
| 1 | Pd(PPh3)4, LiCl, CuCl | SnBu3 | decomposition |
| 2 | Pd2dba3, P(furyl)3, 50 °C | SnBu3 | decomposition |
| 3 | Pd(OAc)2, KOAc, 80 °C | H | dimerization |
| 4 | PdCl2(MeCN)2, DMF, heat | SnBu3 | decomposition |
| 5 | PdCl2(PPh3)2, toluene, heat | SnBu3 | decomposition |
| 6 | CuTC, NMP | SnBu3 | ca. 5% desired product |
| 7 | Pd(PPh3)4, CuTC, [Ph2PO2][NBu4] |
SnBu3 | 55% desired product |
Compound 65 was used without further purification from the previous step.
With sufficient quantities of 68 in hand, chemoselective reduction of the C16–C17 olefin was attempted. No reaction was observed when palladium on carbon was used as the hydrogenation catalyst, and when the more active platinum catalyst was employed, nonselective hydrogenation of both olefins occurred. A chemoselective hydrogenation was eventually effected by using Ra-Ni,50 but this led to the wrong stereochemical outcome of the reduction, giving 69. This result stood in stark contrast to hydrogenation conducted on steroids possessing a more conventional trans C/D ring configuration, where reduction from the α face is typically observed. Clearly, the cis configuration of 68 rendered its convex face more accessible for hydrogenation. Alternatively, a directing effect of the C14 hydroxyl group could also be invoked to explain the stereochemical outcome. A radical-based coupling51 approach employing iodide 71 and lactones 66, 72, and 73 was also attempted but only deiodination of 71 was observed (Scheme 10).
Scheme 10. Attempted Radical Coupling on Iodide 71.
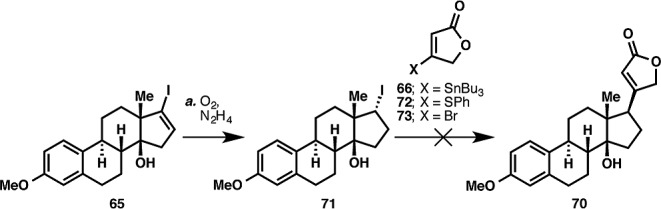
Reagents and conditions: (a) O2 (1 atm), N2H4, EtCO2H, EtOH, 100 °C, 2.5 h.
Another approach was then developed based on the idea of using a butenolide anion equivalent to effect a nucleophilic addition onto the C17 carbon from the more accessible β-face. A hydrazone-boronic acid coupling protocol52 disclosed by Barluenga and co-workers in 2009 was viewed as a highly attractive option owing to the minimal number of concession steps to be performed either before or after the coupling, and thus, the potential brevity of the overall sequence. A C2-substituted furanboronic acid was identified as a suitable butenolide equivalent (Scheme 11), and this compound was accessed by subjecting furan 74 to Ir-catalyzed C–H silylation employing a procedure developed by Falck and co-workers.53 Treatment with NaIO4 unmasked the free boronic acid to give 76. Gratifyingly, coupling of this boronic acid and tosylhydrazone 77 proceeded to give 78 with the correct stereochemical configuration, albeit in modest yield. Conversion of the substituted furan moiety to butenolide 70 could be readily achieved by treatment with basic AcOOH solution.54
Scheme 11. Successful Installation of the Butenolide Moiety via a Hydrazone–Boronic Acid Coupling Strategy.

Reagents and conditions: (a) [Ir(OMe)(cod)]2 (0.05 equiv), dtbpy (0.1 equiv), 2-norbornene (1.5 equiv), PhMe2SiH (1.5 equiv), 80 °C, 8 h, 43%; (b) NaIO4 (3 equiv), HCl (1 M, 0.7 equiv), 4:1 THF:H2O, 23 °C, 2 h, 91%; (c) TsNHNH2 (1.5 equiv), dioxane, 110 °C, 5 h, 64%; (d) K2CO3 (2.2 equiv), dioxane, 110 °C, 5 h, ca. 20%; (e) AcOOH (32 wt % in H2O), NaOAc (5 equiv), CH2Cl2, 23 °C, 24 h, ca. 70%.
Completion of Ouabagenin Synthesis
Having identified a viable method for installation of the butenolide moiety, we turned our efforts back to the synthesis of ouabagenin (1). In the conversion of ouabageninone to the corresponding tosylhydrazone, we encountered the first hurdle: concomitant removal of all the protecting groups was observed upon conversion to the corresponding hydrazone. This problem was rectified by the use of TrisNHNH2, which allowed formation of hydrazone 79 at ambient temperature (Scheme 12). Submitting this hydrazone to the coupling conditions with boronic acid 76, however, led to formation of a complex mixture of products, none of which could be identified as the desired coupled product 81a.
Scheme 12. Attempted Hydrazone–Boronic Acid Coupling on Protected Ouabageninone 62.

Reagents and conditions: (a) TrisNHNH2 (1.5 equiv), CH2Cl2, 23 °C, 10 h, 80%; (b) 76 (2 equiv), K2CO3 (2.2 equiv), dioxane, 110 °C, 5 h; (c) TMSOTf (3 equiv), Et3N (4 equiv), 23 °C, 1 h; (d) Ac2O (3 equiv), pyridine (7 equiv), DMAP (1 equiv), DMF, 40 °C, 20 h, 66%; (e) MOMCl (2.4 equiv), iPr2EtN (3.5 equiv), DMAP (1 equiv), CH2Cl2, 0 to 23 °C, 24 h, 62%.
Positing that the free hydroxyl group at C11 is incompatible with the transient diazo species, we decided to convert it to the corresponding TMS ether (80), acetate (82) and MOM ether (83). Unfortunately, subjection of these compounds to the Barluenga coupling conditions again led to formation of a complex mixture of products. Several additives, notably fluoride salts55 as well as different protecting groups at the C11 hydroxyl moiety were also tried, but none of these strategies led to any improvement to the outcome of the reaction.
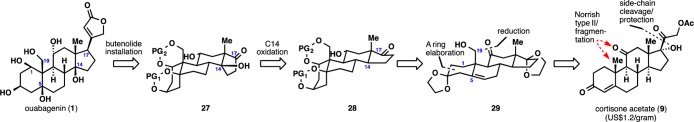
These failures led us to reinvestigate the original Stille coupling/chemoselective reduction route (Scheme 13). Thus, ketone 62 was converted to the corresponding vinyl iodide and then subjected to the modified Stille cross-coupling conditions to deliver dienoate 85. As was observed in the estrone model system, hydrogenation of 85 resulted in reduction from the undesired convex face of the molecule. Use of other conditions led to a nonselective reduction outcome, none of which could be identified as the desired product. We eventually discovered that the use of in situ-generated Co2B56 led to chemoselective formation of tetrasubstituted olefin 86. Extensive screening of organic bases and superbases (Table S1, Supporting Information) again led to the formation of an epimeric mixture of enoates, where our desired product was only present in minor amounts. Gratifyingly, we discovered that heating 86 in the presence of Barton’s base (BTMG)57 leads to the formation of a 3:1 mixture of enoates in favor of the desired product. To complete the synthesis of ouabagenin (1), this product was treated with concentrated HCl to effect global removal of the protecting groups. Overall, the synthesis of ouabagenin (1) was achieved in 20 steps and in 0.6% yield from adrenosterone (30) or in 21 steps and in 0.5% yield from cortisone acetate (9).
Scheme 13. Completion of the Synthesis of Ouabagenin (1).
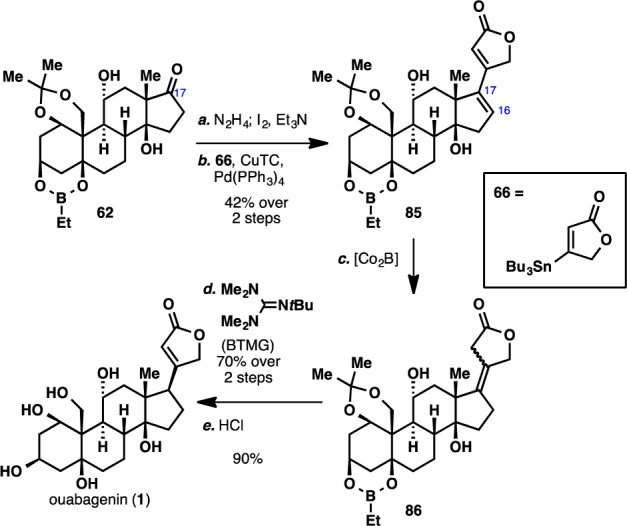
Reagents and conditions: (a) N2H4 (10 equiv), Et3N (10 equiv), 4:1 CH2Cl2:EtOH, 50 °C, 5 h; I2 (3 equiv), Et3N (4 equiv), THF, 10 min; (b) 66 (4 equiv), [Ph2PO2][NBu4] (4 equiv), Pd(PPh3)4 (0.15 equiv), CuTC (3 equiv), DMF, 23 °C, 2 h, 42% over two steps; (c) CoCl2·6H2O (2.5 equiv), NaBH4 (5 equiv), EtOH, 0 to 23 °C, 20 min; (d) BTMG (1.5 equiv), C6H6, 100 °C, 10 min, 70% over two steps; (e) conc. HCl (2 equiv), MeOH, 23 °C, 30 min, 90%.
Synthesis of Novel C19-Hydroxylated Analogues of Corticosteroid Drugs
As an extension of this work, we applied some of the methodologies developed in this campaign to the preparation of C19-hydroxylated analogues of corticosteroid drugs. Topical glucocorticoids represent the treatment of choice for inflammatory dermal diseases58 and when used properly, only limited systemic adverse events are observed, which make them a safe and effective therapy. However, long-term use of topical treatment on large body surface areas can be associated with systemic side effects partly due to mineralocorticoid antagonism of the glucocorticoids, which causes fluid-electrolyte imbalance and hypertension.59 All therapeutically used glucocorticoids possess this undesired mineralocorticoid receptor (MR) antagonism in addition to their glucocorticoid receptor (GR) agonism, and some SAR information on these two receptors has been reported for a range of steroids.60 However, only very limited SAR knowledge is available for glucocorticoids modified at the C19 position, presumably due to the difficulty in procuring meaningful quantities of these C19-modified compounds.
To investigate the pharmacological impact of installing a C19-hydroxy function in the glucocorticoid scaffold, analogue 90 became our initial target compound (Figure 5A). Its synthesis commenced with global protection of the ketone moieties of cortisone acetate (9) to afford 87. Following the procedure delineated in the synthesis of ouabagenin (1), iodide 88 was prepared. Deketalization and iodide hydrolysis afforded enone 89 in a straightforward manner. A chemoselective reduction of the C11 ketone followed by deprotection of the side chain under forcing conditions furnished the desired analogue 90 (for details, see the Supporting Information). To probe the overall effect of the hydroxyl moieties at the C19 and C11 positions, analogues bearing the epimeric hydroxyl group (91) and no hydroxyl group (92) at C11 were also synthesized from intermediate 89 (for details, see the Supporting Information).
Figure 5.

(A) Syntheses of glucocorticoid analogues 90, 91, 92, 97, and 98. aReagents and conditions: (a) HCHO (37%), HCl (conc.), CHCl3, 23 °C, 61%; (b) ethylene glycol, TsOH (cat.), benzene, reflux, 60%; (c) hν, Vycor filter, 60 h, 57%; (d) hν, N-iodosuccinimide (3.0 equiv), Li2CO3 (3.5 equiv), MeOH:PhCH3 = 1:20, 23 °C, 20 min; (e) 2:10:1 TFA:CH2Cl2:H2O, 0 °C, 2 h, 67% over 2 steps; (f) AgF (3.8 equiv), 10:1 MeCN:H2O, 23 °C, 98%; (g) NaBH4 (0.9 equiv), 1:1 CH2Cl2:MeOH, 0 °C, 2 h, 72%; (h) HCl (6 N), TFA, 23 °C, 30 min, 53%; (i) MsCl (1.3 equiv), Et3N (2.0 equiv), DMAP (0.1 equiv), CH2Cl2, 0 to 23 °C, 40 min; LiCl (1.3 equiv), DMF, 60 °C, 3 h, 81%; (j) TMSCl (4.0 equiv), NaI (40.0 equiv), Ac2O, 0 °C, 1 h; Selectfluor (1.3 equiv), MeCN, 0 °C, 1 h, 49%; for 97, (k) (EtCO)2O (1.5 equiv), TMSOTf (0.45 equiv), CH2Cl2, 0 °C, 1 h; (l) HCl (6 N), 75 °C, 5 h, 44% over 2 steps, and for 98, (k) (EtCO)2O (1.5 equiv), TMSOTf (0.45 equiv), CH2Cl2, 0 °C, 1 h, 91%; (l) HCl (6 N), μW, 120 °C, 15 min, 31%. (B) GR binding and anti-inflammatory efficacy of steroid analogues and reference compounds. bGR, glucocorticoid receptor. cIL-12B, interleukin-12B; MR, mineralocorticoid receptor. dThe number in parentheses is the corresponding efficacy value. eThe abbreviation “nd” indicates that examination is not done.
As can be seen in Figure 5B, the GR binding data for 92 showed a total lack of activity (IC50 > 10 000 nM), suggesting that introduction of a hydroxyl group at C19 alone cannot compensate for loss of the critical C11 hydroxyl. However, the data for 90 confirm that introduction of the C19 hydroxyl while keeping the C11 hydroxyl—in the same orientation as in hydrocortisone—only results in a 10-fold loss of affinity compared to the latter. This proves that the C19 hydroxyl is generally tolerated in steroidal GR agonists. The fact that 91, the C11 epimer of 90, is completely inactive in the GR binding assay further underscores the critical role of the presence and orientation of the C11 hydroxyl moiety. We were then very pleased to see that the GR binding of 90 also translated into anti-inflammatory efficacy in a cellular assay (LPS-induced IL-12B released from primary human peripheral blood mononuclear cells), albeit with low potency.
In light of these data and the ∼1000-fold difference in cellular potency (IL-12B) between clobetasole propionate and hydrocortisone, we next elected to explore the effect of incorporating two D-ring clobetasol side chains (chloroketone and propionate ester moieties) on 90. The synthesis started from intermediate 89 (Figure 5A). A chemoselective reduction of the C11 ketone in the presence of the A-ring enone moiety initially proved to be challenging. A preliminary solution was found by first globally reducing the two ketones, followed by allylic oxidation of the C3 hydroxyl group to regenerate the enone moiety. Eventually, it was found that a modified NaBH4 reduction protocol could effect a chemoselective C11 ketone reduction while leaving the A-ring enone intact to arrive at 93. It was serendipitously found that a methylene transfer to the C11 and C19 diol from the side chain of 93 could be effected under acidic conditions to afford 94, which interestingly achieved the desired protection by concomitant deprotection. With diol 94 in hand, a chlorine atom was introduced at the C21 position.61 Installation of a propionate ester62 at C17 and deprotection with HCl then completed the synthesis of analogue 97. We were encouraged to see the increase in GR binding affinity (∼15-fold) and cellular potency (∼30-fold) of 97 compared to 90 (Figure 5B). Furthermore, 97 showed an interesting selectivity profile against the mineralocorticoid receptor where it exhibited neither antagonistic nor agonistic effect when tested up to 1 μM (data not shown), suggesting that the C19 position potentially could be a trigger region for obtaining selectivity for the glucocorticoid receptor over the mineralocorticoid receptor. Finally, further refinement of 97 by introduction of a fluorine atom63 at C6 (to give analogue 98) provided a two-fold improvement in both the GR affinity and cellular potency, which is in agreement with published SAR for compounds bearing no hydroxyl group at C19.64
Conclusion
Stereochemically defined functional adornment of a steroid system was accomplished by following a “redox-relay” approach. This simplifying strategy allowed for the sequential installation of four hydroxyl units that underscore the complexity of ouabagenin (1). First, solid-state photochemistry and cyclobutanol fragmentation achieved hydroxylation at the C19 position of a steroid skeleton, which is a key motif in both ouabagenin (1) and C19-hydroxylated steroid analogues of cortisone acetate (9). Then, many redox-relay events were conducted, involving directed epoxidation, diepoxide fragmentation, and a Saegusa oxidation/Mukaiyama hydration sequence. Unexpected difficulty was encountered when installing the requisite butenolide moiety in 1, which was overcome by careful examination of an estrone model system. Finally, adapting the C19-hydroxylation method to related steroid systems has resulted in a series of hydroxylated analogues for structure–activity relationship studies. Evaluation of these steroid analogues for glucocorticoid receptor (GR) agonism has shown that the C19-hydroxylated steroid skeleton is a promising scaffold for identifying new anti-inflammatory drug candidates with improved properties, which will motivate our continued efforts in this field.
Acknowledgments
Financial support for this work was provided by LEO Pharma and an unrestricted grant from TEVA. Early studies were supported by the NIH/NCI (CA134785). Bristol-Myers Squibb supported a graduate fellowship to H.R., and the Shanghai Institute of Organic Chemistry supported a postdoctoral fellowship to Q.Z. We thank Prof. Tristan H. Lambert and Mr. Jeff S. Bandar for their generous donation of compound SI-1-6. We are grateful to Prof. Arnold L. Rheingold and Dr. Curtis E. Moore (University of California, San Diego) for X-ray crystallographic analysis, Dr. Gary Siuzdak (The Scripps Research Institute) for mass spectrometry assistance, Mr. José Antonio Romero Revilla for technical assistance, and Dr. Yoshihiro Ishihara for assistance in the preparation of this manuscript.
Supporting Information Available
Experimental details, spectra, and X-ray crystallography. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ H.R. and Q.Z. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Levi A. J.; Boyett M. R.; Lee C. O. Prog. Biophys. Mol. Biol. 1994, 62, 1–54. [DOI] [PubMed] [Google Scholar]; b Albrecht H. P.; Geiss K. H.. Cardiac Glycosides and Synthetic Cardiotonic Drugs: Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2005. [Google Scholar]; c Bagrov A. Y.; Shapiro J. I.; Fedorova O. V. Pharmacol. Rev. 2009, 61, 9–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero C. P.; Medarde M.; San Feliciano A. Molecules 2000, 5, 51–81. [Google Scholar]
- a Krenn L.; Kopp B. Phytochemistry 1998, 48, 1–29. [DOI] [PubMed] [Google Scholar]; b Steyn P. S.; van Heerden F. R. Nat. Prod. Rep. 1998, 397–413. [DOI] [PubMed] [Google Scholar]
- a Arnaud A. Compt. Rendu 1888, 106, 1011. [Google Scholar]; b Jacobs W. A.; Bigelow N. M. J. Biol. Chem. 1932, 96, 647–658. [Google Scholar]
- Mannich C.; Siewert G. Ber. Bunsen-Ges. 1942, 75, 737–750. [Google Scholar]
- Schoner W.; Bauer N.; Muller-Ehmsen J.; Kramer U.; Hambarchian N.; Schwinger R.; Moeller H.; Kost H.; Weitkamp C.; Schweitzer T.; Kirch U.; Neu H.; Grunbaum E.-G. Ann. N.Y. Acad. Sci. 2003, 986, 678–684. [DOI] [PubMed] [Google Scholar]
- a Kawamura A.; Guo J.; Itagaki Y.; Bell C.; Wang Y.; Haupert G. T. Jr.; Magil S.; Gallagher R. T.; Berova N.; Nakanishi K. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 6654–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kawamura A.; Abrell L. M.; Maggiali F.; Berova N.; Nakanishi K.; Labutti J.; Magil S.; Haupert G. T. Jr.; Hamlyn J. M. Biochemistry 2001, 40, 5835–5844. [DOI] [PubMed] [Google Scholar]
- a Graf W.; Berner H.; Berner-Frenz L.; Gössinger M. E.; Imhof R.; Wehrli H. Helv. Chim. Acta 1970, 53, 2267–2275. [DOI] [PubMed] [Google Scholar]; b Imhof R.; Gössinger M. E.; Graf W.; Schnuriger W.; Wehrli H. Helv. Chim. Acta 1971, 54, 2275–2285. [DOI] [PubMed] [Google Scholar]; c Imhof R.; Gössinger M. E.; Graf W.; Berner H.; Berner-Frenz L.; Wehrli H. Helv. Chim. Acta 1972, 55, 1151–1153. [DOI] [PubMed] [Google Scholar]; d Gössinger M. E.; Imhof R.; Wehrli H. Helv. Chim. Acta 1972, 55, 1545–1560. [Google Scholar]; e Imhof R.; Gössinger M. E.; Graf W.; Berner-Frenz L.; Berner H.; Schaufelberger R.; Wehrli H. Helv. Chim. Acta 1973, 56, 139–162. [DOI] [PubMed] [Google Scholar]
- Yoshii E.; Oribe T.; Tumura K.; Koizumi T. J. Org. Chem. 1978, 43, 3946–3950. [Google Scholar]
- Wiesner K.; Tsai T. Y. R. Pure Appl. Chem. 1986, 58, 799–810. [Google Scholar]
- Kabat M. M. J. Org. Chem. 1995, 60, 1823–1827. [Google Scholar]
- Wiesner K.; Tsai T. Y. R.; Jäggi F. J.; Tsai C. S. J.; Gray G. D. Helv. Chim. Acta 1982, 65, 2049–2060. [Google Scholar]
- Yoshii E.; Oribe T.; Koizumi T.; Hayashi I.; Tumura K. Chem. Pharm. Bull. 1977, 25, 2249–2256. [Google Scholar]
- Stork G.; West F.; Lee H. Y.; Isaacs R. C. A.; Manabe S. J. Am. Chem. Soc. 1996, 118, 10660–10661. [Google Scholar]
- Jung M. E.; Yoo D. Org. Lett. 2011, 13, 2698–2701. [DOI] [PubMed] [Google Scholar]
- Mukai K.; Urabe D.; Kasuya S.; Aoki N.; Inoue M. Angew. Chem., Int. Ed. 2013, 52, 5300–5304. [DOI] [PubMed] [Google Scholar]
- a Trudeau S.; Deslongchamps P. J. Org. Chem. 2004, 69, 832–838. [DOI] [PubMed] [Google Scholar]; b Zhang H.; Reddy M. S.; Phoenix S.; Deslongchamps P. J. Angew. Chem., Int. Ed. 2008, 47, 1272–1275. [DOI] [PubMed] [Google Scholar]; c Reddy M. S.; Zhang H.; Phoenix S.; Deslongchamps P. Chem.—Asian J. 2009, 4, 725–741. [DOI] [PubMed] [Google Scholar]
- a Deng W.; Jensen M. S.; Overman L. E.; Rucker P. V.; Vionnet J. P. J. Org. Chem. 1996, 61, 6760–6761. [DOI] [PubMed] [Google Scholar]; b Overman L. E.; Rucker P. V. Tetrahedron Lett. 1998, 39, 4643–4646. [Google Scholar]
- Jung M. E.; Pizzi G. Org. Lett. 2003, 5, 137–140. [DOI] [PubMed] [Google Scholar]
- Corey E. J.; Cheng X.-M.. The Logic of Chemical Synthesis; Wiley: New York, 1989; p 17. [Google Scholar]
- Wüst F.; Carlson K. E.; Katzenellenbogen J. A. Steroids 2003, 68, 177–191. [DOI] [PubMed] [Google Scholar]
- Loewenthal H. J. E. Tetrahedron 1959, 6, 269–303. [Google Scholar]
- Čeković Ž. Tetrahedron 2003, 59, 8073–8090. [Google Scholar]
- Ohuchi R.; Shibata K.; Yamakoshi N.; Mori H. Chem. Pharm. Bull. 1981, 29, 43–50. [Google Scholar]
- Woodward R. B. J. Am. Chem. Soc. 1940, 62, 1208–1211. [Google Scholar]
- Vijayarahavan B.; Chauhan S. M. S. Tetrahedron Lett. 1990, 31, 6223–6226. [Google Scholar]
- Ellis P. E. Jr.; Lyons J. E. Coord. Chem. Rev. 1990, 105, 181–183. [Google Scholar]
- Nali M.; Rindone B.; Tollar S.; Valletta L. J. Mol. Catal. 1987, 41, 349–354. [Google Scholar]
- Roller P.; Djerassi C. J. Chem. Soc. (C) 1970, 1089. [Google Scholar]
- Wehrli H.; Heller M. S.; Schaffner K.; Jeger O. Helv. Chim. Acta 1961, 44, 2162–2173. [Google Scholar]
- Ng D.; Yang Z.; Garcia-Garibay M. A. Org. Lett. 2004, 6, 645–647. [DOI] [PubMed] [Google Scholar]
- Gudmundsdottir A. D.; Lewis T. J.; Randall L. H.; Scheffer J. R.; Rettig S. J.; Trotter J.; Wu C.-H. J. Am. Chem. Soc. 1996, 118, 6167–6184. [Google Scholar]
- a Snider B. B.; Vo N. H.; Foxman B. M. J. Org. Chem. 1993, 58, 7228–7237. [Google Scholar]; b Keaton K. A.; Phillips A. J. Org. Lett. 2007, 9, 2717–2719. [DOI] [PubMed] [Google Scholar]; c Wietzerbin K.; Bernadou J.; Meunier B. Eur. J. Inorg. Chem. 2000, 7, 1391–1406. [Google Scholar]
- a Kalvoda J.; Heusler K. Synthesis 1971, 501–526. [Google Scholar]; b Fransisco C. G.; Freire R.; Hernández R.; Medina M. C.; Suárez E. Tetrahedron Lett. 1983, 24, 4621–4624. [Google Scholar]; c Dorta R. L.; Francisco C. G.; Freire R.; Suárez E. Tetrahedron Lett. 1988, 29, 5429–5432. [Google Scholar]
- Nishimura T.; Ohe K.; Uemura S. J. Org. Chem. 2001, 66, 1455–1465. [DOI] [PubMed] [Google Scholar]
- Barluenga J.; González-Bobes F.; Murguía M. C.; Ananthoju S. R.; González J. M. Chem.—Eur. J. 2004, 10, 4206–4213. [DOI] [PubMed] [Google Scholar]
- McDonald C. E.; Holcomb H.; Leathers T.; Ampadu-Nyarko F.; Frommer J. Jr. Tetrahedron Lett. 1990, 31, 6283–6286. [Google Scholar]
- Hrycko S.; Morand P.; Lee F. L.; Gabe E. J. J. Org. Chem. 1998, 53, 1515–1919. [Google Scholar]
- Nicolaou K. C.; Montagnon T.; Baran P. S. Angew. Chem., Int. Ed. 2002, 41, 993–996. [DOI] [PubMed] [Google Scholar]
- Larock R. C.Comprehensive Organic Transformations; Wiley: New York, 1989; pp 505–508. [Google Scholar]
- Lei X.; Zaarur N.; Sherman M. Y.; Porco J. A. Jr. J. Org. Chem. 2005, 70, 6474–6483. [DOI] [PubMed] [Google Scholar]
- Narayan S.; Muldoon J.; Finn M. G.; Fokin V. V.; Kolb H. C.; Sharpless K. B. Angew. Chem., Int. Ed. 2005, 44, 3275–3279. [DOI] [PubMed] [Google Scholar]
- Garlaschelli L.; Mellerio G.; Vidari G. Tetrahedron Lett. 1989, 30, 597–600. [Google Scholar]
- a Kelly R. W.; Sykes P. J. J. Chem. Soc. (C) 1968, 416–421. [PubMed] [Google Scholar]; b Egner U.; Fritzmeier K.-H.; Halfbrodt W.; Heinrich N.; Kuhnke J.; Müller-Fahrnow A.; Neef G.; Schöllkopf K.; Schwede W. Tetrahedron 1999, 55, 11267–11274. [Google Scholar]; c Cleve A.; Neef G.; Ottow E.; Scholz S.; Schwede W. Tetrahedron 1995, 51, 5563–5572. [Google Scholar]
- Isayama S.; Mukaiyama T. Chem. Lett. 1989, 18, 1071–1074. [Google Scholar]
- Barker T. J.; Boger D. L. J. Am. Chem. Soc. 2012, 134, 13588–13591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Leggans E. K.; Barker T. J.; Duncan K. K.; Boger D. L. Org. Lett. 2012, 14, 1428–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lo J. C.; Yabe Y.; Baran P. S. J. Am. Chem. Soc. 2014, 136, 1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Iwasaki K.; Wan K. K.; Oppedisano A.; Crossley S. W. M.; Shenvi R. A. J. Am. Chem. Soc. 2014, 136, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton D. H. R.; O’Brien R. E.; Sternhell S. J. Chem. Soc. 1962, 470–476. [Google Scholar]
- Fürstner A.; Funel J.-A.; Tremblay M.; Bouchez L. C.; Nevado C.; Waser M.; Ackerstaff J.; Stimson C. C. Chem. Commun. 2008, 2873–2875. [DOI] [PubMed] [Google Scholar]
- Shi J.; Shigehisa H.; Guerrero C. A.; Shenvi R. A.; Li C.-C.; Baran P. S. Angew. Chem., Int. Ed. 2009, 48, 4328–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almirante N.; Cerri A. J. Org. Chem. 1997, 62, 3402–3404. [DOI] [PubMed] [Google Scholar]
- Barluenga J.; Tomás-Gamasa M.; Aznar F.; Valdés C. Nat. Chem. 2009, 1, 494–499. [DOI] [PubMed] [Google Scholar]
- Lu B.; Falck J. R. Angew. Chem., Int. Ed. 2008, 47, 7508–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanis S. P.; Head D. B. Tetrahedron Lett. 1984, 25, 4451–4454. [Google Scholar]
- Pérez-Aguilar M. C.; Valdés C. Angew. Chem., Int. Ed. 2012, 51, 5953–5957. [DOI] [PubMed] [Google Scholar]
- Chung S.-K. J. Org. Chem. 1979, 44, 1014–1016. [Google Scholar]
- Barton D. H. R.; Elliott J. D.; Géro S. D. J. Chem. Soc., Perkin Trans. 1 1982, 2085–2090. [Google Scholar]
- Katz M.; Gans E. H. J. Pharm. Sci. 2008, 97, 2936–2947. [DOI] [PubMed] [Google Scholar]
- Fried J.; Borman A. Vitam. Horm. 1958, 16, 303–374. [DOI] [PubMed] [Google Scholar]
- Grossman C.; Scholz T.; Rochel M.; Bumke-Vogt C.; Oelkers W.; Pfeiffer A. F.; Diederich S.; Bahr V. Eur. J. Endocrinol. 2004, 151, 397–406. [DOI] [PubMed] [Google Scholar]
- Lai G.; Tan P.-Z.; Ghoshal P. Synth. Commun. 2003, 33, 1727–1732. [Google Scholar]
- a Procopiou P. A.; Baugh S. P. D.; Flack S. S.; Inglis G. G. A. Chem. Commun. 1996, 2625–2526. [Google Scholar]; b Procopiou P. A.; Baugh S. P. D.; Flack S. S.; Inglis G. G. A. J. Org. Chem. 1998, 63, 2342–2347. [Google Scholar]
- Sankar Lal G. J. Org. Chem. 1993, 58, 2791–2796. [Google Scholar]
- Grossman C.; Scholz T.; Rochel M.; Bumke-Vogt C.; Oelkers W.; Pfeiffer A. F.; Diederich S.; Bahr V. Eur. J. Endocrinol. 2004, 151, 397–406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.