
Figure 3.
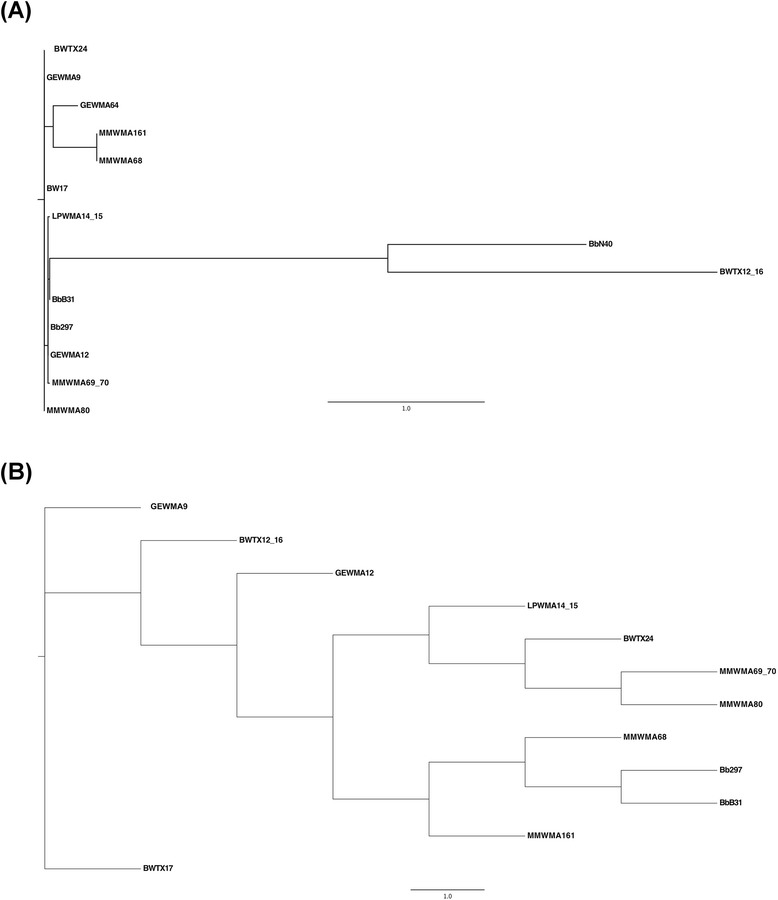
Neighbor-Joining phylogenetic tree of the flaB sequences analysed in this study. For the phylogenetic analyses, ClustalW2 [8] generated preliminary multiple sequence alignments for both the DNA sequences (A) and their respective putative amino acid sequences (B, obtained through the NCBI’s blastx program). These alignments were fed into jModeltTest 2.1.7 [9] and ProtTest 3 [10], programs that perform maximum likelihood (ML) optimization to assess the best-fit models for nucleotide substitution and amino acid substitution, respectively. These results informed the parameter sets to be used in RAxML v1.1 [11], a randomized, accelerated ML phylogenetic tree search program. These ML searches were conducted through 100 inferences of 100 distinct, randomized trees using the general time-reversible (GTR) model with gamma distributed rate heterogeneity for nucleotide data and the LG model with rate heterogeneity for amino acid data. The phylogenetic trees were visualized using FigTree v1.4.2 [12]. Figure Xa demonstrates that BWTX12-16 possesses a widely divergent nucleotide sequence from that of the other sequences, yet Figure Xb shows that the BWTX12-16 amino acid sequence is nested with the other sequences.