

Abstract
Background
Estrogen receptor (ER) positive breast cancer can often be treated by hormone therapy; however a certain population of ER-positive patients become resistant to hormone therapy after long-term hormone treatment. Ethinylestradiol (EE2) is a derivative of estrogen, which has shown promising effects in these patients.
Methods
We successfully obtained tissue samples from 6 patients undergoing EE2 treatment and examined 13 well-known breast cancer-related factors by immunohistochemistry. Of the 6 patients, 5 responded but one patient did not.
Results
Before EE2 treatment, staining for both ER and androgen receptor (AR) was strong in the nucleus, and the progesterone receptor (PgR) was almost no staining. EE2 treatment significantly down-regulated ER and up-regulated PgR while nuclear and cytosolic AR were oppositely down- and up-regulated, respectively. Cytosolic staining of BRCA1 was significantly up-regulated by EE2 whereas nuclear staining tended to decrease. Individual comparisons suggested less induction of PgR and decreasing AKT but increasing pAKT in the non-responder following EE2 treatment.
Conclusions
Our observations revealed that EE2 activated ER downstream genes; however it did not stimulate cell growth. This suggests that hormone resistant cells might receive growth signals from a non-genomic pathway and this may be reflected in their sensitivity to EE2 treatment.
Electronic supplementary material
The online version of this article (doi:10.1186/s40064-015-0851-8) contains supplementary material, which is available to authorized users.
Keywords: Ethinylestradiol, Estrogen receptor, Metastatic breast cancer, Endocrine therapy, Estrogen treatment resistance
Background
Estrogen receptor (ER) positive breast cancer is considered to be a low risk form of the disease and anti-estrogen treatment is generally applied even though this disease can be recurrent, if not life-threatening (NCCN guidelines® http://www.nccn.org/).
There are several different types of anti-estrogenic agents; Tamoxifen is an antagonist of ER, and has been widely used to treat cancer in both pre- and post-menopausal women. Aromatase inhibitors (AIs) are the representative anti-estrogenic agents in post-menopausal women, and are the first choice for patients after menopause. There are several different AI agents available; the non-steroidal AIs: anastrozole and letorozole, and the steroidal AI: exemestane. Fulvestrant is an estrogen receptor antagonist with no agonist effects, which works by down-regulating the estrogen receptor (Kansra et al. 2005). These agents generally inhibit ER function by either decreasing production of estrogen or by blocking the ER itself.
Ethinyl estradiol (EE2) is a derivative of 17β-estradiol (E2), which is the major endogenous estrogen in humans, and exhibits estrogenic activity. EE2 is used in many formulations of combined oral contraceptive pills (Hatcher RA 2004). In addition it has also been prescribed for patients with prostate cancer for many years, because administration of EE2 causes a reduction of androgen production (Dorner et al. 1985).
Very interestingly, EE2 administration has been shown to be effective against the recurrence of ER positive breast cancer after long-term anti-estrogen treatment (Iwase et al. 2013; Ellis et al. 2009). It was believed that estrogen stimulates the growth and development of ER-positive breast cancer, so this finding is quite the opposite to our consensus and the effects are something of a paradox.
To investigate this effect in more detail, we set up a clinical trial to evaluate EE2 treatment in our department. We registered 18 patients who were prescribed EE2 treatment after long-term AI treatment, and so far successfully obtained 23 tissue samples from 6 patients. Using immunohistochemistry, we analyzed the expression of known breast cancer-related genes in this study and examined the effects of EE2.
Results
ER, progesterone receptor (PgR) and Ki67 staining were observed only in the nucleus, while human EGFR-related 2 (Her2), Fas, transforming growth factor beta receptor 1 (TGFβR1) and insulin-like growth factor I receptor beta (IGF1Rb) were found on the cell membrane and phosphoinositide 3-kinase (PI3K) was localized in the cytosol. Androgen receptor (AR), breast cancer susceptibility gene I (BRCA1), protein kinase B (AKT) and phosphorylated AKT (pAKT) staining were observed in both the nucleus and the cytosol. Representative ER, PgR and AR staining are shown in Figure 1. Nuclear and cytosolic staining of each sample was evaluated by histo-score (HS) and cell membrane staining was categorized into 4 groups as described in Materials and methods. Results from all 18 samples are shown in Table 1.
Figure 1.
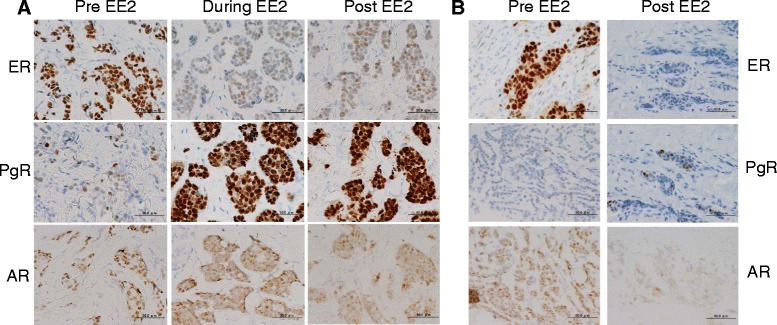
Representative staining of ER, PgR and AR in patients pre, during and post EE2 treatment. A: Staining in patient 4, who responded to EE2. Before EE2 treatment, ER staining was high, PgR staining was low and AR staining was only observed in the nucleus. After administration of EE2, ER decreased, PgR was increased and AR in the nucleus reduced while cytosolic staining increased. B: Staining in patient 3, who did not respond to EE2. Before EE2 treatment, high ER, low PgR and high nuclear AR were observed, while after EE2, ER was dramatically decreased as in patient 4. However, PgR was not induced and nuclear AR was decreased and not translocated to the cytosol.
Table 1.
Staining results of each samples
| ER | PgR | Ki67 | BRCA1 | AR | IGF1Rb | P13K | AKT | pAKT | TUNEL | HER2 | Fas | TGFßR1 | GF1Rb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | nuc | nuc | nuc | nuc | cyto | nuc | cyto | cyto | nuc | cyto | nuc | cyto | nuc | cyto | m | m | m | m | m | |
| 1 | pre | 158 | 8 | 10 | 121 | 0 | 94 | 0 | 80 | 0 | 46 | 18 | 95 | 5 | 90 | 25 | 1 | 0 | 0 | 3 |
| post | 120 | 10 | 10 | 106 | 95 | 5 | 60 | 70 | 0 | 56 | 0 | 90 | 5 | 0 | 0 | 1 | 10 | 1 | 1 | |
| 2 | pre | 156 | 0 | 20 | 97 | 0 | 124 | 50 | 80 | 0 | 106 | 10 | 42 | 0 | 0 | 0 | 1 | 0 | 2 | 1 |
| post | 110 | 137 | 40 | 4 | 0 | 3 | 0 | 0 | 0 | 109 | 46 | 85 | 0 | 0 | 9 | 1 | 0 | 1 | 3 | |
| 3 | pre | 140 | 6 | 20 | 55 | 0 | 111 | 0 | 90 | 0 | 100 | 105 | 0 | 0 | 40 | 6 | 1 | 0 | 1 | 3 |
| post | 0 | 30 | 50 | 39 | 0 | 5 | 0 | 100 | 0 | 100 | 31 | 0 | 133 | 90 | 6 | 3 | 0 | 1 | 1 | |
| 4 | pre | 156 | 17 | 10 | 116 | 0 | 106 | 0 | 0 | 0 | 88 | 40 | 90 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| during | 11 | 203 | 20 | 122 | 0 | 18 | 95 | 0 | 0 | 100 | 85 | 100 | 0 | 0 | 0 | 1 | 0 | 0 | 2 | |
| post | 10 | 122 | 30 | 100 | 100 | 15 | 98 | 0 | 0 | 100 | 95 | 95 | 0 | 0 | 3 | 0 | 0 | 1 | 2 | |
| 5 | pre | 172 | 0 | 5 | 100 | 0 | 128 | 0 | 0 | 0 | 15 | 0 | 95 | 16 | 90 | 2 | 2 | 0 | 2 | 1 |
| pre | 198 | 0 | 3 | 100 | 0 | 109 | 0 | 70 | 0 | 100 | 10 | 80 | 5 | 0 | 0 | 1 | 0 | 1 | 1 | |
| pre | 117 | 0 | 20 | 101 | 0 | 88 | 0 | 0 | 0 | 100 | 90 | 90 | 0 | 24 | 9 | 1 | 0 | 1 | 1 | |
| pre | 162 | 0 | 15 | 98 | 0 | 112 | 0 | 85 | 0 | 100 | 70 | 90 | 0 | 0 | 3 | 1 | 0 | 1 | 1 | |
| pre | 203 | 0 | 10 | 98 | 0 | 120 | 0 | 20 | 0 | 80 | 50 | 80 | 0 | 15 | 0 | 1 | 1 | 1 | ||
| during | 89 | 141 | 5 | 45 | 0 | 101 | 100 | 90 | 0 | 95 | 19 | 60 | 0 | 0 | 0 | 2 | 0 | 1 | 1 | |
| post | 103 | 0 | 20 | 98 | 98 | 100 | 98 | 0 | 0 | 100 | 19 | 95 | 0 | 95 | 0 | 2 | 0 | 1 | 1 | |
| 6 | pre | 205 | 161 | 10 | 80 | 0 | 200 | 90 | 100 | 0 | 98 | 50 | 90 | 46 | 0 | 6 | 1 | 0 | 1 | 3 |
| during | 104 | 170 | 5 | 5 | 0 | 107 | 99 | 0 | 0 | 100 | 3 | 90 | 0 | 0 | 6 | 1 | 0 | 1 | 1 | |
Nuclear and cytosolic staining were evaluated by HistoScore. Membrance staining was clasiffied as 0, 1+, 2+ and 3+ following the recommendations for Her2 staining.
nuc: nuclear, cyto: cytosol, m: membrane.
Comparison of staining between groups at different time-points during EE2 treatment
Statistical comparisons were performed in two ways; one was to compare between Pre EE2 and (During+Post) EE2 treatment (Table 2A), the other comparison was between all 3 groups, Pre, During and Post EE2 treatment (Table 2B). In both comparisons, the expression of 4 genes appeared to be regulated by EE2 treatment. ER staining was quite strong before EE2 treatment; however it was significantly down-regulated by EE2 treatment. In contrast virtually no PgR staining was detectable before EE2 treatment, but it was significantly up-regulated after treatment. Before EE2 treatment, strong AR staining could be observed but was almost completely restricted to the nucleus; however nuclear AR staining was decreased and cytosolic AR was increased by EE2. BRCA1 staining was observed only in the nucleus before and during EE2 treatment, however EE2 significantly increased cytosolic staining of BRCA1 while nuclear staining tended to decrease. Staining of other proteins was not changed by EE2 treatment.
Table 2.
Statistical evaluation of nuclear and cytosol staining between different time points
| A | Pre EE2 (n=10) mean (S.E.) | During + Post EE2 (n=8) mean (S.E.) | P-value | B | Pre EE2 (n=10) mean (S.E.) | During EE2 (n=3) mean (S.E.) | Post EE2 (N=5) mean (S.E.) | P-value | ||
|---|---|---|---|---|---|---|---|---|---|---|
| ER | nuc | 166.7 (9.0) | 68.3 (18.2) | 0.0006** | ER | nuc | 166.7 (9.0) | 68.0 (28.8) | 68.6 (26.1) | 0.0024** |
| PgR | nuc | 19.2 (15.8) | 113.1 (24.1) | 0.01* | PgR | nuc | 19.2 (15.8) | 171.3 (17.9) | 78.2 (26.8) | 0.0113* |
| Ki67 | nuc | 12.3 (1.96) | 22.5 (5.82) | 0.2 | Ki67 | nuc | 12.3 (1.96) | 10.0 (5.0) | 30.0 (7.07) | 0.052 |
| BRCA1 | nuc | 96.6 (5.79) | 64.8 (16.7) | 0.3 | BRCA1 | nuc | 96.6 (5.79) | 57.3 (34.3) | 69.4 (20.3) | 0.56 |
| cyto | 0 (0) | 36.6 (17.8) | 0,047* | cyto | 0 (0) | 0 (0) | 58.6 (23.9) | 0.0126* | ||
| AR | nuc | 119.2 (9.79) | 44.2 (17.2) | 0.039* | AR | nuc | 119.2 (.979) | 75.3 (28.7) | 25.6 (18.7) | 0.0065** |
| cyto | 14.0 (9.79) | 68.75 (15.7) | 0.0097** | cyto | 14.0 (9.79) | 98.0 (1.52) | 51.2 (22.0) | 0.0092** | ||
| PI3K | cyto | 83.3 (9.39) | 95.0 (5.73) | 0.31 | PI3K | cyto | 83.3 (9.3) | 98.3 (1.66) | 93.0 (9.41) | 0.53 |
| AKT | nuc | 44.3 (11.3) | 37.2 (12.6) | 0.72 | AKT | nuc | 44.3 (11.3) | 35.6 (25.0) | 38.2 (16.0) | 0.9 |
| cyto | 75.2 (9.69) | 76.8 (11.8) | 0.58 | cyto | 75.2 (9.69) | 83.3 (12.0) | 73.0 (18.3) | 0.81 | ||
| pAKT | nuc | 7.2 (4.5) | 17.2 (16.5) | 0.63 | pAKT | nuc | 7.2 (4.5) | 0 (0) | 27.6 (26.3) | 0.44 |
| cyto | 25.9 (11.4) | 23.1 (15.1) | 0.57 | cyto | 25.9 (11.4) | 0 (0) | 37.0 (22.6) | 0.36 | ||
| IGT1Rb | cyto | 52.5 (13.3) | 32.5 (16.1) | 0.37 | IGT1Rb | cyto | 52.5 (13.3) | 30.0 (30.0) | 34.0 (21.4) | 0.64 |
| TUNEL | 5.1 (2.42) | 3.0 (1.26) | 0.74 | TUNEL | 5.1 (2.44) | 2.0 (2.0) | 3.6 (1.74) | 0.75 | ||
The Wilcoxon test/the Kruskal-Wallis test was used to analyze the significance of differences between groups.
nuc: nuclear, cyto: cytosol.
*<0.05, **<0.01.
When expression of membrane proteins; Her2, Fas, TGFβR1 and IGF1Rb were analyzed, no differences were observed between groups (Data not shown).
Comparison of staining between individuals at different time-points during EE2 treatment
The therapeutic effect and duration of EE2 treatment were: patient 1: responder 14 m, patient 2: responder 14 m, patient 3: non-responder 4 m, patient 4: responder 12 m, patient 5: responder 8 m, and patient 6: responder still receiving ongoing treatment for more than 14 m. To investigate differences in the regulation of selected genes underlying the different therapeutic effects, we compared differences in expression between Pre- and (During+Post) treatment with the exception of membrane staining. Results are shown in Figure 2. This study was not evaluated statistically because there was only one patient who did not respond to EE2, however, this patient appeared to show a different pattern of PgR expression to the responders, with no increase after EE2 treatment even though ER expression was drastically reduced, AKT expression seemed to decrease whereas pAKT expression increased after treatment both in the nucleus and the cytosol but especially in the nucleus.
Figure 2.
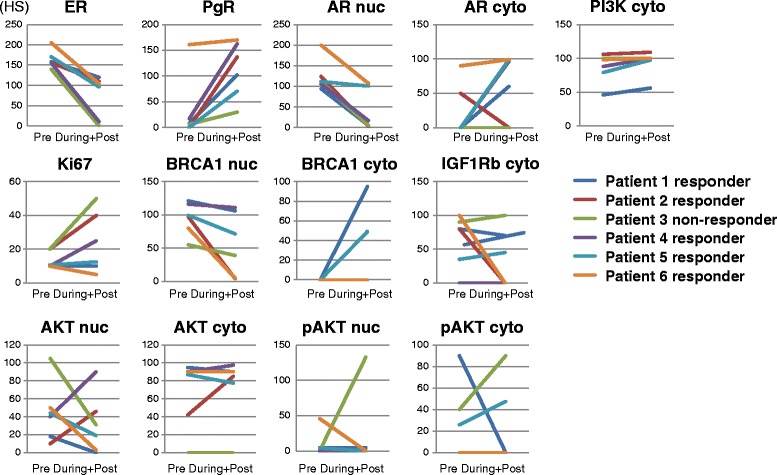
Comparison of expression between Pre and (During+Post) EE2 treatment in individual patients. Patients 1,2,4,5 and 6 responded to EE2 treatment whereas patient 3 did not. The average HS was calculated for a patient if they had several samples in a group.
Discussion
Hormone therapy is important in breast cancer treatment and around 70% of patients are categorized as suitable for hormone therapy (American Cancer Society 2013http://www.cancer.org/cancer/breastcancer/detailedguide/breast-cancer-treating-hormone-therapy). Hormone treatment can be administered orally or by one shot injection, which has less influence on bodily functions so that patients can continue daily life as they are used to (Iwase 2008). There are a variety of different medicines currently available, including anastrozole, letrozole and exemestane as AIs; tamoxifen and raloxifene as estrogen receptor modulators; and fulvestrant as an ER downregulator. Consequently, even if a patient fails to respond to one type of therapy, they can be switched to another medicine with a different structure or method of action and if that also fails, they can again be switched to another medication. Hormone therapy allows patients to maintain a better quality of life for a certain duration before chemotherapy is applied (Carlson et al. 2012).
In breast cancer samples, ER and PgR expression status are evaluated by immunohistochemistry. If a sample shows more than 1% staining for one of these receptors, it is classified as hormone receptor-positive and hormone treatment is applicable (Hammond et al. 2010). Comprehensive gene analysis has allowed breast cancers to be categorized according to their intrinsic subtype and Luminal A, a group with high expression of the ER, is considered to be highly responsive to hormone therapy (Sorlie et al. 2001). Therefore, ER and PgR status are very important for the application of hormone therapy, especially ER status. However, approximately 30% of ER-positive cases do not respond favorably to hormone therapy (Rubens and Hayward 1980) and in addition a subset of the population may become resistant after long-term hormone treatment even though they remain ER positive (Jordan and Ford 2011).
Recently, it was reported that EE2 administration was effective in treating hormone therapy-resistant ER-positive breast cancer patients (Ellis et al. 2009; Iwase et al. 2013). EE2 is a derivative of estrogen, therefore this treatment sounds in opposition to endocrine therapy, which is considered to act by blocking estrogen function. This paradoxical therapy can postpone the necessity for administration of chemotherapy because after EE2 treatment, normal anti-estrogenic agents become effective again even if treatment with the same medication failed before.
The very best point to start EE2 treatment is not yet understood. In a trial of EE2 by Ellis et al., eligible patients were those with metastatic breast cancer treated with AIs at least 24 weeks progression-free survival, or relapse after two or more years of adjuvant AIs (Ellis et al. 2009). In our study, our inclusion criteria were: postmenopausal women with breast cancer (not less than 50 years old) who had progressive disease, and who were receiving sequential treatments with AIs or Selective estrogen receptor modulators (SERMs) (the university hospital medical information network (UMIN) Clinical Trials Registry (UMIN000002831) http://www.umin.ac.jp/ctr/index.htm). It would be a great benefit for ER-positive patients if we could distinguish between hormone therapy insensitivity and EE2 sensitivity. So far 18 patients have been registered for EE2 treatment in our department after long-term Als treatment, and have shown very interesting results; the response rate was 50% and the clinical benefit was 56% (Iwase et al. 2013). Furthermore, we successfully obtained 23 tissue samples from 6 patients. Therefore, first of all, we focused on 13 genes and factors, which are related to cancer progression and performed immunostaining to see how these genes were regulated during EE2 treatment in this study.
The genes which we selected could be divided into 4 categories, hormone-related factors: ER, PgR and AR; growth/proliferative factors: Her2, IGF1Rb, TGFβR1 and Ki67; intracellular signaling factors: AKT, pAKT and PI3K; and apoptosis-related factors: BRCA1, Fas and TUNEL.
Before EE2, i.e. under estrogen-depleted conditions, ER and AR staining was quite strong while virtually no PgR staining was observed, as we had expected. In a comparison among samples of pre-, during and post-treatment or between pre- and (during+post)-treatment, only ER, PgR, AR and BRCA1 showed any significant difference between the treatment groups. Comparison among pre-, during- and post-treatment or between pre- and (during+post)-treatment showed that ER was significantly down-regulated by EE2 treatment whereas PgR was up-regulated (Table 2A,B). Nuclear AR staining was down-regulated whereas cytosolic AR was up-regulated by EE2. ER, PgR and AR are members of the nuclear receptor superfamily and are closely related to estrogen. PgR is well-known as a down-stream gene of ER (Lin et al. 2004) therefore this result strongly suggests that EE2 activates ER signaling. AR has a strong relationship with ER either directly (Panet-Raymond et al. 2000; Peters et al. 2009) or by regulation of their ligands. Transportation and translocation of the AR to the nucleus is performed by its ligand androgen (Callaway et al. 1982). Decreasing androgen concentrations during EE2 treatment could be a reason for this observation; androgen levels were rather high due to AI treatment but decreased to relatively low levels after EE2 treatment as has been observed in prostate cancer treatment (Brunton 2006. The other notable finding was that cytosolic staining of BRCA1 was significantly up-regulated by EE2 whereas nuclear staining tended to decrease (Table 2). BRCA1 and BRCA2 are tumor suppressor genes and these mutations were strongly associated with hereditary breast cancer (Yoshida and Miki 2004; Duncan et al. 1998). BRCA1 is responsible for repairing DNA and when normally expressed it helps repair damaged DNA or destroys cells if the DNA cannot be repaired. BRCA1 staining was mainly observed in nuclei but was also present in the cytosol. BRCA1 nuclear staining was frequently reduced in breast tumor tissue compared to normal tissue. The protein stained in the cytosol was considered to be a mutated form of BRCA1 (Kashima et al. 2000; Tulchin et al. 2013) and loss of nuclear BRCA1 expression is associated with a highly proliferative tumor phenotype (Jarvis et al. 1998). Estrogen treatment upregulates BRCA1 expression but has no effect on cellular localization (Romagnolo et al. 1998). Our results showed that when EE2 treatment ends i.e. in the post-EE2 group, tumors became resistant to EE2 and started to grow again as shown by the Ki67 index which tended to increase after treatment. We were unable to identify the precise reason for BRCA1 translocation; however it may be related to tumor re-growth.
One interesting finding was that EE2 activated ER signaling, as we observed in PgR regulation; however, EE2 did not stimulate cell growth. Of the 6 patients in this study, 5 patients responded indicating that EE2 suppressed tumor growth. ER signaling occurs not only via the classical ER genomic pathway through the estrogen responsive element (ERE), but also by cross-talk between growth factor receptors, the so called non-genomic pathway, and might involve acquisition of hormone therapy resistance (Johnston 2010). Comparison between patients suggested the possibility of differences in the regulation of PgR and AKT/pAKT in the non-responsive patient (Figure 2). It has been reported that long-term estrogen depletion involves a possible contribution of the Akt pathway to the phosphorylation of ER (Fujiki et al. 2014) in breast cancer cells. We could speculate; 1st that EE2 might act in the same manner as E2; however, transcriptional circumstances are changed during long-term hormone treatment; 2nd that EE2 could show differences in conformation and induce recruitment of different co-factors as other SERMs do. Transcriptional pathways include not only the direct genomic pathway but also membrane or non-genomic pathways. Stimulation of another pathway or an imbalance of activation could affect the response to EE2 and hormone resistance. The results of gene expression profiling show wide variations between patients and we need more cases and samples to conclude whether long term estrogen depletion causes activation or inactivation of estrogen receptor signaling (Martin et al. 2005; Martin et al. 2003), Non-responder might have dysfunction of estrogen-ER signaling and might be stimulated cell proliferation by non-genomic pathway.
The biology of estrogen-depleted cells was investigated by cell culture, which indicated that estrogen depleted estrogen dependent cells increased ER expression and became sensitive to estrogen. These cells were able to grow at lower doses of estrogen (Martin et al. 2005). Additionally, treatment with estrogen within the normal range did not increase proliferation but induced apoptosis in estrogen-depleted cells (Martin et al. 2005). Recently it was reported that genomic mutation of the ER itself could occur during long-term hormone therapy (Jeselsohn et al. 2014). This mutated ER then becomes hypersensitive to estrogen and is able to grow even with super low amounts of estrogen, suggesting that this is one of the possible mechanisms by which these cells develop resistance to hormone therapy. Ellis et al. investigated the effects of different concentrations of EE2, 6 mg or 30 mg per day (Ellis et al. 2009) and reported that the effect of 6 mg was similar to that obtained with a dose of 30 mg. Therefore, the lower dosage of 6 mg was sufficient to examine its function further. In our study, all 18 patients were administered 6 mg per day and showed a response rate of 50% and a clinical benefit of 56% (Iwase et al. 2013).
Conclusions
EE2 is a truly effective and beneficial agent; however we need to accumulate further knowledge to understand how to apply this medicine: its dosage, application and criteria. Additionally, the mechanism underlying the effects of EE2 acting through the ER is still not clearly understood.
In this study, the total number of samples was too small to analyze and reach a valid conclusion concerning the mechanism of hormone therapy resistance. We will continue to enter the data from the other patients in the EE2 trial. In the future, it will be obligatory to perform large scale gene expression profiling because an unknown gene or gene family may be involved in changing critical characteristics. Further investigation is awaited.
Materials and methods
Patients and samples
This EE2 trial was approved by the institutional review board of Kumamoto University Hospital and registered to UMIN center (UMIN000002831). This study was informed to all patients and we obtained consent to participate in the study and consent to publish. The criteria for registration have previously been described in detail (Iwase et al. 2013). Briefly, all patients had received prior sequential hormone therapies including chemotherapy. Each patient’s pretreatment history is shown in Table 3.
Table 3.
Characteristics of patients and the timing of tissue collection
| Patient number/age | Previous treatment | EE2 response/biopsy site | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LHRHa | LHRHa | LHRHa | |||||||||||||||||
| 1 | + Tam | + ANA | + L | TS-1 | VNR | PTX | E | EE2 | L | EE2 | |||||||||
| 10m | 12m | 3m | 12m | 21m | 3m | 4m | 14m | 5m | Responder liver, local | ||||||||||
| 59 | ^ | ^ | |||||||||||||||||
| B | A | ||||||||||||||||||
| 2 | TS-1 + | Responder lymph node | |||||||||||||||||
| DTX | ANA | E | EC | DTX | XC | L | EE2 | L | EE2 | ||||||||||
| 15m | 13m | 5m | 13m | 24m | 6m | 7m | 14m | 6m | |||||||||||
| 62 | ^ | ^ | |||||||||||||||||
| B | A | ||||||||||||||||||
| 3 | H + | Non-responder local | |||||||||||||||||
| FEC | XT | ANA | E | EE2 | FUL | PTX | |||||||||||||
| 3m | 12m | 8m | 2m | 4m | 3m | ||||||||||||||
| 56 | ^ | ^ | ^ | ^ | ^ | ||||||||||||||
| B | B | B | A | A | |||||||||||||||
| E | MPA | Tam | L | TS-1 | ANA | Tor | EE2 | L | Responder skin | ||||||||||
| 4 | 8m | 9m | 2m | 13m | 8m | 2m | 12m | ||||||||||||
| 64 | ^ | ^ | ^ | ^ | |||||||||||||||
| B | B | D | A | ||||||||||||||||
| AC | H | L | Tor | E+H | PTX | L | EE2 | FuL | Responder local | ||||||||||
| 5 | 8m | 7m | 8m | 6m | 1m | 4m | 3m | 8m | |||||||||||
| 58 | ^ | ^ | ^ | ^ | ^ | ^ | ^ | ^ | |||||||||||
| B | B | B | B | B | B | D | A | ||||||||||||
| L | ANA | XC | TS-1 | ANA | Tor | PTX | ANA | EE2 | Responder local | ||||||||||
| 6 | 16m | 3m | 9m | 3m | 2m | 7m | 11m | 3m | 14m -- | ||||||||||
| 83 | ^ | ^ | |||||||||||||||||
| B | B | ||||||||||||||||||
^: the time point when tissue was obtained.
B: before EE2 treatment D: During EE2 treatment; A: After EE2 treatment.
Sample shown in bold letter were used in this study.
Abbreviations: ANA: anastozole; AC: doxorubicine + cyclophosphamide; DTX: docetaxel; E: exemestane; EC: Epirubicine + cydophoshamide; FEC: cydoshsphamide + epirubine+5-FU; FUL: fulvestrant; H: herceptine; L: letrozole; MPA: medroxyprogesterone; PTX: paclitaxel; Tam: tamoxifen; Tor: toremifeme; VNR: vinorelubin; XC: capecitabine + cyclophosphamide; XT: capecitabine + docetaxel.
A total of 23 tissue samples were obtained from 6 patients; however, 4 pre-treatment samples and one post-treatment sample were not evaluated in this study to avoid the complicating effects of chemotherapy. Therefore, 18 tissues from 6 patients were used in this study. All patients had been administered EE2 after long-term treatment with multiple anti-hormone agents. Tissue samples were collected by core needle biopsy from metastatic lesions of patients who had undergone EE2 treatment at certain clinical points, as shown in Table 3. Of the 18 samples, 10 samples were obtained before EE2 treatment, 3 were collected during treatment and 5 were obtained after treatment. All samples were used for the immunohistochemical (IHC) study to compare expression during this time period.
Antibody, immunohistochemical methods and evaluations
A total of 13 different staining procedures were performed in this study, including immunostaining for 11 breast cancer-related genes plus one antibody to detect phosphorylated protein and TdT-mediated dUTP nick end labeling (TUNEL). These 13 targeted proteins or markers are functionally categorized into 6 groups: nuclear receptors (ERα, PgR, AR); growth factors (Her2, IGF1Rb, TGFβR1); tumor suppressor genes (BRCA1); cell proliferation (Ki-67, TUNEL); apoptosis related (Fas); intracellular signal transduction (AKT, pAKT, PI3K). Information on all the antibodies is shown in Table 3.
All formalin-fixed, paraffin-embedded specimens were cut into 4-μm sections, deparaffinized, heated 3 times for 5 min each in citrate buffer (pH 7) in a 1,000 W microwave for antigen retrieval and incubated for 10 min in distilled water containing 3% hydrogen peroxide. The primary antibody was applied after blocking, and incubated at 4°C overnight. Detection and visualization was performed by several methods as indicated in Table 4, according to the manufactures’ protocol. As a negative control, parallel sections were immunostained without exposure to primary antibodies. No immunoreactivity was observed in these sections.
Table 4.
List of antibodies and methods of visualization
| Antibody | Source | Dilution | Visualization | |
|---|---|---|---|---|
| ER | 1D5 (Dako) | Mouse monoclonal | 1:50 | Histofine Simple stain MAX-PO° (Nichirei) |
| PgR | PgR636 (Dako) | Mouse monoclonal | 1:800 | Histofine Simple stain MAX-PO° (Nichirei) |
| AR | NCL-AR-318 (Leica) | Mouse monoclonal | 1:100 | VECTASTAIN Elite ABC (Vector) |
| Ki67 | MIB-1 (Dako) | Mouse monoclonal | 1:50 | I-VIEW DAB universal kit (Roche) |
| BRCA1 | MS110 (Abcam) | Mouse monoclonal | 1:200 | Histofine Simple stain MAX-PO° (Nichirei) |
| IGF1Rb | #3027 (CST) | Rabbit polyclonal | 1:600 | VECTASTAIN Elite ABC (Vector) |
| TGFβR1 | 8A11 (Abcam) | Mouse monoclonal | 1:100 | VECTASTAIN Elite ABC (Vector) |
| Fas | C18C12 #4233 (CST) | Rabbit monoclonal | 1:480 | SignalStain° boost (CST) |
| AKT | 11E7 #4685 (CST) | Rabbit monoclonal | 1:100 | SignalStain° boost (CST) |
| pAKT (Ser473) | D9E #4060 (CST) | Rabbit monoclonal | 1:50 | SignalStain° boost (CST) |
| PI3K p85 | M253 (Abcam) | mouse monoclonal | 1:200 | VECTASTAIN Elite ABC (Vector) |
| Her2 | anti-HER2 (4B5) (Roche) | Rabbit monoclonal | I-VIEW DAB universal kit (Roche) | |
| TUNEL | TUNEL Apoptosis Detection Kit (Millipore) | |||
This study was reported according to the Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK) criteria (McShane et al. 2005). Nuclear staining and cytoplasmic staining were independently scored by HS. The HS represented the product of the staining intensity (0: negative, 1: weak, 2: moderate and 3: strong) and the percentage of positive cells (0–100%) for each sample, with a maximum HS of 300. We counted approximately 100 cancer cells in five randomly-chosen microscopic fields. Cytomembrane staining was evaluated according to the recommendation for Her2 staining (Wolff et al. 2013), with staining categorized into scores of 0, 1+, 2+ and 3+.
Statistical analysis
The nuclear or cytosolic staining of each protein was statistically analyzed. The Wilcoxon signed-rank test was used for comparisons between 2 groups and the Kruskal-Wallis test was used for 3 groups. Membrane staining was analyzed using Student’s t-test between groups. P values < 0.05 were considered a significant result. All analyses were performed using JMP software version 10.0.1 for Windows (SAS institute Japan, Tokyo, Japan).
Acknowledgements
The authors are thankful to Y. Azakami for excellent technical support. This work was supported by a Grant in Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan #26461952 (YO).
Abbreviations
- AI
Aromatase inhibitor
- AKT
Protein kinase B
- AR
Androgen receptor
- BRCA1
Breast cancer susceptibility gene I
- E2
17β-estradiol
- EE2
Ethinyl estradiol
- ER
Estrogen receptor
- ERE
Estrogen responsive element
- Her2
Human EGFR-related 2
- HS
Histo-score
- IGF1Rb
Insulin-like growth factor I receptor beta
- pAKT
Phosphorylated AKT
- PgR
Progesterone receptor
- PI3K
Phosphoinositide 3-kinase
- REMARK
Recommendations for Tumor Marker Prognostic Studies
- SERMs
Selective estrogen receptor modulators
- TGFβR1
Transforming growth factor beta receptor 1
- TUNEL
TdT-mediated dUTP nick end labeling
- UMIN
The University Hospital Medical Information Network
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YO participated in the design of the study, carried out the evaluation of immunostaining and drafted the manuscript. TaT carried out the immunostaining and its evaluation and performed the statistical analysis and coordination and helped to draft the manuscript. YY, MY-I, MH, AS, SF and TeT contributed to collect human breast cancer samples and obtained clinical information. HI participated in the design of the study, and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Yoko Omoto, Email: yomoto@kumamoto-u.ac.jp.
Takashi Takeshita, Email: henamon@gmail.com.
Yutaka Yamamoto, Email: ys-yama@triton.ocn.ne.jp.
Mutsuko Yamamoto-Ibusuki, Email: mibusuki@kumamoto-u.ac.jp.
Mitsuhiro Hayashi, Email: mhayashi@kumamoto-u.ac.jp.
Aiko Sueta, Email: aikos0214@yahoo.co.jp.
Saori Fujiwara, Email: mirecout_5963@yahoo.co.jp.
Tetsuya Taguchi, Email: ttaguchi@koto.kpu-m.ac.jp.
Hirotaka Iwase, Email: hiwase@kumamoto-u.ac.jp.
References
- American Cancer Society (2013) Hormone therapy for breast cancer. http://www.cancer.org/cancer/breastcancer/detailedguide/breast-cancer-treating-hormone-therapy. Accessed 20 Oct 2014.
- Brunton LL. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 11. New York: The McGraw-Hill Companies, Inc.; 2006. [Google Scholar]
- Callaway TW, Bruchovsky N, Rennie PS, Comeau T. Mechanisms of action of androgens and antiandrogens: effects of antiandrogens on translocation of cytoplasmic androgen receptor and nuclear abundance of dihydrotestosterone. The Prostate. 1982;3(6):599–610. doi: 10.1002/pros.2990030609. [DOI] [PubMed] [Google Scholar]
- Carlson RW, Allred DC, Anderson BO, Burstein HJ, Edge SB, Farrar WB, Forero A, Giordano SH, Goldstein LJ, Gradishar WJ, Hayes DF, Hudis CA, Isakoff SJ, Ljung BM, Mankoff DA, Marcom PK, Mayer IA, McCormick B, Pierce LJ, Reed EC, Smith ML, Soliman H, Somlo G, Theriault RL, Ward JH, Wolff AC, Zellars R, Kumar R, Shead DA, National Comprehensive Cancer N Metastatic breast cancer, version 1.2012: featured updates to the NCCN guidelines. Journal of the National Comprehensive Cancer Network. 2012;10(7):821–829. doi: 10.6004/jnccn.2012.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner G, Schnorr D, Stahl F, Rohde W. Successful treatment of prostatic cancer with the orally active depot estrogen ethinylestradiol sulfonate (Turisteron) Exp Clin Endocrinol. 1985;86(2):190–196. doi: 10.1055/s-0029-1210486. [DOI] [PubMed] [Google Scholar]
- Duncan JA, Reeves JR, Cooke TG. BRCA1 and BRCA2 proteins: roles in health and disease. Molecular Pathology. 1998;51(5):237–247. doi: 10.1136/mp.51.5.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, Dickler MN, Silverman P, Fleming GF, Kommareddy A, Jamalabadi-Majidi S, Crowder R, Siegel BA. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA. 2009;302(7):774–780. doi: 10.1001/jama.2009.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki N, Konno H, Kaneko Y, Gohno T, Hanamura T, Imami K, Ishihama Y, Nakanishi K, Niwa T, Seino Y, Yamaguchi Y, Hayashi S. Estrogen response element-GFP (ERE-GFP) introduced MCF-7 cells demonstrated the coexistence of multiple estrogen-deprivation resistant mechanisms. Journal of Steroid Biochemistry and Molecular Biology. 2014;139:61–72. doi: 10.1016/j.jsbmb.2013.08.012. [DOI] [PubMed] [Google Scholar]
- Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, Hicks DG, Lester S, Love R, Mangu PB, McShane L, Miller K, Osborne CK, Paik S, Perlmutter J, Rhodes A, Sasano H, Schwartz JN, Sweep FC, Taube S, Torlakovic EE, Valenstein P, Viale G, Visscher D, Wheeler T, Williams RB, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28(16):2784–2795. doi: 10.1200/JCO.2009.25.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher RA NA. Contraceptive Technology, vol 18th. New York: Ardent Media; 2004. Combined Hormonal Contraceptive Methods; pp. 391–460. [Google Scholar]
- Iwase H. Current topics and perspectives on the use of aromatase inhibitors in the treatment of breast cancer. Breast cancer. 2008;15(4):278–290. doi: 10.1007/s12282-008-0071-y. [DOI] [PubMed] [Google Scholar]
- Iwase H, Yamamoto Y, Yamamoto-Ibusuki M, Murakami KI, Okumura Y, Tomita S, Inao T, Honda Y, Omoto Y, Iyama KI. Ethinylestradiol is beneficial for postmenopausal patients with heavily pre-treated metastatic breast cancer after prior aromatase inhibitor treatment: a prospective study. British journal of cancer. 2013;109(6):1537–1542. doi: 10.1038/bjc.2013.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis EM, Kirk JA, Clarke CL. Loss of nuclear BRCA1 expression in breast cancers is associated with a highly proliferative tumor phenotype. Cancer Genetics and Cytogenetics. 1998;101(2):109–115. doi: 10.1016/S0165-4608(97)00267-7. [DOI] [PubMed] [Google Scholar]
- Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo JA, Cristofanilli M, Gomez H, Arteaga CL, Giltnane J, Balko JM, Cronin MT, Jarosz M, Sun J, Hawryluk M, Lipson D, Otto G, Ross JS, Dvir A, Soussan-Gutman L, Wolf I, Rubinek T, Gilmore L, Schnitt S, Come SE, Pusztai L, Stephens P, Brown M, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20(7):1757–1767. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SR. New strategies in estrogen receptor-positive breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(7):1979–1987. doi: 10.1158/1078-0432.CCR-09-1823. [DOI] [PubMed] [Google Scholar]
- Jordan VC, Ford LG. Paradoxical clinical effect of estrogen on breast cancer risk: a "new" biology of estrogen-induced apoptosis. Cancer Prev Res. 2011;4(5):633–637. doi: 10.1158/1940-6207.CAPR-11-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansra S, Yamagata S, Sneade L, Foster L, Ben-Jonathan N. Differential effects of estrogen receptor antagonists on pituitary lactotroph proliferation and prolactin release. Mol Cell Endocrinol. 2005;239(1–2):27–36. doi: 10.1016/j.mce.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Kashima K, Oite T, Aoki Y, Takakuwa K, Aida H, Nagata H, Sekine M, Wu HJ, Hirai Y, Wada Y, Yamamoto K, Hasegawa K, Sonoda T, Maruo T, Nagata I, Ohno M, Suzuki M, Kobayashi I, Kuzuya K, Takahashi T, Torii Y, Tanaka K. Screening of BRCA1 mutation using immunohistochemical staining with C-terminal and N-terminal antibodies in familial ovarian cancers. Japanese Journal of Cancer Research. 2000;91(4):399–409. doi: 10.1111/j.1349-7006.2000.tb00959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Strom A, Vega VB, Kong SL, Yeo AL, Thomsen JS, Chan WC, Doray B, Bangarusamy DK, Ramasamy A, Vergara LA, Tang S, Chong A, Bajic VB, Miller LD, Gustafsson JA, Liu ET. Discovery of estrogen receptor alpha target genes and response elements in breast tumor cells. Genome biology. 2004;5(9):R66. doi: 10.1186/gb-2004-5-9-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LA, Farmer I, Johnston SR, Ali S, Dowsett M. Elevated ERK1/ERK2/estrogen receptor cross-talk enhances estrogen-mediated signaling during long-term estrogen deprivation. Endocrine-related cancer. 2005;12(Suppl 1):S75–84. doi: 10.1677/erc.1.01023. [DOI] [PubMed] [Google Scholar]
- Martin LA, Farmer I, Johnston SR, Ali S, Marshall C, Dowsett M. Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. J Biol Chem. 2003;278(33):30458–30468. doi: 10.1074/jbc.M305226200. [DOI] [PubMed] [Google Scholar]
- McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM, Statistics Subcommittee of the NCIEWGoCD Reporting recommendations for tumor marker prognostic studies (REMARK) Journal of the National Cancer Institute. 2005;97(16):1180–1184. doi: 10.1093/jnci/dji237. [DOI] [PubMed] [Google Scholar]
- Panet-Raymond V, Gottlieb B, Beitel LK, Pinsky L, Trifiro MA. Interactions between androgen and estrogen receptors and the effects on their transactivational properties. Mol Cell Endocrinol. 2000;167(1–2):139–150. doi: 10.1016/S0303-7207(00)00279-3. [DOI] [PubMed] [Google Scholar]
- Peters AA, Buchanan G, Ricciardelli C, Bianco-Miotto T, Centenera MM, Harris JM, Jindal S, Segara D, Jia L, Moore NL, Henshall SM, Birrell SN, Coetzee GA, Sutherland RL, Butler LM, Tilley WD. Androgen receptor inhibits estrogen receptor-alpha activity and is prognostic in breast cancer. Cancer Res. 2009;69(15):6131–6140. doi: 10.1158/0008-5472.CAN-09-0452. [DOI] [PubMed] [Google Scholar]
- Romagnolo D, Annab LA, Thompson TE, Risinger JI, Terry LA, Barrett JC, Afshari CA. Estrogen upregulation of BRCA1 expression with no effect on localization. Molecular Carcinogenesis. 1998;22(2):102–109. doi: 10.1002/(SICI)1098-2744(199806)22:2<102::AID-MC5>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Rubens RD, Hayward JL. Estrogen receptors and response to endocrine therapy and cytotoxic chemotherapy in advanced breast cancer. Cancer. 1980;46(12 Suppl):2922–2924. doi: 10.1002/1097-0142(19801215)46:12+<2922::AID-CNCR2820461435>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulchin N, Ornstein L, Dikman S, Strauchen J, Jaffer S, Nagi C, Bleiweiss I, Kornreich R, Edelmann L, Brown K, Bodian C, Nair VD, Chambon M, Woods NT, Monteiro AN. Localization of BRCA1 protein in breast cancer tissue and cell lines with mutations. Cancer Cell Int. 2013;13(1):70. doi: 10.1186/1475-2867-13-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, Allred DC, Bartlett JM, Bilous M, Fitzgibbons P, Hanna W, Jenkins RB, Mangu PB, Paik S, Perez EA, Press MF, Spears PA, Vance GH, Viale G, Hayes DF, American Society of Clinical O, College of American P Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31(31):3997–4013. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Science. 2004;95(11):866–871. doi: 10.1111/j.1349-7006.2004.tb02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]